

Abstract
CacyBP/SIP [calcyclin-binding protein/Siah-1 [seven in absentia homolog 1 (Siah E3 ubiquitin protein ligase 1)] interacting protein] is a multifunctional protein whose activity includes acting as an ERK1/2 phosphatase. We analyzed dimerization of mouse CacyBP/SIP in vitro and in mouse neuroblastoma cell line (NB2a) cells, as well as the structure of a full-length protein. Moreover, we searched for the CacyBP/SIP domain important for dimerization and dephosphorylation of ERK2, and we analyzed the role of dimerization in ERK1/2 signaling in NB2a cells. Cell-based assays showed that CacyBP/SIP forms a homodimer in NB2a cell lysate, and biophysical methods demonstrated that CacyBP/SIP forms a stable dimer in vitro. Data obtained using small-angle X-ray scattering supported a model in which CacyBP/SIP occupies an anti-parallel orientation mediated by the N-terminal dimerization domain. Site-directed mutagenesis established that the N-terminal domain is indispensable for full phosphatase activity of CacyBP/SIP. We also demonstrated that the oligomerization state of CacyBP/SIP as well as the level of post-translational modifications and subcellular distribution of CacyBP/SIP change after activation of the ERK1/2 pathway in NB2a cells due to oxidative stress. Together, our results suggest that dimerization is important for controlling phosphatase activity of CacyBP/SIP and for regulating the ERK1/2 signaling pathway.—Topolska-Woś, A. M., Shell, S. M., Kilańczyk, E., Szczepanowski, R. H., Chazin, W. J., Filipek, A. Dimerization and phosphatase activity of calcyclin-binding protein/Siah-1 interacting protein: the influence of oxidative stress.
Keywords: CacyBP/SIP, ERK1/2, MAPK phosphatase, protein structure, NB2a cells
CacyBP/SIP was originally discovered in Ehrlich ascites tumor cells as an S100A6-binding protein (1, 2) and then found in human cells as an interacting partner of Siah-1 (3). CacyBP/SIP is highly expressed in brain and spleen (2), and within brain tissue, it is mainly localized in neuronal cells (4).
Although CacyBP/SIP was first identified as S100A6 ligand, it is now well established that it interacts with other members of the S100 family, as well as other proteins, including Siah-1 and S-phase kinase-associated protein 1 (Skp1) (3), tubulin (5), actin (6), and tropomyosin (7). Recently, it has been shown that CacyBP/SIP binds ERK1/2 and acts as a phosphatase toward these kinases (8, 9), which leads to down-regulation of the transcription factor ETS domain-containing protein (ELK-1) in vitro and in the nuclear fraction of NB2a cells.
CacyBP/SIP is composed of an N-terminal helix-turn-helix, central CS (domain present in CHORD containing proteins and in Sgt1), and C-terminal disordered SGS (Sgt1-specific) domains (10, 11). The modular multidomain structure of CacyBP/SIP made crystallization of the full-length protein difficult. However, high-resolution structures of the N-terminal domain were determined by NMR for mouse CacyBP/SIP (PDB code: 1YSM) and by X-ray crystallography for human CacyBP/SIP (PDB code: 2A26). An X-ray crystal structure of the CS domain of human CacyBP/SIP is also available (PDB code: 1X5M). Moreover, structural and biophysical analyses of CacyBP/SIP domains interacting with Siah-1 and Skp1 have been reported (10, 12, 13).
The oligomerization state of CacyBP/SIP has been proposed to be a dimer based on the crystal structure of the N-terminal domain of human CacyBP/SIP (12, 13). However, narrow line widths of this domain in NMR spectra of the mouse protein suggested the domain is monomeric in solution (10). Here, we report a systematic analysis in vitro of the oligomerization state of full-length mouse CacyBP/SIP and investigate its influence on the interaction with, and phosphatase activity toward, ERK1/2. These studies were extended to examine the properties of mouse CacyBP/SIP as a phosphatase in NB2a cells under conditions of oxidative stress that cause activation of the ERK1/2 signaling pathway.
MATERIALS AND METHODS
Culture of NB2a cells and preparation of protein lysates
Mouse neuroblastoma NB2a cells were grown in Eagle's Minimum Essential Medium containing 10% (v/v) fetal bovine serum, 25 mM sodium bicarbonate, penicillin (100 μg/ml), streptomycin (100 μg/ml), gentamycin (50 μg/ml), and fungizone (0.25 μg/ml). The media were changed every 2 to 3 days, and cells were passaged when confluent.
The cytoplasmic and nuclear fractions from NB2a cells were obtained using the NE-PER kit (nuclear and cytoplasmic extraction reagents; Thermo Fisher Scientific, Rockford, IL, USA) according to the manufacturer’s instruction. The total protein lysate was obtained by suspending samples in a buffer containing: 0.1% (v/v) Triton X-100, 20 mM Tris, pH 7.5, 50 mM KCl, 3 mM MgCl2, and protease inhibitors (Complete Mini EDTA-free; Roche, Indianapolis, IN, USA). Cells were homogenized mechanically by running 30 times through the needle. Afterward, the solution was centrifuged at 10,000 g for 10 minutes at 4°C. Protein concentration was estimated by the Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA) and samples containing 50 μg proteins from each fraction were analyzed by SDS-PAGE and Western blot.
Cell transfection
NB2a cells (70–80% confluent) were transfected with 8 μg of the expression plasmids: p3xFLAG-CMV-10-CacyBP/SIP or p3xFLAG-CMV-10 (as a control) using Lipofectamine2000 (Invitrogen, Camarillo, CA, USA) according to the manufacturer's protocol. After 24 hours, cells were harvested and the total protein lysate was prepared as described above.
H2O2 treatment
To induce an oxidative stress, differentiated NB2a cells were treated with hydrogen peroxide (H2O2). For that the medium devoid of antibiotics was supplemented with 0.002% (v/v) of H2O2. Cells were harvested after 3 hours to prepare samples for analysis. For the control, NB2a cells were incubated for 3 hours in medium without antibiotics and H2O2.
Coimmunoprecipitation
A 40 μl aliquot of 50% slurry of the anti-FLAG agarose beads (Sigma-Aldrich, St. Louis, MO, USA) equilibrated with immunoprecipitation (IP) buffer [10 mM Tris-HCl, pH 7.5, 50 mM KCl, 3 mM MgCl2, 0.1% (v/v) Triton X-100] was incubated 800 μg total protein lysate (input) for 2 hours at 4°C. After extensive washing in IP buffer, the bound proteins were eluted with 0.1 M glycine, pH 3.5, and analyzed by SDS-PAGE and Western blot.
Cross-linking experiment
Protein (50 μg) from cytoplasmic and nuclear fractions or from total protein lysate was incubated at 25°C for 30 minutes with a chemical zero-length cross-linking agent, EDC [1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride; Sigma-Aldrich], at final concentration of 2.5 mM. Reaction was quenched by addition of cold acetone (−20°C). After centrifugation at 10,000 g for 10 minutes, the pellet was suspended in loading buffer with 10 mM DTT and heated at 95°C for 15 minutes before SDS-PAGE and Western blot.
Immunofluorescence microscopy
NB2a cells grown on coverslips pretreated with 50 μg/ml poly-l-lysine (Sigma-Aldrich) were fixed with 3% (w/v) paraformaldehyde in PBS buffer (137 mM NaCl, 2.7 mM KCl, 4.3 mM NaH2PO4, 1.4 mM KH2PO4) for 20 minutes at room temperature (RT). The coverslips then were washed with PBS, incubated with 50 mM NH4Cl in PBS at RT for 10 minutes, and washed with PBS. Cells were then permeabilized for 5 minutes with 0.1% (v/v) Triton X-100 in PBS and washed again in PBS. For blocking, an incubation with 5% (v/v) FBS in PBS for 1 hour at RT was applied. After washing with PBS, coverslips were incubated with rabbit anti-CacyBP/SIP polyclonal antibody [Cell Signaling Technology, Danvers, MA, USA; diluted 1:50 in PBS containing 0.5% (v/v) FBS] at RT for 1 hour and afterward washed with PBS for 10 minutes. Then, either Alexa Fluor 488 goat anti-mouse or Alexa Fluor 594 goat anti-rabbit IgG antibodies [diluted 1:200 in PBS containing 0.5% (v/v) FBS; Molecular Probes, Eugene, OR, USA] were added and incubation was prolonged for 1 hour. After washing with PBS, incubation with DAPI (1:20,000) was carried out for 5 minutes, and then coverslips were immobilized on glass slides with Mowiol mounting solution. Cells were analyzed under Zeiss 780 Confocal Microscope (Carl Zeiss GmbH, Jena, Germany) with ×63 oil objective in the Laboratory of Confocal Microscopy (Nencki Institute of Experimental Biology, Warsaw, Poland).
Proximity ligation assay
For visualization of CacyBP/SIP homodimerization, the proximity ligation assay (PLA) (In situ PLA Technology, Olink Bioscience, Uppsala, Sweden) was used. NB2a cells were grown as described above for immunofluorescence microscopy. The reaction with primary polyclonal antibody (rabbit anti-CacyBP/SIP, 1:50) was carried out for 1.5 hours at 37°C. After washing, the incubation with anti-rabbit PLA plus and minus probes (1:4) was conducted for 2 hours at 37°C in a humidity chamber. All following steps were performed according to the manufacturer’s protocol and with the reagents and media provided in the PLA-kit. Simultaneously to the PLA, the standard immunofluorescence was applied to monitor the nuclear translocation of CacyBP/SIP upon H2O2 treatment.
Plasmids, protein expression, and purification
Plasmids p3xFLAG-CMV-10-CacyBP/SIP and p3xFLAG-CMV-10 for NB2a cell transfection or pET28-CacyBP/SIP and pET30a-ERK2 for bacteria transformation were prepared as described previously (5, 8, 14). Similarly, pET28a encoding CacyBP/SIP fragments covering residues 1–77, 74–178, 178–229 were prepared as described earlier (5, 10). Several mutants with mutations in the N-terminal domain of CacyBP/SIP were designed and prepared using site-directed mutagenesis. For PCR reaction the Phusion polymerase was used with primers listed in Table 1. The PCR products were treated with DpnI endonuclease to remove the template. All constructs were verified by sequence analysis. All His6-tagged CacyBP/SIP mutants were expressed in Escherichia coli BL21 strain and purified by gravity flow at RT using TALON Co2+ metal affinity chromatography (Clontech, Mountain View, CA, USA), followed by removal of the His6-tag with thrombin. Proteins were dialyzed against 150 mM NaCl, 20 mM Tris-HCl, pH 8.4, if necessary enriched with 5% (v/v) of glycerol and concentrated using centrifugal filters (Amicon; Millipore, Temecula, CA, USA). Size-exclusion chromatography (SEC) was applied as a last step of purification.
TABLE 1.
List of primers used for preparation of CacyBP/SIP mutants
| Mutant | Mutation | Template | Primers |
|---|---|---|---|
| CacyBP/SIP D11A/E14A | D11A | pET28a CacyBP/SIP | 5′-ttgcagaaagccctagaagaggtcaaagta-3′ |
| 5′-tactttgacctcttctagggctttctgcaa-3′ | |||
| E14A | pET28a CacyBP/SIP D11A | 5′-aaagccctagaagcggtcaaagtattgctg-3′ | |
| 5′-cagcaatactttgaccgcttctagggcttt-3′ | |||
| CacyBP/SIP D11H/E14K | D11H | pET28a CacyBP/SIP | 5′-caatactttgaccttttctaggtgtttctgcaactcttccaaaacggaagc-3′ |
| E14K | 5′-gcttccgttttggaagagttgcagaaacacctagaaaaggtcaaagtattg-3′ | ||
| CacyBP/SIP K25A/R26A | K25A | pET28a CacyBP/SIP | 5′-gtccactagggcaagactacgtgatac-3′ |
| 5′-gtatcacgtagtcttgccctagtggac-3′ | |||
| R26A | pET28a CacyBP/SIP K25A | 5′-gtacactagggcagcactacgtgatac-3′ | |
| 5′-gtatcacgtagtgctgccctagtggac-3′ | |||
| CacyBP/SIP K25E/R26E | K25E | pET28a CacyBP/SIP | 5′-ggaaaagtccactagggaagaactacgtgatactc-3′ |
| R26E | 5′-gagtatcacgtagttcttccctagacttttcc-3′ | ||
| CacyBP/SIP D11A/E14A/K25A/R26A | K25A | pET28a CacyBP/SIP D11A/E14A | 5′-caaagtattgctggaaaagtccactagggcagcactacgtgatactcttacaagtgaaaag-3′ |
| R26A | 5′-cttttcacttgtaagagtatcacgtagtgctgccctagtggacttttccagcaagcaatactttg-3′ | ||
| CacyBP/SIP S22D/T23D | S22D | pET28a CacyBP/SIP | 5′-agtattgctggaaaaggacactaggaaaagact-3′ |
| 5′-gtagtcttttcctagtgtccttttccagcaata-3′ | |||
| T23D | pET28a CacyBP/SIP S22D | 5′-agtattgctggaaaaggacgataggaaaagact-3′ | |
| 5′-gtagtcttttcctatcgtccttttccagcaata-3′ |
Substituted nucleotides are shown in boldface.
SDS-PAGE and Western blot
Gel electrophoresis with 12% (w/v) polyacrylamide containing 0.1% (w/v) SDS was performed by the method of Laemmli (15). Proteins were transferred electrophoretically onto nitrocellulose and identified using appropriate rabbit polyclonal anti-CacyBP/SIP or rabbit monoclonal anti-p-ERK1/2 antibodies (both from Cell Signaling, diluted 1:1000). After washing with TBS-T buffer [50 mM Tris pH 7.5, 200 mM NaCl, 0.05% (v/v) Tween 20] blots were allowed to react with secondary antibodies, either goat anti-mouse IgG (1:10,000; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) or goat anti-rabbit IgG (1:10,000; MP Biomedicals, Irvine, CA, USA) conjugated to horseradish peroxidase. After 3 washes with TBS-T and 2 washes with TBS (50 mM Tris pH 7.5, 200 mM NaCl) blots were developed with the ECL chemiluminescence kit (Bio-Rad), followed by exposure against Kodak X-ray film. The intensity of the protein bands was quantified using Ingenius instrument (Syngene Ingenious, Cambridge, UK) and analyzed using ImageJ software [U.S. National Institutes of Health (NIH) freeware] with PARP-1 (mouse monoclonal antibody; diluted 1:5,000; Alexis Biochemicals, San Diego, CA, USA) as a reference protein for the nuclear fraction and β-actin (mouse monoclonal horseradish peroxidase-linked; diluted 1:5000; Sigma-Aldrich) for the cytoplasmic fraction or total protein lysate.
2-Dimensional electrophoresis
Protein (200 µg) from total protein lysate, cytoplasmic, or nuclear fraction of neuroblastoma NB2a cells, precipitated with TCA and washed with cold acetone, was resuspended in loading buffer containing 8 M urea, 2% (w/v) (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate), 50 mM DTT, 0.2% (v/v) ampholine (Bio-Lyte pH 3-10; Bio-Rad). Samples were applied on the linear 7 cm long pH gradient (3–10) strips (Bio-Rad). After 1 h of incubation at RT, strips were covered with mineral oil (Bio-Rad) and incubated for 16 hours at RT. After a hydration step strips were applied into the Protean IEF Cell instrument (Bio-Rad) and isoelectrofocusing was carried out for 20 h with the maximal voltage of 10 kVh. The final voltage was 4000 V. Strips were then washed for 30 minutes in buffer containing 375 mM Tris-HCl pH 8.8, 6 M urea, 2% (w/v) SDS, 20% (v/v) glycerol, and 2% (w/v) DTT. Washing was performed for the next 30 min in buffer containing 2% (w/v) iodoacetamide instead of DTT. Incubation was followed by standard SDS-PAGE (12% polyacrylamide gel) and Western blot analysis.
Analytical SEC and multiangle light scattering
Analytical size-exclusion chromatography was performed using GE Healthcare (Uppsala, Sweden) Superdex 200 analytical gel filtration columns (total resin volume of 24 ml or 120 ml) with a flow rate of 0.5 or 1 ml/min, respectively. To analyze the resulting peaks, appropriate fractions were collected and concentrated using centrifugal filters (Amicon; Millipore), if necessary. Alternatively, SEC-multiangle light scattering (MALS) was applied in which SEC was coupled with the DAWN HELEOS MALS Instrument (Wyatt Technology, Santa Barbara, CA, USA). For SEC-MALS the Superdex 200 2.4 ml column was used with flow rate of 0.05 ml/min. ASTRA (Wyatt Technologies) software was used for data processing. The average molar mass (Mn) was determined to estimate the monodispersity of the peaks.
Phosphatase activity assay
Recombinant ERK2 was autophosphorylated for 30 min at 30°C in the presence of kinase buffer (Cell Signaling) and supplemented with 0.2 mM ATP. To remove the excess ATP and phosphatase inhibitors from kinase buffer, samples were diluted and concentrated to the starting volume using centrifugal filters (Amicon; Millipore). The efficiency of ATP removal was tested by mass spectrometry (Center for Mass Spectrometry, Vanderbilt University, TN, USA). Dephosphorylation of ERK2 by recombinant CacyBP/SIP was carried out for 40 minutes at 37°C in a buffer containing 150 mM NaCl and 20 mM Tris-HCl at pH 8.4. Samples containing 1 µg ERK2 and an equal molar amount of CacyBP/SIP were collected every 10 minutes and the reaction was stopped by addition of loading buffer. Afterward, proteins were separated by SDS-PAGE and analyzed by Western blot.
ELISA binding assay
CacyBP/SIP or its mutants were immobilized onto 96-well microtiter plate in 50 µl of the kinase buffer (Cell Signaling). After 1.5 hours of incubation at RT the solution was removed and wells were washed with the coating buffer (25 mM HEPES, pH 7.6, 100 mM KCl). The remaining adsorption sites were blocked for 1 hour at RT with the coating buffer supplemented with 2 mg/ml bovine serum albumin (BSA). After rinsing the wells with the coating buffer, the increasing amounts of purified recombinant ERK2 in the kinase buffer were applied on wells, to follow the stoichiometric ratio to CacyBP/SIP. After 1.5 hours of incubation at RT, wells were washed with washing buffer (Cell Signaling) and the primary antibody against ERK1/2 (Cell Signaling) in the kinase buffer was applied at 1:1000 dilution. After overnight incubation at 4°C, wells were washed with the washing buffer and anti-rabbit IgG-horseradish peroxidase conjugated antibody (Cell Signaling) in kinase buffer (1:10,000). After 1 hour of incubation at RT wells were washed with the washing buffer and colorimetric detection with the TMB peroxidase EIA substrate kit (Bio-Rad) was applied. The absorbance was measured at 450 nm using the Bio-Rad microplate reader. As a control, plate was coated with BSA, instead of CacyBP/SIP.
Analytical ultracentrifugation, sedimentation velocity, and equilibrium
Sedimentation velocity and equilibrium assays were performed using a Beckman-Coulter Optima XL-I instrument (Beckman Coulter, Fullerton, CA, USA) equipped with an AN-Ti 60 rotor. Absorbance scans were recorded at 280 nm. All experiments were performed at 4°C.
The sedimentation velocity experiment was performed at ∼15,800 × g using Epon double-sector cells, filled with 0.40 ml CacyBP/SIP and 0.41 ml dialysis buffer (150 mM NaCl, 20 mM Tris-HCl at pH 8.4). The samples were equilibrated in a centrifuge at 4°C for 1 hour. Data were analyzed with the continuous distribution of coefficients c(s) model using the SEDFIT (National Institutes of Health [NIH], Bethesda, MD, USA) (16). Partial specific volume of the protein, the density and the viscosity of the buffer were calculated with the SEDNTERP program (University of New Hampshire, Durham, NH, USA) (17).
Sedimentation equilibrium data, for 3 different protein concentrations, were collected in 2-channel cells with sapphire windows. The samples were equilibrated at 4°C at a desired speed and the run equilibrium was monitored for scans collected every 3 hours, by test approach to equilibrium procedure, from SEDFIT program (NIH) (18). Data were acquired every 0.001 cm with 20 replications using a step scan mode at speeds of 1100 and 1800 × g. Data were analyzed with SEDPHAT program (U.S. National Institutes of Health) (18).
Fourier transform infrared spectroscopy
The analysis of the distribution of secondary structure in CacyBP/SIP wild type and mutants was performed using Fourier transform infrared spectroscopy (FT-IR). The FT-IR spectra were collected using a Tensor 27 CONFOCHECK (Bruker Optik GmbH, Ettlingen, Germany) spectrometer equipped with a BioATRCell unit and under constant flow of nitrogen. For each spectrum 120 scans were collected at a temperature of 25°C. The reference spectrum was recorded in identical conditions using the standard buffer (150 mM NaCl, 20 mM Tris at pH 8.4). All spectra were collected, processed, and deconvoluted using Bruker OPUS software (version 7.2).
Small-angle X-ray scattering
Small-angle X-ray scattering (SAXS) data were collected at the Structurally Integrated Biology for Life Sciences (SIBYLS) beamline 12.3.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory (Berkeley, CA, USA). Scattering measurements were performed at 15°C in 20 μl volume samples in a buffer containing 150 mM NaCl, 20 mM Tris-HCl at pH 8.4, and 5% (v/v) glycerol. A Hamilton robot was used for loading samples from a 96-well plate into a helium-purged sample chamber. Data were collected on the original SEC fractions from each run, as well as concentration series from fractions sampled from the later eluting half of each SEC elution peak (∼2- to 8-fold concentration). Fractions before the SEC void volume were used for buffer subtraction of the original SEC fractions.
SAXS experiments were performed using an X-ray beam from a multilayer monochromator of 12 keV (λ 1.3 Å) covering the following momentum transfer range: 0.011 Å−1 < q < 0.322 Å−1, where q is defined as q = 4π sin (θ/2)/λ with scattering angle θ and wavelength λ. The multilayer monochromator provides increased X-ray flux, allowing stronger signals for lower protein concentrations. Sequential exposures (0.5, 1, 2, 5, and 0.5 s) were taken, and data were monitored for radiation-dependent aggregation. All SAXS data were collected using the MarCCD 165 detector in a fast frame transfer mode and reduced via normalization to the incident beam intensity.
Sequence, modeling, and graphical analyses
Sequence alignment was performed using the Protein BLAST server. SAXS data were analyzed using PRIMUS from version 3.0 ATSAS 2.0 (19). The ATSAS package was used for Guinier, Kratky, probability distribution function [P(r)], and CRYSOL plots (European Molecular Biology Laboratory, Hamburg, Germany). Ab initio envelopes were calculated with the GASBOR and DAMAVER programs (both from the European Molecular Biology Laboratory) (20, 21). A homology model was generated using YASARA Structure (Yasara Biosciences GmbH, Vienna, Austria) and PyMOL (DeLAno Scientific, Palo Alto, CA, USA) with 1YSM and 1X5M (PDB) as templates, and Modeler (9v4) interface in Chimera (22, 23) for ab initio modeling.
RESULTS
Oligomerization state of CacyBP/SIP in NB2a cells
To determine the oligomeric state of CacyBP/SIP, we performed coimmunoprecipitation assay. Because CacyBP/SIP is found at a high levels in brain cells and in different neuroblastoma (4); therefore, we chose neuroblastoma NB2a cells for our experiments. The cells were transfected with either vector encoding CacyBP/SIP with an N-terminal 3xFLAG tag or with empty vector as a control. Total protein lysate was mixed with anti-FLAG resin and the eluted proteins analyzed by Western blot using rabbit anti-CacyBP/SIP antibody. A 30 kDa band corresponding to the endogenous CacyBP/SIP was observed in the input from both control and CacyBP/SIP-3xFLAG transfected cells (Fig. 1A), and an additional 33 kDa band corresponding to the fusion protein was observed in NB2a cells transfected with plasmid encoding CacyBP/SIP-3xFLAG. Interestingly, after immunoprecipitation with anti-FLAG resin, both bands were observed in the elution from NB2a lysates expressing CacyBP/SIP-3xFLAG (Fig. 1A, right panel), and no band was observed in the elution of cells transfected with empty plasmid, as the endogenous protein has no affinity to the anti-FLAG resin (Fig. 1A, left panel). These results indicate that the CacyBP/SIP-3xFLAG fusion protein may interact with the endogenous CacyBP/SIP.
Figure 1.

Presence of CacyBP/SIP dimer in NB2a total protein lysate. A) Coimmunoprecipitation of the endogenous CacyBP/SIP with CacyBP/SIP-3xFLAG fusion protein on anti-FLAG agarose resin (right panel). Control experiment after transfection with empty plasmid (left panel). B) Cross-linking of NB2a total protein lysate, analyzed by Western blot using anti-CacyBP/SIP antibody.
Chemical cross-linking of NB2a cell lysate was used to further examine whether CacyBP/SIP dimerizes in cells. NB2a total protein lysate was incubated with or without the zero-length cross-linking agent EDC prior to Western blot analysis. Probing with a polyclonal antibody against CacyBP/SIP revealed that apart from band corresponding to CacyBP/SIP monomer, upon addition of EDC, an additional band corresponding to the expected molecular weight of the CacyBP/SIP dimer was observed (Fig. 1B). Taken together these results suggest that CacyBP/SIP forms homodimer in cells.
Mouse CacyBP/SIP dimerizes through the N-terminal helical hairpin domain
Recombinant CacyBP/SIP protein was purified from E. coli and analyzed by SEC-MALS. Analysis indicated the sample was highly pure and free of soluble aggregates. The average molecular mass of the protein was calculated to be approximately 53 kDa with less than 10% variability in mass across the width of the peak for several independent samples (Fig. 2A). These results are consistent with the existence of CacyBP/SIP dimer and imply the sample was monodispersed. Although dimerization of CacyBP/SIP did not depend on protein concentration, we found a tendency to aggregate at the higher protein concentrations.
Figure 2.

Analysis of CacyBP/SIP dimer under in vitro conditions. A) SEC-MALS for CacyBP/SIP. LS5, light-scattering detector; UV, UV absorption detector; dRI, differential refractive index detector; QELS, quasielastic light-scattering detector. B) Analytical ultracentrifugation—sedimentation velocity analysis of CacyBP/SIP scanned at 280 nm. C) Analytical ultracentrifugation—sedimentation equilibrium data of CacyBP/SIP. The global fit to 6 data sets collected using protein at 3 different concentrations (3.2, 1.6, and 0.8 mg/ml). D) SEC-MALS for CacyBP/SIP dimerization domain.
We next used analytical ultracentrifugation to directly interrogate the dimerization of CacyBP/SIP. SEDFIT analysis of data acquired from sedimentation velocity experiments yielded an average molecular mass of ∼52 kDa (Fig. 2B), further confirming that CacyBP/SIP forms a dimer. The high value of the frictional ratio (∼2.5) derived from these data indicates that the CacyBP/SIP has significant nonglobular character, consistent with a modular domain architecture. Figure 2C shows the global fit of sedimentation equilibrium data at 3 different concentrations, equilibrated at speeds of 1100 (squares) and 1800 g (circles). The fit of the data provided low and randomly distributed residuals with a molecular mass of 52.2 kDa, consistent with that determined by SEC-MALS.
CacyBP/SIP consists of a central globular CS domain flanked by a partially globular N- and a disordered C-terminal domain (10). To determine which domain or domains mediate CacyBP/SIP dimerization, we purified the separated N-terminal (1–77), CS (74–178), and C-terminal (178–229) domains and characterized them by SEC-MALS. Monodisperse samples were obtained for both the N-terminal and CS domains (Fig. 2D). In contrast, the C-terminal domain displayed significant polydispersity and a tendency to aggregate, consistent with our previous report (10). The average molecular mass of each separated domain was determined by SEC-MALS (Table 2). The average molecular mass of the N-terminal domain was 16.8 kDa, approximately twice the expected mass calculated from the protein sequence. The average molecular mass determined for the CS domain (14.2 kDa) was consistent with the mass calculated from the sequence of the monomeric domain. The molecular mass of the C-terminal domain (13.3 kDa) was also ∼2-fold the mass calculated from the sequence but its high polydispersity and unstructured nature suggest this was rather due to nonspecific self-association than dimerization. Together, these results imply that the N-terminal domain mediates dimerization of mouse CacyBP/SIP.
TABLE 2.
Parameters calculated from SEC-MALS for CacyBP/SIP fragments
| CacyBP/SIP 1-77 (N-terminal domain) | CacyBP/SIP 74-178 (CS domain) | CacyBP/SIP 178-229 (C-terminal domain) | |
|---|---|---|---|
| Polydispersity | 1.00–1.01 | 1.00–1.01 | 1.2–1.5 |
| Molecular mass from sequence (kDa) | 8.8 | 14.2 | 6.1 |
| Molecular mass from SEC-MALS (kDa) | 16.8 | 12.2 | 13.3 |
| Measurement error (%) | ±2.3 | ±3 | ±4.5 |
Measurement error represents the sample-to-sample variation.
The structure of the mouse CacyBP/SIP dimer was further investigated by SAXS, a powerful method to determine the architecture of molecules in solution. After optimization, the final analysis was performed on scattering data from the protein concentration of ∼0.8 mg/ml and the shortest exposure of 0.5 s (Fig. 3), as CacyBP/SIP exhibited a tendency to aggregate at higher concentrations and was sensitive to radiation damage at longer exposure times. Guinier analysis (Fig. 3A, inset) indicated that the selected sample was free of aggregation, and provided a radius of gyration of 48 Å. Dimensionless Kratky analysis of the raw scattering data (Fig. 3B) suggests that CacyBP/SIP contains a mixture of globular domains and disordered regions. The Porod volume was estimated to be 1.7 × 105 Å3, consistent with a molecular mass of 55 kDa, corresponding to that of a CacyBP/SIP dimer. The probability distribution function P(r), displayed a broad main peak with a maximum distance (Dmax) of 182 Å (Fig. 3C). An Ab initio molecular envelope was generated from the SAXS data using GASBOR and suggests an extended architecture of CacyBP/SIP dimer consisting of a central mass flanked by long extensions (Fig. 3D).
Figure 3.

Structure of CacyBP/SIP dimer estimated by SAXS. A) Raw scattering intensity data for CacyBP/SIP from a 0.5 s exposure at ∼1 mg/ml. The Guinier plot is shown in the inset. B) Dimensionless Kratky plot for CacyBP/SIP dimer. C) P(r) function for CacyBP/SIP dimer. D) Ab initio molecular envelope derived from the P(r) function after averaging of 10 models.
A series of models was generated to aid in analyzing the CacyBP/SIP dimer architecture. Consistent with previous studies of proteins with flexibly linked domains (24–26), no single model fit well to the experimental data. The Rg and Dmax values for our models are summarized in Supplemental Table 1, and P(r) functions for each model are compared with the experimental data in Supplemental Fig. 1. The analyses of the models suggest that the globular domains occupy a compact, as opposed to a fully extended, antiparallel configuration with the linker between the N-terminal and CS domains flexible enough to allow the protein to sample a range of architectures.
CacyBP/SIP has 2 functional ERK1/2 phosphatase domains
Amino acid sequence analysis revealed that CacyBP/SIP contains 2 kinase interaction motifs (KIMs) (Fig. 4A), one in the N-terminal domain and one, as previously reported, in the C-terminal domain (9). Multisequence alignment shows that the presence of 2 KIMs might be characteristic for the MAP kinase phosphatase (MKP) family of dual-specificity phosphatases (Fig. 4B). Because we have previously reported that the C-terminal domain of CacyBP/SIP binds to and dephosphorylates ERK2 (8, 9), here we examined whether the N-terminal domain also interacts with ERK2. For that we have performed a standard ELISA assay with full-length CacyBP/SIP, each of the isolated domains, and BSA as a negative control (Fig. 4C). As expected, we found that in addition to the C-terminal domain, the N-terminal domain of CacyBP/SIP binds to ERK2. In addition, the CS domain binds to ERK2, presumably because it contains a modified KIM sequence (Fig. 4A, blue dash line). The ELISA assays provide a binary yes-or-no answer for binding, so these results indicate that all 3 domains interact with ERK2.
Figure 4.

Analysis of CacyBP/SIP functional organization. A) CacyBP/SIP sequence analysis. Three KIM sequences (underlined), one each in N terminus, CS domain, and C terminus of CacyBP/SIP. X, any amino acid; ɸ, hydrophobic residue: Leu, Ile, Val, ɸB, not obligatory residue. B) Multisequence alignment for CacyBP/SIP and dual-activity phosphatases from MKP family. KIM sequences are underlined. C) ELISA assay for ERK2 binding with full-length CacyBP/SIP wild-type (WT) and domain fragments. D) Phosphatase activity assay for full-length CacyBP/SIP and its fragments, analyzed by Western blot using anti-p-ERK1/2 antibody. E) Densitometric analysis of the activity assay. Statistical analysis was performed for 5 independent experiments.
The presence of a KIM in the N-terminal domain of CacyBP/SIP suggests that this domain could, apart from binding, exhibit phosphatase activity toward ERK2. We therefore monitored the dephosphorylation of p-ERK2 by Western blot after incubation with full-length CacyBP/SIP and the isolated N-terminal (1–77), CS (74–178), and C-terminal (178–229) domains (Fig. 4D). The amount of p-ERK2 at each time point was quantified by densitometry (Fig. 4E). Full-length CacyBP/SIP efficiently dephosphorylated p-ERK2 with almost complete loss of p-ERK2 signal at 40 min. Conversely, the CS domain had no activity toward ERK2. Both the N-terminal and C-terminal domains dephosphorylated p-ERK2 but the rate of ERK2 dephosphorylation by each separate domain was lower than that observed for the full-length protein. These results demonstrate that in addition to the previous association of the C-terminal domain with phosphatase activity, the N-terminal domain also possesses phosphatase activity and that both contribute to the overall activity of full-length CacyBP/SIP. The binding of the CS domain to ERK2 is not required for CacyBP/SIP phosphatase activity, but presumably this interaction contributes to the avidity and efficiency of the enzymatic activity of the protein.
The influence of the N-terminal domain on dimerization and ERK1/2 phosphatase activity
The KIM sequence in the N-terminal domain of CacyBP/SIP is located within the helical hairpin dimerization domain. This prompted us to investigate the interrelationship between the KIM, CacyBP/SIP phosphatase activity on ERK1/2, and dimerization. As a first step, we examined homology models of mouse CacyBP/SIP N-terminal dimerization domain to look for key residues that could be mutated to inhibit dimerization. This analysis identified 4 surface-exposed amino acids at the dimer interface that partially overlap with the N-terminal KIM sequence (Asp11, Glu14, Lys25, and Arg26) and form pairs of salt bridges (Asp11:Lys25, Glu14:Arg26) (Fig. 5A).
Figure 5.

Analysis of CacyBP/SIP dimer formation. A) Three-dimensional homology model of full-length CacyBP/SIP with the putative residues involved in the dimerization. N-term, N-terminal domain; CS, CS domain; C-term, C-terminal domain. B) Comparison of SEC chromatograms for CacyBP/SIP mutants within the N-terminal domain. Peak corresponding to the protein dimer is marked in red, protein monomer in green. C) Effect of mutations within the N-terminal domain of full-length CacyBP/SIP on its phosphatase activity and ERK1/2 binding. The activity assay for full-length CacyBP/SIP and its mutants analyzed by Western blot developed using anti-p-ERK1/2 antibody. D) Densitometric analysis for the phosphatase activity assay of full-length CacyBP/SIP and its mutants within the N-terminal domain. Statistical analysis was performed for 5 independent experiments.
To determine whether these electrostatic interactions contribute significantly to the stabilization of the dimer, both charge neutralization and charge reversal mutations were generated for each residue and the mutants were examined by SEC. The D11A/E14A double mutation resulted in a partial disruption of the CacyBP/SIP dimer as indicated by the presence of a small shoulder in the SEC trace corresponding to the mass of the monomer (Fig. 5B). The D11A/E14A mutation also had a significant impact on protein stability, as indicated by the presence of higher molecular weight species eluting at a volume corresponding to soluble aggregate. In contrast, the K25A/R26A double mutation had no significant effect on either protein stability or the monomer/dimer ratio. The D11A/E14A/K25A/R26A tetra-mutant had a significant destabilizing effect on CacyBP/SIP, but exhibited no measureable change in the monomer/dimer ratio (Fig. 5B). FT-IR experiments performed for CacyBP/SIP wild-type and mutant proteins indicated that the changes in the monomer/dimer ratio were not due to a change in the structure relative to the wild-type protein (Table 3).
TABLE 3.
Content of α-helical and β-sheet structures in wild-type CacyBP/SIP and its mutants calculated from FT-IR measurements
| CacyBP/SIP | Secondary structures estimation (%) |
|||
|---|---|---|---|---|
|
α-Helix |
β-Sheet |
|||
| 1st readout | 2nd readout | 1st readout | 2nd readout | |
| Wild-type | 28 | 28 | 30 | 30 |
| D11A/E14A | 28 | 28 | 34 | 33 |
| D11H/E14K | 26 | 26 | 33 | 32 |
| K25A/R26A | 27 | 26 | 34 | 34 |
| K25E/R26E | 25 | 26 | 34 | 34 |
| D11A/E14A/K25A/R26A | 27 | 27 | 34 | 34 |
| S22D/T23D | 25 | 24 | 34 | 34 |
Next, we tested the charge reversal mutations, which introduce repulsion between dimer subunits. The D11H/E14K mutation behaved as expected with a more significant effect on the monomer/dimer ratio than the D11A/E14A mutation (Fig. 5B). The D11H/E14K mutation also appeared to have less of a detrimental effect on protein stability than D11A/E14A, as reflected in a smaller amount of soluble aggregates. In contrast, the K25E/R26E charge reversal mutant had no effect on CacyBP/SIP monomer/dimer ratio or protein stability as observed for the charge neutralization mutants (Fig. 5B). Taken together, these results suggest that Asp11 and Glu14 play a role in maintaining the overall stability and supporting the dimerization of CacyBP/SIP, and the residues Lys25 and Arg26 are not essential. The presence of the basic residue Arg24 immediately preceding Lys25-Arg26 may be relevant in this context.
The results obtained for the Asp11 and Glu14 mutation prompted us to investigate whether CacyBP/SIP dimer relies on electrostatic interactions. To this end, we performed a series of SEC experiments on the full-length protein with increasing ionic strength. Remarkably, despite a number of specific salt bridges in the crystal structure of the human N-terminal helical hairpin domain (and the corresponding homology model of the mouse protein), we found no evidence of ionic strength-dependent electrostatic screening effecting dimerization over the range from 100 to 1000 mM NaCl (Supplemental Fig. 2). These results suggest that if the salt bridge network predicted by the homology model is present, it is strongly supported by hydrophobic interactions and that both contribute to overall CacyBP/SIP dimer stability.
Given that the D11H/E14K mutation altered the monomer/dimer ratio and stability of CacyBP/SIP, we tested its effect on CacyBP/SIP phosphatase activity against p-ERK2. The results showed that the D11H/E14K charge reversal mutation reduced phosphatase activity by approximately 10–15%, whereas D11A/E14A charge neutralization had no significant effect on the ability of CacyBP/SIP to dephosphorylate p-ERK2 (Fig. 5C, D). We also analyzed the Lys25 and Arg26 mutations, which are directly within the N-terminal KIM and expected to interfere with the phosphatase activity. Indeed, both charge neutralization and charge reversal mutations of residues K25 and R26 resulted in a complete loss of phosphatase activity, similar to that observed for the substitution of all 4 residues by alanine (D11A/E14A/K25A/R26A) (Fig. 5C, D). It is well established that CacyBP/SIP can be post-translationally modified and it is conceivable that post-translational modification could alter the monomer/dimer equilibrium (and possibly the phosphatase activity). Ser22 and Thr23 represent 2 interesting potential phosphorylation sites because they are in the dimerization domain and immediately adjacent to the N-terminal KIM. To test this hypothesis, we prepared the phospho-mimic double mutant S22D/T23D. SEC analysis showed that the monomer/dimer ratio was shifted significantly toward the monomeric state such that the CacyBP/SIP monomer was dominant (Fig. 6A). The identity of the monomer and dimer peaks was confirmed by SEC-MALS (Fig. 6B). We next analyzed the ERK1/2 phosphatase activity and found that this mutant is at least as active and possibly even more active than the wild-type protein (Fig. 5C, D).
Figure 6.
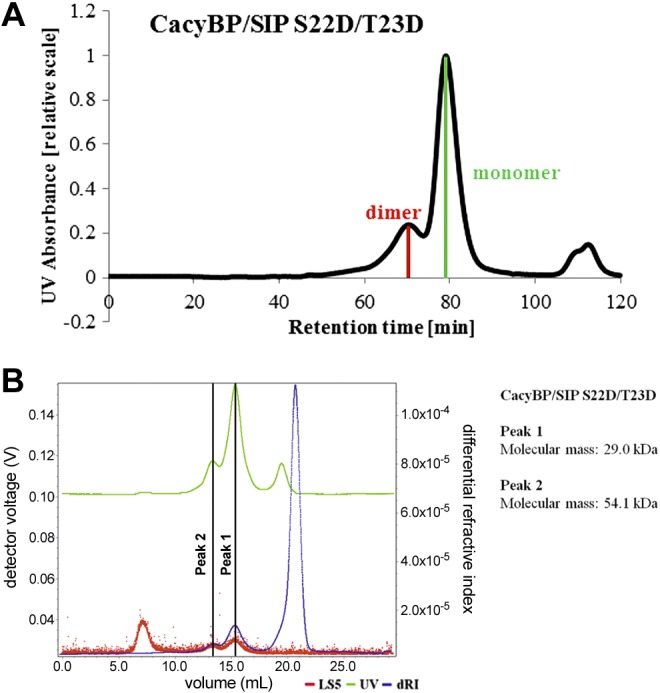
Analysis of CacyBP/SIP S22D/T23D mutant. A) SEC chromatogram for CacyBP/SIP S22D/T23D mutant. B) SEC-MALS analysis of the molecular mass of the main species of CacyBP/SIP S22D/T23D mutant.
In summary, our results show that the residues in the N-terminal KIM are essential for the phosphatase activity of CacyBP/SIP and suggest this activity may be modulated by dimerization. Moreover, we have obtained evidence suggesting both dimerization and phosphatase activity may be altered by phosphorylation within the KIM of the N-terminal domain.
CacyBP/SIP is sensitive to oxidative stress in NB2a cells
The in vitro analyses described here suggested a link between CacyBP/SIP dimerization and phosphatase activity toward ERK1/2. To search for the possible roles of CacyBP/SIP dimerization in the ERK1/2 kinase cascade, cell-based analyses were performed. Because the ERK1/2 pathway is specifically activated upon oxidative stress (27), experiments were performed with differentiated neuroblastoma NB2a cells treated with H2O2.
To determine whether CacyBP/SIP responds to oxidative stress, we used confocal immunofluorescence microscopy to examine its subcellular distribution in NB2a cells. In the untreated NB2a cells, CacyBP/SIP was present predominantly in the cytoplasm as expected (Fig. 7A, upper panels). In contrast, a significant amount of the protein was detected in the nuclei of NB2a cells treated with H2O2 (Fig. 7A, lower panels). Nuclear translocation of CacyBP/SIP was also quantified based on the immunofluorescent staining (Fig. 1A), presented as a ratio of cytoplasmic/nuclear protein (Fig. 1B). Because the immunofluorescent staining seemed to be more intense upon H2O2-treatment, subcellular fractionation experiments were performed to quantify the extent of redistribution of CacyBP/SIP under oxidative stress. Total protein lysate, and cytoplasmic and nuclear fractions were prepared from untreated or H2O2-treated NB2a cells and then analyzed by Western blot. The relative amounts of CacyBP/SIP were quantified by densitometry (Fig. 7C). As expected, treatment of NB2a cells with H2O2 resulted in an ∼40% increase of CacyBP/SIP level in total protein lysate. Fractionation of NB2a cells revealed that the level of nuclear CacyBP/SIP increased by ∼35% after H2O2 treatment in comparison to the untreated cells, whereas addition of H2O2 had only modest effect on the cytoplasmic level of CacyBP/SIP (Fig. 7D). Taken together, these results suggest that both expression and nuclear import of CacyBP/SIP are up-regulated in response to oxidative stress.
Figure 7.

Subcellular localization and properties of CacyBP/SIP in control and H2O2-treated NB2a cells. A) Immunofluorescence performed for CacyBP/SIP localization in untreated and H2O2-treated NB2a cells. CacyBP/SIP is marked in red, nuclei in blue. B) Statistical representation of CacyBP/SIP nuclear translocation (presented in A) shown as the ratio of cytoplasmic to nuclear immunofluorescence staining intensity in untreated (gray bar) and H2O2-treated (black bar) NB2a cells. Quantification was performed for all cells shown in panel A. C) Densitometric analysis of CacyBP/SIP level in the total protein lysate. Quantification was performed for the results obtained by Western blot from 3 independent experiments. D) Densitometric analysis of CacyBP/SIP level in the cytoplasmic and nuclear fractions in control and stress conditions. Quantification was performed for the results obtained by Western blot from 3 independent experiments. E) Colocalization of CacyBP/SIP with p-ERK1/2 in untreated and H2O2-treated NB2a cells. CacyBP/SIP is stained in red, p-ERK1/2 in green, while nuclei in blue. Colocalization of CacyBP/SIP and p-ERK1/2 is visible as yellow. F) Proximity ligation assay for homodimerization detection. Red PLA signal represents homodimers of CacyBP/SIP, and nuclei are marked in blue. G) Two-dimensional electrophoresis for CacyBP/SIP forms in different cellular compartments analyzed with Western blot using anti-CacyBP/SIP antibody.*P < 0.05; **P < 0.01; ***P < 0.001.
Next, we tested whether the nuclear translocation of CacyBP/SIP is linked to its phosphatase activity toward ERK1/2. Hence, we performed the immunofluorescent staining for both CacyBP/SIP and p-ERK1/2 in untreated and H2O2-treated NB2a cells. The data showed that CacyBP/SIP and p-ERK1/2 are located mainly in the cytoplasm of untreated cells. Oxidative stress causes translocation to the nucleus, with both proteins colocalizing in this cellular compartment (Fig. 7E). Thus, CacyBP/SIP follows its target protein to the nucleus where dephosphorylation presumably takes place, suggesting that CacyBP/SIP translocation is linked to its function as a phosphatase.
To test whether nuclear translocation and phosphatase activity correlate with dimerization of CacyBP/SIP, we used a PLA adapted for detection of homodimers in cells (28). In untreated cells the red PLA signal was observed in the cytoplasm only (Fig. 7F, upper panel), consistent with our confocal immunofluorescence microscopy results (Fig. 7A). Upon treatment with H2O2, the red PLA signal is still found mainly in the cytoplasm (Fig. 7F, lower panel). Note, that single signals appeared in the nucleus of H2O2-treated cells, although their number was significantly lower than in the cytoplasm. Because 2 different PLA probes were used for 1 primary antibody, this method reports on CacyBP/SIP homodimerization only at a qualitative level. Nevertheless, the results suggest that the prevalent form of CacyBP/SIP in the cytoplasm of NB2a cells is a dimer, but that under conditions of oxidative stress the most prevalent form of the protein in the nucleus may be a monomer.
According to recent reports, CacyBP/SIP activity might be regulated by phosphorylation (29–31); therefore, we examined whether post-translational modifications of CacyBP/SIP correlate with its subcellular redistribution. We performed 2-dimensional electrophoresis. In untreated cells, 3 major forms of CacyBP/SIP were resolved in total protein lysate (Fig. 7G). One form migrating near pH 8 most likely represents unmodified CacyBP/SIP, which has a pI of 7.6. Two additional forms were observed migrating near neutral pH and pH 4. The most acidic form of CacyBP/SIP with pI ∼ 4 likely corresponds to multiphosphorylated protein (31). Upon treatment with H2O2 only 2 forms of CacyBP/SIP were resolved: the unmodified protein at pH 8 and the acidic form resolving between pH 4–5 (Fig. 7G). These results indicate that oxidative stress induces a change in the post-translational modifications of CacyBP/SIP, presumably in the phosphorylation of CacyBP/SIP.
We next tested whether changes in post-translational modification of CacyBP/SIP correlated with its subcellular distribution. The 3 major forms of CacyBP/SIP were observed in the cytosolic fractions of untreated cells, whereas only neutral and basic were found in the nucleus (Fig. 7G). Following treatment with H2O2 the highly acidic form disappeared from the cytoplasm, whereas neutral form of CacyBP/SIP in the nucleus increased significantly. These results suggest that changes in phosphorylation correlate with the subcellular redistribution of CacyBP/SIP in response to oxidative stress.
DISCUSSION
CacyBP/SIP plays an important role in multiple cellular processes, including cell differentiation, cytoskeleton reorganization, and down-regulation of MAP kinase cascade (4, 5, 9, 31). In this work, we have shown that full-length mouse CacyBP/SIP forms a dimer in vitro and in NB2a cells. Moreover, by applying SAXS, we have obtained evidence indicating that the structural architecture of full-length CacyBP/SIP is an extended antiparallel dimer with considerable conformational flexibility.
The SAXS analysis revealed a substantial similarity in the spatial architecture of CacyBP/SIP to that of the homologous protein Sgt1 (32). Similar to CacyBP/SIP, Sgt1 is composed of 3 domains (α-helical TPR, β-sheet sandwich CS and unstructured SGS domains) and was shown that plant Sgt1 dimerizes through its N-terminal TPR domain. Moreover, despite its larger size (229 residues for mouse CacyBP/SIP versus 373 for plant Sgt1), Sgt1 displays Dmax values and an elongated shape of the molecular envelope similar to those of CacyBP/SIP (32). Interestingly, Sgt1 exhibits a monomer/dimer equilibrium that strongly depends on the ionic strength, whereas under similar conditions, no evidence for CacyBP/SIP monomer was found and the oligomerization state was insensitive to ionic strength. Moreover, it has been suggested that dimerization of Sgt1 might be inhibited by oxidation of cysteine residues within the TPR domain (32, 33), which is not the case for CacyBP/SIP.
CacyBP/SIP and Sgt1 have both been shown to be components of multiprotein E3 ubiquitin ligases (3, 34), and their high flexibility and structural modularity are likely useful properties for their putative roles as scaffold/adapter proteins. It has been suggested that the principal role of CacyBP/SIP in SCFTBL1 ubiquitin ligase is to bring various components of the complex into physical proximity, orchestrating the correct distance and orientation of the ligase to β-catenin (10). A similar scaffold/adapter function might also explain the role of CacyBP/SIP in the tubulin-actin-tropomyosin complex (6, 7).
The ensemble of our data suggests a possible model that links different CacyBP/SIP functions, post-translational modifications and changes in subcellular localization, and dimerization. CacyBP/SIP is a functional component of a complex E3 ubiquitin ligase (3, 35), and also serves as an ERK1/2 phosphatase (9, 31). Our results show that in response to oxidative stress CacyBP/SIP translocates into the nucleus. We also found that CacyBP/SIP clearly exists as a dimer in the cytoplasm, but indirect evidence has been obtained suggesting that CacyBP/SIP may be monomeric in the nucleus. Moreover, the D22/T23 mutant shows that a shift toward the monomeric state at least maintains and may in fact increase CacyBP/SIP phosphatase activity. Previous studies have demonstrated that the phosphatase activity of CacyBP/SIP is restricted to the nucleus (9, 31), but protein ubiquitination activity is predominantly in the cytosol (35, 36). This suggests that redistribution of CacyBP/SIP into different subcellular locations driven by post-translational modifications acquired in response to different cellular stimuli may be coupled to a functional switch. It is intriguing to consider the possibility that the post-translational modifications, in particular phosphorylation, may alter the monomer/dimer equilibrium and in turn regulate phosphatase activity. Based on our accumulated evidence, we postulate that the monomer of CacyBP/SIP might be an active form of the protein toward ERK1/2. Experiments to test and refine this model will be reported in due course.
We found that CacyBP/SIP nuclear translocation is accompanied by apparent changes in its phosphorylation state. The pattern of CacyBP/SIP forms detected in NB2a cells upon H2O2-treatment is notably similar to the pattern observed in brain tissue of AD patients (31, 37). CacyBP/SIP is subject to multiple changes upon oxidative stress and it is known that AD is characterized by an increase in the cellular levels of reactive oxygen species (38, 39). These interesting correlations provide additional motivation for further studies of the function and regulation of CacyBP/SIP under conditional of cellular stress.
Supplementary Material
Acknowledgments
The authors thank Dr. U. Wasik (Nencki Institute of Experimental Biology, Warsaw, Poland) for help with pET28 CacyBP/SIP S22D/T23D plasmid preparation and Prof. A. Dziembowski (Institute of Biochemistry and Biophysics, Warsaw) for equipment support. This work was supported by the European Union through the European Regional Developmental Funds within the scope of the International PhD Studies in Neurobiology MPD4-502 (to A.M.T.W.), U.S. National Institutes of Health, National Institute of General Medical Sciences Grant R01-GM075156 (to W.J.C.), National Science Centre Grant NZ1/00595 (to A.F.), and by statutory funds from the Nencki Institute of Experimental Biology (Warsaw, Poland). S.M.S. is supported by Postdoctoral Fellowship 119569-PF-11-271-01-DMC from the American Cancer Society. The X-ray scattering technology and applications to the determination of macromolecular shapes and conformations were performed at the SIBYLS beamline (12.3.1) at the Advanced Light Source, Lawrence Berkeley National Laboratory, U.S. Department of Energy (DOE) program Integrated Diffraction Analysis Technologies (IDAT). Project subject carried out with use of Centre for Preclinical Research and Technology (CePT) infrastructure financed by the European Union - the European Regional Development Fund within the Operational Programme “Innovative Economy” for 2007–2013.
Glossary
- BSA
bovine serum albumin
- CacyBP/SIP
calcyclin-binding protein/Siah-1 interacting protein
- CS domain
domain present in CHORD containing proteins and in Sgt1
- Dmax
maximum peak distance
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- FT-IR
Fourier transform infrared spectroscopy
- IP
immunoprecipitation
- KIM
kinase interaction motif
- MALS
multiangle light scattering
- MKP
MAP kinase phosphatase
- NB2a
mouse neuroblastoma cell line
- PLA
proximity ligation assay
- P(r)
probability distribution function
- RT
room temperature
- SAXS
small-angle X-ray scattering
- SEC
size-exclusion chromatography
- SGS
Sgt1-specific
- Siah-1
seven in absentia homolog 1 (Siah E3 ubiquitin protein ligase 1)
- Skp1
S-phase kinase-associated protein 1
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Filipek A., Wojda U. (1996) p30, a novel protein target of mouse calcyclin (S100A6). Biochem. J. 320, 585–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Filipek A., Kuźnicki J. (1998) Molecular cloning and expression of a mouse brain cDNA encoding a novel protein target of calcyclin. J. Neurochem. 70, 1793–1798 [DOI] [PubMed] [Google Scholar]
- 3.Matsuzawa S. I., Reed J. C. (2001) Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol. Cell 7, 915–926 [DOI] [PubMed] [Google Scholar]
- 4.Jastrzebska B., Filipek A., Nowicka D., Kaczmarek L., Kúznicki J. (2000) Calcyclin (S100A6) binding protein (CacyBP) is highly expressed in brain neurons. J. Histochem. Cytochem. 48, 1195–1202 [DOI] [PubMed] [Google Scholar]
- 5.Schneider G., Nieznanski K., Kilanczyk E., Bieganowski P., Kuznicki J., Filipek A. (2007) CacyBP/SIP interacts with tubulin in neuroblastoma NB2a cells and induces formation of globular tubulin assemblies. Biochim. Biophys. Acta 1773, 1628–1636 [DOI] [PubMed] [Google Scholar]
- 6.Schneider G., Nieznanski K., Jozwiak J., Slomnicki L. P., Redowicz M. J., Filipek A. (2010) Tubulin binding protein, CacyBP/SIP, induces actin polymerization and may link actin and tubulin cytoskeletons. Biochim. Biophys. Acta 1803, 1308–1317 [DOI] [PubMed] [Google Scholar]
- 7.Jurewicz E., Ostrowska Z., Jozwiak J., Redowicz M. J., Lesniak W., Moraczewska J., Filipek A. (2013) CacyBP/SIP as a novel modulator of the thin filament. Biochim. Biophys. Acta 1833, 761–766 [DOI] [PubMed] [Google Scholar]
- 8.Kilanczyk E., Filipek S., Jastrzebska B., Filipek A. (2009) CacyBP/SIP binds ERK1/2 and affects transcriptional activity of Elk-1. Biochem. Biophys. Res. Commun. 380, 54–59 [DOI] [PubMed] [Google Scholar]
- 9.Kilanczyk E., Filipek S., Filipek A. (2011) ERK1/2 is dephosphorylated by a novel phosphatase—CacyBP/SIP. Biochem. Biophys. Res. Commun. 404, 179–183 [DOI] [PubMed] [Google Scholar]
- 10.Bhattacharya S., Lee Y. T., Michowski W., Jastrzebska B., Filipek A., Kuznicki J., Chazin W. J. (2005) The modular structure of SIP facilitates its role in stabilizing multiprotein assemblies. Biochemistry 44, 9462–9471 [DOI] [PubMed] [Google Scholar]
- 11.Lee Y. T., Dimitrova Y. N., Schneider G., Ridenour W. B., Bhattacharya S., Soss S. E., Caprioli R. M., Filipek A., Chazin W. J. (2008) Structure of the S100A6 complex with a fragment from the C-terminal domain of Siah-1 interacting protein: a novel mode for S100 protein target recognition. Biochemistry 47, 10921–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuzawa S., Li C., Ni C. Z., Takayama S., Reed J. C., Ely K. R. (2003) Structural analysis of Siah1 and its interactions with Siah-interacting protein (SIP). J. Biol. Chem. 278, 1837–1840 [DOI] [PubMed] [Google Scholar]
- 13.Santelli E., Leone M., Li C., Fukushima T., Preece N. E., Olson A. J., Ely K. R., Reed J. C., Pellecchia M., Liddington R. C., Matsuzawa S. (2005) Structural analysis of Siah1-Siah-interacting protein interactions and insights into the assembly of an E3 ligase multiprotein complex. J. Biol. Chem. 280, 34278–34287 [DOI] [PubMed] [Google Scholar]
- 14.Filipek A., Jastrzebska B., Nowotny M., Kuznicki J. (2002b) CacyBP/SIP, a calcyclin and Siah-1-interacting protein, binds EF-hand proteins of the S100 family. J. Biol. Chem. 277, 28848–28852 [DOI] [PubMed] [Google Scholar]
- 15.Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 16.Schuck P. (2003) On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 320, 104–124 [DOI] [PubMed] [Google Scholar]
- 17.Laue T. M., Shah B. D., Ridgeway T. M., Pelletier S. L. (1992) Computer-aided interpretation of analytical sedimentation data for proteins. In Analytical ultracentrifugation in biochemistry and polymer science (Harding S. E., Rowe A. J., Horton J. C., eds.), pp. 90–125, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 18.Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 36, 1277–1282 [Google Scholar]
- 20.Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sali A., Potterton L., Yuan F., van Vlijmen H., Karplus M. (1995) Evaluation of comparative protein modeling by MODELLER. Proteins 23, 318–326 [DOI] [PubMed] [Google Scholar]
- 23.Fiser A., Do R. K., Sali A. (2000) Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brosey C. A., Yan C., Tsutakawa S. E., Heller W. T., Rambo R. P., Tainer J. A., Ivanov I., Chazin W. J. (2013) A new structural framework for integrating replication protein A into DNA processing machinery. Nucleic Acids Res. 41, 2313–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johs A., Harwood I. M., Parks J. M., Nauss R. E., Smith J. C., Liang L., Miller S. M. (2011) Structural characterization of intramolecular Hg(2+) transfer between flexibly linked domains of mercuric ion reductase. J. Mol. Biol. 413, 639–656 [DOI] [PubMed] [Google Scholar]
- 26.Pretto D. I., Tsutakawa S., Brosey C. A., Castillo A., Chagot M. E., Smith J. A., Tainer J. A., Chazin W. J. (2010) Structural dynamics and single-stranded DNA binding activity of the three N-terminal domains of the large subunit of replication protein A from small angle X-ray scattering. Biochemistry 49, 2880–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruffels J., Griffin M., Dickenson J. M. (2004) Activation of ERK1/2, JNK and PKB by hydrogen peroxide in human SH-SY5Y neuroblastoma cells: role of ERK1/2 in H2O2-induced cell death. Eur. J. Pharmacol. 483, 163–173 [DOI] [PubMed] [Google Scholar]
- 28.Gullberg M., Andersson A. C. (2009) Highly specific detection of phosphorylated proteins by Duolink. Nat. Methods 6, DOI:10.1038/nmeth.f.267 [Google Scholar]
- 29.Filipek A., Jastrzebska B., Nowotny M., Kwiatkowska K., Hetman M., Surmacz L., Wyroba E., Kuznicki J. (2002a) Ca2+-dependent translocation of the calcyclin-binding protein in neurons and neuroblastoma NB-2a cells. J. Biol. Chem. 277, 21103–21109 [DOI] [PubMed] [Google Scholar]
- 30.Wu J., Tan X., Peng X., Yuan J., Qiang B. (2003) Translocation and phosphorylation of calcyclin binding protein during retinoic acid-induced neuronal differentiation of neuroblastoma SH-SY5Y cells. J. Biochem. Mol. Biol. 36, 354–358 [DOI] [PubMed] [Google Scholar]
- 31.Kilanczyk E., Wasik U., Filipek A. (2012) CacyBP/SIP phosphatase activity in neuroblastoma NB2a and colon cancer HCT116 cells. Biochem. Cell Biol. 90, 558–564 [DOI] [PubMed] [Google Scholar]
- 32.Taube M., Pieńkowska J. R., Jarmołowski A., Kozak M. (2014) Low-resolution structure of the full-length barley (Hordeum vulgare) SGT1 protein in solution, obtained using small-angle X-ray scattering. PLoS ONE 9, e93313.DOI: 10.1371/journalpone0093313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nyarko A., Mosbahi K., Rowe A. J., Leech A., Boter M., Shirasu K., Kleanthous C. (2007) TPR-Mediated self-association of plant SGT1. Biochemistry 46, 11331–11341 [DOI] [PubMed] [Google Scholar]
- 34.Peart J. R., Lu R., Sadanandom A., Malcuit I., Moffett P., Brice D. C., Schauser L., Jaggard D. A., Xiao S., Coleman M. J., Dow M., Jones J. D., Shirasu K., Baulcombe D. C. (2002) Ubiquitin ligase-associated protein SGT1 is required for host and nonhost disease resistance in plants. Proc. Natl. Acad. Sci. USA 99, 10865–10869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dimitrova Y. N., Li J., Lee Y. T., Rios-Esteves J., Friedman D. B., Choi H. J., Weis W. I., Wang C. Y., Chazin W. J. (2010) Direct ubiquitination of beta-catenin by Siah-1 and regulation by the exchange factor TBL1. J. Biol. Chem. 285, 13507–13516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liani E., Eyal A., Avraham E., Shemer R., Szargel R., Berg D., Bornemann A., Riess O., Ross C. A., Rott R., Engelender S. (2004) Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 101, 5500–5505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wasik U., Schneider G., Mietelska-Porowska A., Mazurkiewicz M., Fabczak H., Weis S., Zabke C., Harrington C. R., Filipek A., Niewiadomska G. (2013) Calcyclin binding protein and Siah-1 interacting protein in Alzheimer’s disease pathology: neuronal localization and possible function. Neurobiol. Aging 34, 1380–1388 [DOI] [PubMed] [Google Scholar]
- 38.Behl C. (1999) Alzheimer’s disease and oxidative stress: implications for novel therapeutic approaches. Prog. Neurobiol. 57, 301–323 [DOI] [PubMed] [Google Scholar]
- 39.Luque-Contreras D., Carvajal K., Toral-Rios D., Franco-Bocanegra D., Campos-Peña V. (2014) Oxidative stress and metabolic syndrome: cause or consequence of Alzheimer’s disease? Oxid. Med. Cell. Longev. 2014, 497802.DOI: 10.1155/2014/497802 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.