

Abstract
The Smoothened (Smo) receptor, a member of class F G protein–coupled receptors, is the main transducer of the Hedgehog (Hh) signaling pathway implicated in a wide range of developmental and adult processes. Smo is the target of anticancer drugs that bind to a long and narrow cavity in the 7-transmembrane (7TM) domain. X-ray structures of human Smo (hSmo) bound to several ligands have revealed 2 types of 7TM-directed antagonists: those binding mostly to extracellular loops (site 1, e.g., LY2940680) and those penetrating deeply in the 7TM cavity (site 2, e.g., SANT-1). Here we report the development of the acylguanidine MRT-92, which displays subnanomolar antagonist activity against Smo in various Hh cell-based assays. MRT-92 inhibits rodent cerebellar granule cell proliferation induced by Hh pathway activation through pharmacologic (half maximal inhibitory concentration [IC50] = 0.4 nM) or genetic manipulation. Using [3H]MRT-92 (Kd = 0.3 nM for hSmo), we created a comprehensive framework for the interaction of small molecule modulators with hSmo and for understanding chemoresistance linked to hSmo mutations. Guided by molecular docking and site-directed mutagenesis data, our work convincingly confirms that MRT-92 simultaneously recognized and occupied both sites 1 and 2. Our data demonstrate the existence of a third type of Smo antagonists, those entirely filling the Smo binding cavity from the upper extracellular part to the lower cytoplasmic-proximal subpocket. Our studies should help design novel potent Smo antagonists and more effective therapeutic strategies for treating Hh-linked cancers and associated chemoresistance.—Hoch, L., Faure, H., Roudaut, H., Schoenfelder, A., Mann, A., Girard, N., Bihannic, L., Ayrault, O. Petricci, E., Taddei, M., Rognan, D., Ruat, M. MRT-92 inhibits Hedgehog signaling by blocking overlapping binding sites in the transmembrane domain of the Smoothened receptor.
Keywords: antagonist, medulloblastoma, molecular modeling, stem cell
Smoothened (Smo), a member of the GPCR superfamily, belongs to class F (Frizzled family) of these receptors (1–3). Smo is the main transducer of the Hedgehog (Hh) signaling pathway responsible for a large range of developmental processes and regulates key physiologic responses in adult tissues, including the maintenance of stem and precursor cells (4, 5). In the absence of Hh ligands, the 12-pass transmembrane protein Patched (Ptc) negatively regulates Smo, presumably through a transporter-like activity modulating the availability of a yet elusive endogenous lipid molecule (6). The binding of Hh to Ptc activates the canonical Hh signaling pathway by translocating Smo to the primary cilium. This initiates a complex signaling cascade mediated by activation of Gli transcription factors and translocation of their active forms to the nucleus leading to gene transcription. The biochemical and molecular mechanisms regulating Smo trafficking at the primary cilium presumably involve multiple states of Smo that might be modulated by Ptc itself (1, 6). The natural and teratogenic alkaloid molecule cyclopamine presumably blocks canonical Hh signaling by binding to the 7-transmembrane (7TM) domain of Smo (7, 8). Following the discovery of this first Smo ligand and the evidence that up-regulation of Hh signaling occurs in multiple types of cancer (9, 10), extensive research is undertaken for developing Smo antagonists (1, 9, 11). This is supported by the recent approval of GDC-0449 (vismodegib) for treating metastatic basal cell carcinoma (BCC) and locally advanced BCC untreatable by surgery and radiation (12, 13). The importance of Smo inhibitors of different chemical classes is also demonstrated by ongoing clinical trials on a large range of metastatic and advanced cancers (1, 9, 11). The recently described high-resolution X-ray structures of the 7TM and the extracellular domains of human Smo (hSmo) highlights both conserved and unique structural features of this atypical GPCR (2, 3, 14, 15). X-ray structures of the hSmo 7TM domain enabled identification of a narrow and deep hydrophobic cavity between the 7TM bundle, the 3 extracellular loops (ECL) and the cysteine-rich domain of the receptor’s amino-terminal tail. Small molecular weight ligands bind to this amino-terminal tail (2, 3). In addition to understanding the binding mode of Smo antagonists to the 7TM, these studies are also important for delineating the complex processes inducing Smo resistance to drug treatment, as seen in the receptor’s mutations (D473H in hSmo) rendering it resistant to GDC-0449 (16) or to LDE225 (17).
In this study, we report the design, putative binding mode, and pharmacologic properties of MRT-92, a novel Smo antagonist that is not sensitive to the D473H mutation and uniquely binds to the entire transmembrane cavity of the Smo receptor.
MATERIALS AND METHODS
Molecular docking
Input structures for MRT compounds were obtained by converting a MarvinSketch v6.1 (ChemAxon Ltd., Budapest, Hungary) 2D sketch into 3D coordinates using the Corina v3.1 software (Molecular Networks GmbH, Erlangen, Germany). The most probable protonation state at pH 7.4, according to ChemAxon’s pKa calculation plug-in (ChemAxon Ltd., Budapest, Hungary), was retained for each ligand. Four input structures for hSmo were retrieved from the Protein Data Bank (PDB; accession codes 4JKV, 4N4W, 4O9R, 4QIM). The cytochrome b652 insert used for crystallization purpose was removed in each structure, leaving the transmembrane domain (4JKV, S190-C550; 4N4W, S190-T553; 4O9R, Q192-R551; 4QIM, S190-T553) for docking. In case of a dimeric structure (4JKV), only chain A was kept. After removing all nonprotein atoms (ligand, ions, solvent, lipids), hydrogen atoms were added to the protein using standard geometries in the SYBYL-X 2.1.1 package (Certera, St. Louis, MO, USA). The binding site in each structure was defined by the sum of residues closer than 4.5 Å from any of the bound antagonists (LY2940680, SANT-1, cyclopamine, Anta XV) heavy atom in the reference 4 X-ray structures: N219ECD, L221 ECD, F222ECD, M2301.32f, I2341.36f, F2742.51f, W2812.58f, M301ECL1, L303ECL1, V3213.32f, LEU3253.36f, M3263.37f, V3293.40f, D384ECL2, V386ECL2, S387ECL2, I389ECL2, F391ECL2, Y394ECL2, K395ECL2, R4005.43f, V4045.47f, I4085.51f, F4626.43f, V4636.44f, T4666.47f, H4706.51f, D4736.54f, Q477ECL3, W480ECL3, E481ECL3, F484ECL3, P513ECL3, L5157.35f, E5187.38f, N5217.41f, L5227.42f, A5247.44f, MET5257.45f, and T5287.48f. A protomol was first defined using standard settings of Surflex-Dock v2.745 (31) from the above-cited list of residues, and further used to dock MRT-92 and MRT-83 using the docking accuracy parameter set (pgeom option) in Surflex-Dock. For every Smo structure, a total of 20 poses was saved for each ligand.
Drugs
Synthesis of MRT-91, MRT-92, MRT-93, MRT-94, and MRT-95 are available upon request. Bodipy cyclopamine (BC) was from Toronto Research Chemicals Inc. (North York, ON, Canada). GSA-10, GDC-0449, LDE225, MRT-83, SAG, ALLO-2, and M25 compounds have been synthesized as described (20, 21). LY2940680 was from Selleckchem (Houston, TX, USA), SANT-1 and XAV939 from Tocris Bioscience (Lille, France), and itraconazole, 17-DMAG, and IWR-1 from Sigma-Aldrich (Saint-Quentin Fallavier, France). ShhN was provided by Dr. D. Baker (Biogen Idec, Boston, MA, USA). [3H]Thymidine was from PerkinElmer Life (Waltham, MA, USA). [3H]MRT-92 (38.1 Ci⋅mmol−1) has been tritiated by RC Tritec (Teufen, Switzerland). SAG was dissolved in ethanol, BC in methanol, and all other compounds in DMSO at a concentration of 10 mM or 2.5 nM for GSA-10. No significant influence of the vehicle (DMSO) was observed in the assays.
Plasmids and mutagenesis
The pRL-TK Renilla luciferase plasmid has been described (23) and is referred to in the text as R. reniformis luciferase. The pRK5 and pRK5-SP-myc-Smo plasmids have been described (44). hSmo mutations (L325F3.36f, V329F3.40f, D384AECL2, S387AECL2, Y394AECL2, R400A5.43f, I408F5.51f, T466F6.47f, D473H6.54f, E518K7.38f, M525G7.45f) were generated by the Imagif platform in Gif-sur-Yvette using a site-directed mutagenesis protocol. The Wnt reporter plasmid M50 Super8xTOPFlash (Tcf/Lef), the pLNC Wnt-3aHA, and the control pRL-TK R. reniformis luciferase were obtained from Addgene.
Antibodies
A previously described polyclonal rabbit antiserum against rat Smo was used (45). The mouse anti-acetylated tubulin antibody, the mouse monoclonal anti–c-Myc antibody, the rabbit anti–γ-tubulin, and the goat anti-mouse FITC antibody were obtained from Sigma-Aldrich. The goat anti-mouse Alexa Fluor 546 antibody was obtained from Invitrogen (Saint-Aubin, France) and the goat anti-rabbit FITC antibody from Merck Millipore (Lyon, France). The horseradish peroxidase–conjugated goat anti-mouse IgG was obtained from Merck Millipore and the anti-rabbit from Sigma-Aldrich.
Cell culture and transfection
Zeocin, geneticin (G418), penicillin–streptomycin, and all cell culture media or products were from Invitrogen except as stated otherwise. HEK293, C3H10T1/2, and NIH3T3 (American Type Culture Collection, Manassas, VA, USA) were cultured in DMEM supplemented with 10% fetal calf serum. The Shh-light2 cells (from Prof. P. A. Beachy, Stanford University, Stanford, CA, USA) and HEK293 cells stably expressing hSmo (HEK-hSmo) were cultured in the same medium supplemented with 0.4 mg/ml G418 and 0.150 mg/ml Zeocin or 0.5 mg/ml G418, respectively. HEK293 cells were transiently transfected by X-tremeGENE 9 from Roche Diagnostics (using 20 µg per T75 flask) with WT or mutant Smo receptors for [3H]MRT-92 binding. Cells were distributed into 6-well plates containing glass coverslips coated with 0.05 mg/ml poly-d-lysine (BD Biosciences, Le Pont de Claix, France) for BC binding, Smo ciliary accumulation, and immunofluorescence experiments, or into 96-well plates for alkaline phosphatase (AP) assay and Gli-dependent luciferase reporter assay.
Gli-dependent luciferase reporter, AP, and ciliary Smo accumulation assays
Shh-light2 cells were incubated for 40 hours with ShhN (5 nM) and the studied compounds. The cell-based bioassay was performed as described previously (18). C3H10T1/2 cells were incubated for 6 days in the presence of SAG (0.1 µM) or GSA-10 (1 µM) and the studied compounds. The cell-based bioassay was performed as described previously (46). The protocol used and data analysis for ciliary Smo accumulation were performed as described previously (19).
Primary cerebellar cultures and medulloblastoma proliferation assay
Isolation of cerebellar granule cell precursors (GCPs) and quantitation of [3H]thymidine incorporation were performed as described previously (47). Shh medulloblastomas were obtained from Ptc+/− mice (003081; The Jackson Laboratory, Bar Harbor, ME, USA) (28) and were serially passaged in nude mice. Shh medulloblastoma cells were isolated and cultured as described (48). Cells from 3 independent Shh medulloblastomas were treated in culture 48 hours and cell viability was measured using the CellTiter-Glo (Promega, Lyon, France). The protocol involving mouse use was performed in accordance with National and European regulation on the protection of animals used for scientific purposes.
Membrane preparation
HEK-hSmo or HEK293 transiently expressing wild-type (WT) or mutant Smo or an empty vector (pRK5) were collected by scraping in PBS. Cell pellets were resuspended at 4°C in 10 volumes of ice-cold buffer HE (50 mM HEPES pH 7.4, 1 mM EDTA) supplemented by a protease inhibitors cocktail (10 µl/ml; Sigma-Aldrich). Cell pellets were homogenized with a Polytron blender for 15 seconds, placed on ice (5 minutes), and homogenized for 15 seconds. After centrifugation (500 g, 30 minutes, 4°C), the supernatant was centrifuged again (48,000 g, 45 minutes, 4°C). A Dounce homogenizer was used to resuspend the final pellet using 2 ml of ice-cold buffer HE. The membrane suspension was passed through a 25-gauge needle, formed into aliquots, and stored at −80°C. The protein concentration was determined by the method of Bradford with bovine serum albumin as standard.
Immunocytochemistry
Detection of Smo protein inside HEK293 and at the cell surface was performed as described previously (46). The Smo N-terminal Myc tag was detected using a mouse monoclonal anti-Myc antibody (1/400). Smo expression (green) was visualized using a fluorescent anti-mouse FITC antibody (1/1000).
Western blot analysis
Western blot analyses were performed as described (21, 49). Nitrocellulose membranes were probed (2 hours) at room temperature with a mouse monoclonal anti-Myc antibody (1/2000) or a rabbit anti–γ-tubulin (1/1000). Horseradish peroxidase–conjugated goat anti-mouse or anti-rabbit IgG was used, respectively, as secondary antibody (1/1000), and immunoreactivity revealed with enhanced chemiluminescence kit ECL (Thermo Scientific, Waltham, MA, USA).
[3H]MRT-92 and BC binding assays
[3H]MRT-92 binding assays were performed in polypropylene tubes. Membranes, radioligand and drugs were diluted in HEPES buffer (50 mM HEPES, 3 mM MgCl2, pH 7.2) supplemented with 0.2% bovine serum albumin to reduce nonspecific binding. Usually, 2 µg or 20 µg of protein, for stably or transiently transfected cells, were incubated with 0.4 nM of [3H]MRT-92 in a final volume of 400 µl. Triplicate incubations were generally carried out at 37°C for 180 min and stopped in ice-cold water followed by rapid filtration (Brandel apparatus) through glass fiber filters (GF/C) pretreated with 0.3% polyethyleneimine. Filters were washed with ice-cold PBS-NaCl (PBS pH 7.4 with 400 mM NaCl). Radioactivity on the filters was quantified using a liquid scintillation counter (PerkinElmer Life). Specific binding to WT or D473H Smo is defined as the binding inhibited by 1 µM GDC-0449 or MRT-83, respectively, and represented more than 95% of total binding. Radioactive saturation experiments were performed with the addition of [3H]MRT-92 over a range of concentrations (0.1–10.5 nM). BC assay was performed as described previously (18). Data were expressed as percentage of fluorescence intensity observed with BC alone.
Data analysis
Means and sem were calculated using Excel 2007 (Microsoft Corp., Redmond, WA, USA). Statistical analysis was performed by Student’s t test. Statistical significance was considered for P ≤ 0.05, P ≤ 0.01, and P ≤ 0.001. Curve fitting, half maximal inhibitory concentration (IC50), and Kd determinations were performed by Prism 4.03 software (GraphPad Software, San Diego, CA, USA). Ki values for compounds were determined from the following equation (50):
where IC50 is their half-inhibition concentration, L is the concentration of [3H]MRT-92, and Kd is the equilibrium dissociation constant of [3H]MRT-92.
RESULTS
Comparative binding mode of antagonists to hSmo transmembrane domain
The X-ray structures of hSmo bound to several antagonists including LY2940680, SANT-1, cyclopamine, and Anta XV, along with the associated mutagenesis experiments (2, 3), revealed 2 types of 7TM-directed antagonists: first, type 1—those binding mostly to ECL, (from here on called site 1), e.g., LY2940680, cyclopamine, Anta XV, GDC-0449, and LDE225; and second, type 2—those penetrating deeply into the 7TM cavity (site 2), e.g., SANT-1 (2, 3) (Fig. 1A, B). These studies have underlined the crucial role of L3253.36f, the rotameric state of which influences the volume of the 7TM cavity and the level of ligand penetration into the crevice. We use the modified Ballesteros-Weinstein residue number specifically adapted for class F GPCR (3). In this scheme, each transmembrane residue is labeled with a superscript x.yyf, denoting the transmembrane helix (x) and the position (yy) with respect to a fully conserved amino acid for class F receptors (for which yy = 50). The letter f indicates that the numbering applies to a class F GPCR.
Figure 1.

Binding of different antagonists (A, LY2940680; B, SANT-1; C, MRT-92) to the transmembrane domain of hSmo (white ribbons). The ECD, the 3 extracellular loops (ECL1, ECL2, ECL3), and the 7 transmembrane helices (I–VII) are labeled, with the exception of helix VI, which is masked for the sake of clarity. The bound ligand is indicated by sticks and rendered by a transparent surface. The inset illustrates the structure of each ligand. D) All 3 ligands are simultaneously rendered in the binding site. A, B) X-ray structures (PDB accession codes 4JKV and 4N4W, respectively) (2, 3). C) Obtained by docking MRT-92 into the SANT-1-bound hSmo structure.
The cleft-closed structure (PDB accession codes 4JKV, 4O9R, 4QIM) accommodates the antagonist close to the ECL upper part (type 1 binding, Supplemental Fig. 1A), whereas the cleft-opened structure (PDB accession code 4N4W) enables the antagonist to deeply penetrates the 7TM cavity, toward more cytoplasmic-proximal residues (type 2 binding, Supplemental Fig. 1B). Although type 1 and type 2 antagonists partially overlap in their bound mode, we wondered whether a third type of antagonists interacting simultaneously with both sites 1 and 2 may exist.
Using virtual screening of commercial compound libraries against a Smo antagonist pharmacophore, we recently identified an acylthiourea (MRT-10) displaying antagonist activity in various Hh assays (IC50 = 0.65 µM for differentiation of the multipotent mesenchymal C3H10T1/2 cells into AP-positive osteoblasts) (18). Next, we developed and optimized acylthiourea, acylurea, and acylguanidine derivatives of MRT-10, which led to the discovery of acylguanidine MRT-83 (Supplemental Fig. 2). MRT-83 exhibited antagonist activity comparable to that of GDC-0449 (Table 1) (19, 20). It inhibited BC binding to HEK-hSmo cells, suggesting that MRT-83 shares interactions with site 1 residues. We could not reproducibly dock MRT-83 in any of the available cleft-closed hSmo structures. However, the cleft-opened conformation (4N4W, SANT-1 bound Smo) accommodated MRT-83 (Supplemental Fig. 1C), in agreement with the observed structure–activity relationships on this last compound (20). The docking study, however, showed that the very bottom part of the pocket (site 2) is still unoccupied, suggesting that further elongating the biaryl moiety of MRT-83 may lead to even more potent analogs.
TABLE 1.
Compared IC50 values for MRT-92, MRT-83, LDE225, and GDC-0449 in Hh cell-based assays
| Compound | IC50 (nM) |
||||
|---|---|---|---|---|---|
| Shh-Light2 (1) | C3H10T1/2 (2) |
GCPs (3) | BC binding (4) | ||
| SAG | GSA-10 | ||||
| MRT-92 | 2.8 ± 0.5 | 5.6 ± 0.4 | 1000 ± 50 | 0.4 ± 0.1 | 8.4 ± 0.9 |
| MRT-83 | 15 ± 2a | 11 ± 3a | 38 ± 7c | 6 ± 1a | 5 ± 1b |
| LDE225 | 12 ± 2 | 20 ± 1a | 92 ± 12c | 3 ± 1 | 8 ± 2 |
| GDC-0449 | 7 ± 1b | 11 ± 1b | 3300 ± 600c | 4 ± 1b | 7 ± 1b |
IC50 values were determined on Gli-dependent luciferase reporter activity induced in (1) Shh-Light2 cells, (2) AP activity in C3H10T1/2 cells upon stimulation by SAG or GSA-10, (3) proliferative activity of cerebellar GCPs measured by [3H]thymidine incorporation, and (4) BC binding to HEK293-hSmo cells. Data are the means ± sem of at least 3 independent experiments, as depicted in Figs. 2 and 3.
Roudaut et al. (19);
Solinas et al. (20);
Gorojankina et al. (21).
Discovery of MRT-92, a Smo antagonist that selectively blocks the Hh canonical pathway
Following our design hypothesis, we synthesized MRT-83 derivatives with longer biaryl moieties (Table 2) and evaluated their potency to block Smo-induced differentiation of the mesenchymal progenitor cells into osteoblasts (21, 22). The Smo agonists SAG and GSA-10 stimulate the differentiation of C3H10T1/2 cells into AP-positive osteoblasts by stabilizing different agonist-bound Smo conformational states (SmoSAG and SmoGSA-10) exhibiting distinct antagonist-binding preferences and pharmacologic properties (21). Among the 5 synthesized analogs, MRT-92 blocked both SAG (0.1 µM) and GSA-10 (1 µM) induced differentiation of C3H10T1/2 cells as measured by the AP response, albeit with a strikingly different potency (Fig. 2A and Table 1). MRT-92 displayed an IC50 of 5.6 nM for SAG induction of AP response, whereas it poorly blocked SmoGSA-10, with an IC50 of 1000 nM. These data indicate that although MRT-92 is a low-affinity SmoGSA-10 antagonist, it selectively blocks SmoSAG-induced AP response in C3H10T1/2 cells with significantly high potency.
TABLE 2.
IC50 values for MRT-83 and derivatives on SAG- and GSA-10 -induced differentiation of C3H10T1/2 cells
| Compound | Structure | IC50 (µM) |
|
|---|---|---|---|
| SmoSAG | SmoGSA-10 | ||
| MRT-83 | 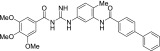 |
0.011 ± 0.003 | 0.038 ± 0.007 |
| MRT-91 | 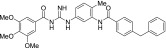 |
0.22 ± 0.02 | 2.6 ± 1.4 |
| MRT-92 | 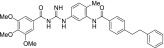 |
0.006 ± 0.001 | 1.0 ± 0.05 |
| MRT-93 | 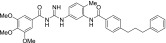 |
0.039 ± 0.009 | 1.4 ± 0.4 |
| MRT-94 | 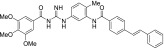 |
0.015 ± 0.004 | 0.7 ± 0.1 |
| MRT-95 | 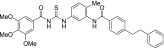 |
0.033 ± 0.006 | 1.5 ± 0.4 |
C3H10T1/2 cell differentiation was stimulated by SAG (SmoSAG) or GSA-10 (SmoGSA-10). Data are means ± sem of at least 3 independent experiments.
Figure 2.
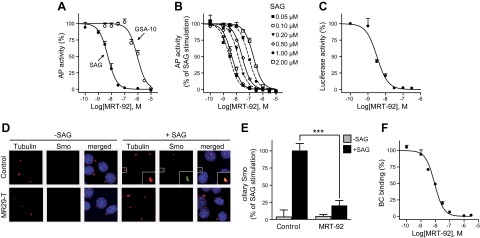
Characterization of MRT-92 in biologic assays. A–C) Activity of MRT-92 on SAG (0.1µM) or GSA-10 (1 µM) induced differentiation of C3H10T1/2 cells as measured from AP activity (A) and on ShhN (5 nM)-induced Gli-dependent luciferase activity in Shh-light2 cells (C). B) MRT-92 competitively blocks SAG-induced AP activity. Its IC50 (2.8 nM) observed with SAG (0.05 µM) increases with higher concentrations of SAG (from 0.05 µM to 2 µM) and was shifted by more than 60-fold at the highest agonist concentration, indicating competitive inhibition. A–C) Inhibition curves were generated using increasing concentrations of MRT-92; values are expressed as percentage of the maximal response induced by SAG, GSA-10, or ShhN, respectively. D, E) NIH3T3 cells untreated or treated with SAG (1 µM) alone or in the presence of MRT-92 (0.3 µM) were stained with antibodies against endogenous Smo (green), acetylated tubulin (red), and DAPI (blue). D) In the absence of SAG, Smo is not detected at the cilium. MRT-92 alone does not induce Smo trafficking to the primary cilium. Cell exposure to SAG induces Smo trafficking to the cilium, which is abolished by MRT-92. E) Quantification of the percentage of Smo-positive cilia after treatment. F) Inhibition of BC (5 nM) binding to HEK-hSmo cells in the presence of increasing concentrations of MRT-92 was visualized using fluorescence microscopy in a representative field. The values are expressed as percentage of the fluorescence detected in control HEK-hSmo incubated with BC alone. Data are means ± sem (n = 3) of a representative experiment over 3 to 5 independent experiments (A–C, F) or of 3 to 5 independent experiments (D, E). ***P ≤ 0.001.
The other acylguanidine or thioacylurea derivatives tested, exhibited a similar micromolar potency toward SmoGSA-10 but were also potent inhibitors at SmoSAG-induced response although with a lower potency than MRT-92. Interestingly, introducing an alkyl linker of increasing size (1 to 3 carbon atoms) between both aryl moieties was first detrimental to potency (MRT-91, 1 carbon linker) and then beneficial when the 2 phenyl moieties are separated by 2 or 3 carbons (Table 2). A saturated 2-carbon linker (MRT-92) seems to be optimal for potency because activity dropped upon further elongation (MRT-93) or unsaturation of the central bond (MRT-94, Table 2). The guanidine moiety of MRT-92 also seems important because the activity of the thiourea analog (MRT-95) was diminished (Table 2). Moreover, MRT-92 showed competitive antagonism to SAG. Increasing concentrations of SAG led to a progressive rightward shift in the MRT-92 inhibition curves with a maximal 60-fold increase in its IC50 (Fig. 2B). These data indicate that MRT-92 shares a common binding site with SAG.
To further explore MRT-92’s potency as an antagonist of the Hh canonical pathway, we determined its antagonist properties in Shh-light2 cells, a NIH3T3 cell line stably transfected with a Gli-dependent firefly luciferase reporter gene used for identifying such antagonists (23). MRT-92 inhibited the ShhN (5 nM)-induced Gli-luciferase reporter transcription and displayed an IC50 of 2.8 nM (Fig. 2C). We then investigated MRT-92 functional properties in multiple cell-based assays. We notably compared its potency to that of chemically different high-affinity hSmo antagonists (MRT-83, GDC-0449, and LDE225; Supplemental Fig. 2) and concluded that MRT-92 is the most potent of all these Smo antagonists (Table 1).
MRT-92 inhibits Smo trafficking at the primary cilium
It is proposed that SAG-induced Smo activation requires Smo accumulation at the primary cilium (4, 19, 24, 25). To examine the mechanism of action of MRT-92, we investigated its ability to modulate the trafficking of endogenous Smo to the primary cilium of NIH3T3 cells (Fig. 2D, E). In DMSO vehicle-treated cells, Smo was not detected at the primary cilium visualized by the acetylated tubulin-positive signal (Fig. 2D). After treatment with SAG (1 µM) for 18 hours, Smo accumulation was induced at the primary cilium, as shown by the colocalization of acetylated tubulin and Smo-positive immunolabelings. MRT-92 (0.3 µM) blocked this effect but did not promote Smo-positive signals when the compound was administrated alone. These data demonstrate that MRT-92 abrogates Smo ciliary translocation promoted by SAG in these cells.
MRT-92 does not block Wnt signaling
Smo displays the highest amino acid sequence identity to Frizzled receptors that are responsible for Wnt signaling (26). We evaluated the ability of MRT-92 to modulate Wnt signaling in HEK293 cells transiently transfected with Tcf/Lef-dependent firefly luciferase reporter together with a R. reniformis luciferase control reporter, in the presence or absence of a Wnt3a plasmid (19). As expected, Wnt3a-induced Wnt signaling was blocked by the Wnt antagonist IWR-1 (10 µM) (27). However, MRT-92 (0.1 µM) did not significantly modify the basal activity level or the Wnt3a-induced Tcf/Lef luciferase activity in these cells (Supplemental Fig. 3), indicating that it does not display significant agonist and antagonist Wnt signaling activity in this assay.
MRT-92 inhibits rat GCPs and Shh medulloblastoma cell proliferation
A hallmark of Smo antagonists is to block proliferation of GCPs induced by Hh pathway activation (1). Thus, we evaluated the ability of MRT-92 to inhibit rat GCPs proliferation in primary culture induced by Smo activation as measured by [3H]thymidine incorporation over basal level. MRT-92 inhibited SAG (0.01 µM)-induced GCPs proliferation with an IC50 of 0.4 nM (Fig. 3A), whereas the other tested Smo antagonists were 7 to 15 times less potent (Table 1). Medulloblastoma formation occurs in Ptc+/− mice because of loss of Ptc and aberrant activation of the Hh pathway in GCPs during cerebellar development (28). We then investigated whether MRT-92 can block the constitutive activity of the Hh pathway observed in primary culture of cells from Shh medulloblastoma as measured by the metabolic activity of these cells, used as an index of their proliferation state. We observed that MRT-92 (0.3 µM) and GDC-0449 (3 µM) inhibited this response by more than 50% but remained unmodified by the Wnt/β catenin inhibitor (XAV939, 3 µM). Moreover, the response was completely abolished by a heat shock protein 90 inhibitor (17-DMAG, 0.5 µM) (Fig. 3B). Altogether, these data demonstrate that MRT-92 is a potent antagonist of GCPs proliferation induced by Hh pathway activation through pharmacologic or genetic manipulation.
Figure 3.

Effects of MRT-92 on GCPs and Shh medulloblastoma cell proliferation. A) Antagonist potency of MRT-92 on SAG (0.01 µM)-induced proliferation of rat cerebellar GCPs. Inhibition curve was generated using increasing concentrations of MRT-92 and the values are expressed as a percentage of the maximal response induced by SAG. B) Antagonist activity of MRT-92 on Ptc+/− medulloblastoma cells’ metabolic activity. MRT-92 (0.3 µM), GDC-0449 (3 µM), 17-DMAG (0.5 µM), or XAV939 (3 µM) was added into the medium of Ptc+/− medulloblastoma culture cells, and metabolic activity was measured 48 hours later. Data are means ± sem (n = 3) of a representative experiment over 4 independent experiments (A) or of 4 independent experiments (B). ***P ≤ 0.001.
Characterization of [3H]MRT-92 binding to HEK-hSmo cell membranes
The high potency of MRT-92 for inhibiting the Hh canonical pathway identified in several Hh-based assays, including inhibition of GCPs, encouraged us to develop a tritiated analog ([3H]MRT-92, Fig. 4A) for further evaluating the mode of action of Smo modulators. At 37°C, [3H]MRT-92 (0.30 nM) bound to HEK-hSmo membranes with an association rate constant (k1) of 0.03 min−1 × nM−1, with equilibrium attained after 150 to 180 min (Fig. 4B). Dissociation occurring in the presence of GDC-0449 (20 µM) followed first-order kinetics with a rate constant (k−1) of 0.007 min−1. The ratio (k−1/k1) gave a Kd of 0.24 ± 0.1 nM. Saturation of specific [3H]MRT-92 binding at equilibrium, defined using 1 µM GDC-0449, was monophasic (nH = 0.93) leading to a linear Scatchard plot. GraphPad Prism analysis of the binding isotherms led to a Kd of 0.30 ± 0.1 nM and a maximal capacity value (Bmax) of 14.2 ± 1.0 pmol/mg of protein. At 0.4 nM, specific binding of [3H]MRT-92 to HEK-hSmo represented 95% of total binding, and nonspecific binding increased linearly from 0.1-10 nM of [3H]MRT-92 probe concentration (Fig. 4C). These data indicate that [3H]MRT-92 is a potent radioligand for labeling hSmo. Binding of [3H]MRT-92 to HEK-hSmo membranes was inhibited in a monophasic manner by a series of Smo antagonists of different structures (Supplemental Fig. 2). These include itraconazole, an antifungal agent with Hh antagonist properties (29), and the Smo agonist SAG. The exception was purmorphamine, another Smo agonist (30) (Table 3 and Supplemental Fig. 4). These experiments demonstrate that all these molecules, except purmorphamine, are potent inhibitors of [3H]MRT-92 binding to hSmo.
Figure 4.
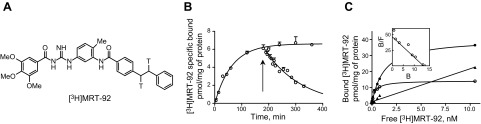
Reversible labeling of the hSmo receptor with [3H]MRT-92. A) Chemical structure of [3H]MRT-92 (T indicates a tritium atom). B) Association and dissociation of [3H]MRT-92 upon addition of 20 µM GDC-0449 (arrow). Computer analysis of the specific binding gave an association rate constant k1 = 3 × 107 M−1⋅min−1 and a dissociation k−1 = 7 × 10−3 min−1. The ratio k−1/k1 gave an equilibrium dissociation constant (Kd) of 0.24 ± 0.1 nM. C) Saturation of [3H]MRT-92 binding to HEK-hSmo cell membranes stably expressing hSmo. Membranes (2 µg protein) were incubated at 37°C for 180 minutes in 0.4 ml of HEPES buffer with [3H]MRT-92 in increasing concentrations. Nonspecific binding was evaluated using 1 µM GDC-0449. Computer analysis of the specific binding gave a Kd of 0.30 ± 0.1 nM and a Bmax of 14.2 ± 1.0 pmol/mg of protein. Inset shows Scatchard plot analysis of the specific binding. Data are means ± sem (n = 3) of a representative experiment over 3 to 5 independent experiments. ●, total binding; ○, specific binding; ▲, nonspecific binding; B, bound [3H]MRT-92; F, free [3H]MRT-92.
TABLE 3.
Potency of Smo modulators using [3H]MRT-92 binding to human WT Smo and D473H6.54f mutant receptors
| Compound |
Ki (nM) |
|
|---|---|---|
| WT | D473H6.54f | |
| MRT-92 | 0.7 ± 0.1 | 0.7 ± 0.2 |
| MRT-83 | 4.6 ± 0.7 | 5.5 ± 0.2 |
| ALLO-2 | 3.8 ± 1.5 | 5.1 ± 2.0 |
| LDE225 | 1.6 ± 0.4 | 108 ± 18*** |
| GDC-0449 | 5.8 ± 1.3 | >1000*** |
| LY2940680 | 0.5 ± 0.2 | 55 ± 0.8*** |
| SANT-1 | 1.1 ± 0.1 | 67 ± 0.9*** |
| M25 | 0.9 ± 0.1 | 61 ± 13*** |
| Itraconazole | 28.3 ± 0.1 | >1000*** |
| SAG | 2.4 ± 0.3 | 45 ± 0.3*** |
| Purmorphamine | >5000 | ND |
Data (means ± sem of 3 to 5 independent experiments) derived from experiments, as shown in Supplemental Fig. 4. ND, not determined.
P ≤ 0.001.
Characterization of the MRT-92 binding site
Evidence from X-ray structures revealed that the binding sites of SAG and cyclopamine are largely overlapping (3, 7) between the extracellular domain (ECD), the 3 ECL, and the upper part of the 7TM helices of hSmo. To delineate the precise binding site of MRT-92, we docked this novel antagonist with Surflex-Dock (31) in all available X-ray structures of hSmo with closed or opened lower 7TM cleft conformation. In agreement with our preliminary docking experiments of the lead compound MRT-83, the initial design hypothesis, and the observed structure–activity relationships (Table 2), we could only dock MRT-92 using the cleft-opened structure (SANT-1-bound form). As anticipated, MRT-92 is predicted to fill the entire 7TM cavity from the upper ECD to its most cytoplasmic-proximal part (Fig. 1C), and it therefore shares characteristics of both type 1 and type 2 Smo antagonists (Fig. 1D). MRT-92 is predicted to tightly occupy the Smo 7TM cavity using 3 main anchoring areas (Fig. 5): 1) aromatic and hydrophobic interactions of the trimethoxyphenyl moiety to the upper ECD (L221ECD) and ECL3 loop (W480ECL3, F484ECL3), 2) a network of 4 hydrogen bonds between the central polar part (acylguanidine, amide) of the ligand and polar residues in the upper extracellular part of the cavity (N219ECD, D384ECL2,Y394ECL2, E5187.38f); and 3) extensive aromatic and hydrophobic contacts in the narrow and deep-penetrating nonpolar part of the cavity (V386ECL2, F391ECL2, Y394ECL2, M2301.32f, H4706.51f, L5227.42f, I4085.51f, T4666.47f, L3253.36f, M5257.45f, V4636.44f, F2742.51f, V3293.40f).
Figure 5.
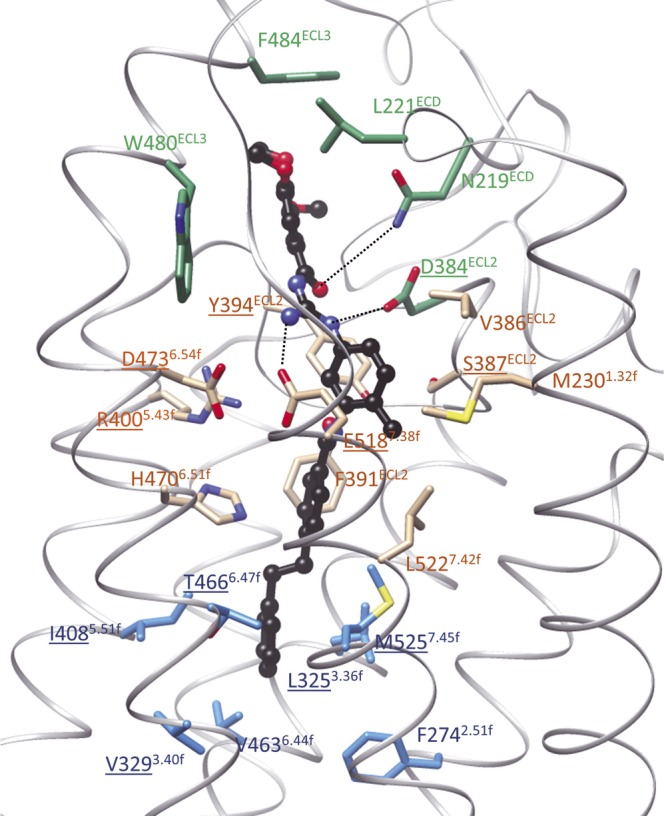
Close-up of the proposed binding mode of MRT-92 to hSmo. The ligand is displayed in ball and sticks using the following color coding: carbon, black; nitrogen, blue, oxygen, red. Smo side chains (carbon atoms) are colored according to their contribution to bind only LY2940680 (green), only SANT-1 (cyan), or both antagonists (tan). Protein nitrogen, oxygen, and sulfur atoms are colored in blue, red, and yellow, respectively. Smo residues are labeled according to the new class F Ballesteros-Weinstein nomenclature (3). Protein–ligand hydrogen bonds are displayed as dashed lines. Amino acids mutated in this study are underlined.
To verify this unique binding mode, we first checked whether MRT-92 could compete with the fluorescent-labeled cyclopamine analog BC, prototypical of type 1 binding (2, 3, 7). MRT-92 indeed blocked BC (5 nM) binding to HEK-hSmo in a dose-dependent manner with an IC50 of 8.4 nM, in the same range the other reference Smo antagonists tested (Fig. 2F and Table 1). These data indicate that MRT-92 binds to hSmo at least at the cyclopamine binding site and therefore presents binding features characteristic of type 1 antagonists.
To precisely identify the amino acids implicated in MRT-92 binding, we mutated 11 amino acids delineating either binding sites 1 and 2 (2, 3, 7, 32) (Fig. 5). For investigating the amino acids implicated in type 1 binding, we mutated into alanine, D384ECL2, S387ECL2, and Y394ECL2 residues, as well as R4005.43f, known to interact with LY2940680 (2). We next characterized the D473H6.54f and E518K7.38f mutants shown to abrogate GDC-0449 binding (2, 16, 32). To further investigate the interaction of the terminal biaryl side chain of MRT-92 with site 2, we engineered 4 mutants with a reduced site 2 volume (L325F3.36f, V329F3.40f, I408F5.51f, and T466F6.47f) and 1 mutated hSmo (M525G7.45f) with an increased site 2 volume (Fig. 5). These mutants and WT hSmo were transiently transfected into HEK293 cells. Their subcellular distribution was visualized using a Myc antibody that recognizes the Myc epitope located at the amino-terminal tail of the WT and mutant receptors. Next, we analyzed Smo immunoreactivity on permeabilized and nonpermeabilized transfected HEK293 cells. Myc immunoreactivity was detected at the cell surface of nonpermeabilized and at the cytoplasmic level of permeabilized HEK293 transfected cells, while no signal was detectable with control pRK5-transfected cells (Fig. 6A). These data suggest that all mutant receptors were addressed to the cell membrane. Western blot analysis under reducing SDS-PAGE conditions identified a major polypeptide migrating with a molecular mass of 86 kDa in whole lysates of membranes of the transfected HEK293 cells. This signal was absent in control pRK5-transfected cell preparations, thereby indicating its specificity. Importantly, all mutant receptors were expressed at a similar level (L325F, V329F, I408F, T466F, D473H) or more robustly (D384A, S387A, Y394A, R500A, E518K, M525G) than the WT receptor (Fig. 6B).
Figure 6.

Functional analysis of human WT and mutant Smo receptors. A) Analysis of hSmo expression by immunocytofluorescence in HEK293 cells. Cells were transfected with the WT or mutant hSmo (D384A, S387A, Y394A, R400A, L325F, V329F, I408F, T466F, M525G, D473H E518K). The N-terminal Myc tag was detected using an anti-Myc antibody on nonpermeabilized (NP) or permeabilized (P) cells. B) Immunoblot analysis of membranes (5 µg) of HEK293 cells transiently transfected with an empty vector (pRK5), or a vector containing WT or mutant hSmo, was performed after SDS-PAGE. Smo proteins were detected using an anti-Myc antibody. Molecular mass marker is shown on the left (kDa). Arrowhead on the right indicates the major band corresponding to the WT or mutant hSmo and to γ-tubulin used as charge control. C) Effect of Smo mutations on [3H]MRT-92 total binding. Membranes (20 µg of protein) of HEK293 cells transiently expressing WT or mutant hSmo were incubated with 0.5 nM [3H]MRT-92. [3H]MRT-92 binding is expressed as percentage of the WT total binding (2000 cpm). Data are means ± sem (n = 3) of 3 to 5 independent experiments. *P ≤ 0.05; ***P ≤ 0.001.
To investigate the contribution of these amino acids to the MRT-92 binding pocket, we performed a [3H]MRT-92 binding assay on membranes from HEK293 transiently transfected with WT and mutant Smo receptors. S387AECL2, I408F5.51f, and D473H6.54f mutations did not modify [3H]MRT-92 binding (Fig. 6C)—data that were further corroborated by inhibition competition assay of the radioligand showing no difference in the IC50 of MRT-92 for WT and these mutants (Supplemental Table 1). Our data suggest that these residues do not play a significant role in MRT-92 recognition. In contrast, [3H]MRT-92 binding to membranes of HEK293 cells expressing D384AECL2, Y394AECL2, R400A5.43f, and E518K7.38f mutations was reduced to the level of the control pRK5-transfected cells, excluding further [3H]MRT-92 binding analysis and demonstrating that these amino acids are required for MRT-92 recognition. For L325F3.36f, V329F3.40f, T466F6.47f, and M525G7.45f mutants, [3H]MRT-92 binding was significantly impaired (Fig. 6C). In agreement with these data, the IC50 for MRT-92 inhibition deduced from the radioligand competition assay were right-shifted 3- to 8-fold (Supplemental Table 1). On the contrary, for the M525G7.45f mutant, the inhibition curve for MRT-93, which displayed a longer alkyl linker, was left shifted, although it was not statistically significant (Supplemental Fig. 5). These data suggest that MRT-93 can still interact at the back of the mutant pocket, whereas MRT-92 interaction is diminished, which is in agreement with the MRT-92 binding model.
We next determined the IC50 for LY2940680 (type 1 antagonist) and SANT-1 (type 2 antagonist) on mutant receptors retaining [3H]MRT-92 binding (Supplemental Table 1). The IC50 for LY2940680 inhibition of [3H]MRT-92 binding was right shifted (3- to 100-fold) for the S387AECL2, L325F3.36f, and D473H6.54f mutants but did not differ from that of WT receptor for the other mutants (Supplemental Table 1). The ability of SANT-1 to inhibit [3H]MRT-92 binding to V329F3.40f and T466F6.47f mutants was abolished, and it was severely impaired for L325F3.40f, I408F5.51f, and M525G7.45f mutants (4- to 140-fold drop of the IC50), but was not modified for the S387AECL2 mutant. Taken together, these data confirm our docking hypothesis that MRT-92-binding mode differs from that of either LY2940680 or SANT-1 by simultaneously occupying binding sites 1 and 2 (Fig. 7).
Figure 7.
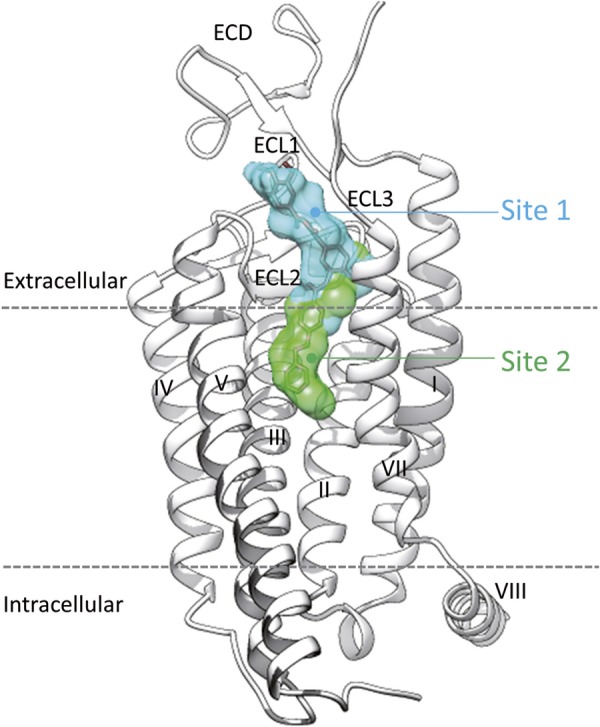
Schematic representation of MRT-92 binding to hSmo. Site 1 (cyan) is occupied by the agonists (SAG, SAG1.5) and antagonists (LY2940680; Anta XV, cyclopamine, KAAD-cyclopamine, GDC-0449), whereas site 2 (green) is only used by the antagonist SANT-1. The novel antagonist MRT-92 (stick model) occupies both sites simultaneously. Smo ECD and loops (ECL1–3) as well as the transmembrane helices are labeled as in Fig. 1. The intracellular helix 8 (VIII) is further indicated with respect to the membrane boundaries (dashed lines).
Differential binding modes of Smo modulators to the GDC-0449 resistant D473H mutant
Because the IC50 of MRT-92 was not modified for the D473H mutant, we next evaluated the properties of [3H]MRT-92 binding to this mutant. Saturation of specific [3H]MRT-92 binding to membranes of HEK293 cells transiently expressing D473H mutant leads to a linear Scatchard plot and a Kd value of 0.50 ± 0.1 nM (Supplemental Fig. 6), which was not significantly different from [3H]MRT-92 Kd for WT hSmo. Thus, we utilized this radioligand assay for investigating whether Hh and Smo modulators of different structures interacted with this naturally occurring mutant. [3H]MRT-92 binding to HEK293 membranes expressing D473H mutant was inhibited in a monophasic manner by MRT-92, MRT-83, and ALLO-2 with potency similar to the WT receptor. However, the dose-response curves for LDE225, LY2940680, SANT-1, M25, and SAG were right shifted (20–100-fold) or abrogated for GDC-0449 and itraconazole (Table 3 and Supplemental Fig. 4).
DISCUSSION
Structural studies of hSmo in complex with various antagonists (2, 3, 7) revealed the existence of 2 type of antagonists: those binding either to the upper 7TM cavity (type 1; e.g., LY2940680, cyclopamine, vismodegib, LDE225) or the bottom part of the 7TM cavity (type 2; e.g., SANT-1). Studying the binding mode of the herein described antagonist, MRT-92, suggests a third binding mode (type 3) bridging the 2 binding sites described above. Moreover, our present study demonstrates that MRT-92 is among the most potent Smo antagonist known so far (Table 1), being almost 10-fold more potent in inhibiting rat GCPs proliferation than GDC-0449 and LDE225, 2 compounds currently in clinical trials for the treatment of medulloblastoma [(17, 33); see also trial numbers NCT00822458, NCT01601184, NCT01239316, NCT01125800, NCT01208831, and NCT00880308, at ClinicalTrials.gov]. MRT-92, like GDC-0449, also blocks the proliferation of medulloblastoma cells from Ptc+/− mice (Fig. 3B), suggesting its potential use in the treatment of medulloblastoma associated to mutation-driven Hh-associated cancers.
Smo antagonists are under intense investigation for a growing number of Hh-dependent tumors (1, 9, 34). However, several clinical trials with Smo antagonists were either discontinued (TAK-441, IPI-926) or led to negative results for patients with metastatic colorectal carcinoma and ovarian cancers treated with GDC-0449 (11, 35, 36). To better understand these failures, it is of utmost importance to relate the clinical efficacy of Smo antagonists to their binding mode and to check whether a highly potent type 3 antagonist like MRT-92 may confer some advantages over the existing type 1 or type 2 Smo antagonists.
Smo can adopt different conformations, both in the cytoplasm and at the primary cilium, and Smo antagonists stabilize distinct conformations in the cilium (1). Cyclopamine drives Smo from the cytoplasm into the cilium in an inactive state, whereas most Smo antagonists acting in the canonical pathway are proposed to block an inactive form of Smo in the cytoplasm. MRT-92 belongs to this class of antagonists. It is suggested that ciliated medulloblastoma with high Hh signaling would respond to treatments that target the primary cilium (37). However, cilia can both mediate and suppress Hh-dependent tumorigenesis (4, 9), and from a therapeutic point of view, it is not yet evident whether a Smo antagonist should block Smo in the cytoplasm or in the primary cilium. MRT-92, like cyclopamine and GDC-0449, displays low sensitivity to block the effects of GSA-10 on Smo. Hence, it would be important to further determine whether the cancer-related effects of Smo clinical candidates and their adverse effects, which include hair loss, muscle spasms, fatigue, and decreases in weight and appetite (9), are associated with inhibition of different Smo conformations such as SmoSAG and SmoGSA-10.
The X-ray structures of hSmo bound with various antagonists and the agonist SAG1.5 (2, 3, 7) demonstrated the existence of a long and narrow ligand binding pocket delineated by the extracellular linker 3 ECL and 7TM residues where Smo modulators bind. The evidence that MRT-92 binding site (Fig. 7) overlaps the binding pocket of both type 1 and type 2 antagonists is supported by 1) its binding inhibition of a known type 1 antagonist (BC; Table 1), 2) the ability of type 1 and type 2 antagonists to potently inhibit [3H]MRT-92 binding (Table 3), 3) the observation that specific mutations affecting type 1 (3 in ECL2 and 1 in TM7) or type 2 (2 in TM3, 1 in TM6, and 1 in TM7) antagonists abolished or significantly reduced [3H]MRT-92 binding to Smo mutants (Supplemental Table 1), and 4) MRT-92 competitively inhibited SAG-induced differentiation of mesenchymal cells. The 7TM cavity can accommodate a large variety of ligand chemotypes (Supplemental Fig. 2) at overlapping binding sites, sometimes upon minor conformational rearrangement of a few side chains, like L3253.36f or E5187.38f (2, 3, 7). L3253.36f is proposed to act as a gatekeeper to close or open a back pocket in the 7TM crevice (3). This plasticity of the 7TM cavity, as revealed by X-ray studies, is a key player in MRT-92 antagonist potency. When its side chain is perpendicular to the 7TM main axis (i.e., bound to LY2940680), the cavity ends at the middle of the helical bundle, therefore forcing the bound antagonist to mainly interact with ECL residues. When its side chain is parallel to the 7TM main axis (i.e., bound to SANT-1), the back pocket is open such that antagonists deeply penetrate into a bigger cavity (800 A2 for SANT-1-bound Smo vs. 700 A2 for LY2940680-bound Smo). Interestingly, the binding of [3H]MRT-92 was only moderately affected by the L325F3.36f mutation, and competition binding experiments revealed that this mutation slightly decreases binding of LY2940680 and of SANT-1 (Supplemental Table 1). In addition, this mutation did not affect the binding of [3H]cyclopamine (3).
Mutations in a single residue (e.g., D473H6.54f) provides the molecular basis for chemoresistance to GDC-0449 (16). The binding of various Smo modulators to the D473H6.54f mutant was rendered difficult by the fact that [3H]cyclopamine does not bind to this mutant excluding further analysis. Here, we propose that MRT-92 will be insensitive to that mutation (Fig. 6), as supported by a similar Kd deduced from saturation experiments of [3H]MRT-92 toward WT and D473H6.54f mutant (Fig. 4 and Supplemental Fig. 6) and a similar Ki for inhibiting [3H]MRT-92 binding to these receptors. This property is also shared by its close analogs MRT-83 (19) and ALLO-2, which inhibited the GDC-0449-resistant D477G mouse Smo mutant and inhibited both BC and [3H]cyclopamine binding (38). We took advantage of this fact to characterize the potency of structurally different Smo modulators. We observed notably that the D473H6.54f mutation drastically impairs the potency of LDE225, GDC-0449, LY2940680, M25, SANT-1, and SAG to inhibit [3H]MRT-92 binding (Supplemental Fig. 4). Our data now support previous results deduced from crystallographic experiments for an important role of D4736.54f, clustered with R4005.43f and E5187.38f, to selectively interact with some type 1 antagonists (GDC-0449, cyclopamine), but which was less appreciated for LY2940680 (2), whereas MRT-92 binds to the GDC-0449-resistant W281L2.58f and V321M3.32f mutants has to be further investigated (39).
The antifungal agent itraconazole, shown to inhibit Hh pathway activation with an IC50 in the micromolar range (40), is in clinical trials for the treatment of BCC associated with this pathway (41). Itraconazole does not compete to the BC binding site, and it is hypothesized to act on Smo indirectly (40). We now show that this molecule inhibits [3H]MRT-92 binding to hSmo with high potency (Ki = 28 nM), providing the first direct evidence that this molecule is a potent Smo antagonist. However, we found that inhibition of [3H]MRT-92 by itraconazole was sensitive to the D473H6.54f mutation, whereas this molecule was shown to inhibit WT and GDC-0449-resistant D477G mouse Smo with the same potency (29). Thus, further work is required to delineate whether itraconazole belongs to type 1 to 3 antagonists or to yet another type.
Although a large number of Smo antagonists are developed, only a limited number of agonists are characterized (1, 42). SAG inhibits [3H]MRT-92 binding to hSmo, whereas purmorphamine did not. These data suggest a different binding mode for SAG and purmorphamine because the latter was shown to interact at the level of the cyclopamine binding site (30). Recent evidence argues that cyclopamine and GDC-0449, 2 molecules displaying antagonist activity toward canonical Hh signaling, also display agonist activity toward Smo in a 3T3-L1 model of adipose tissue (43). Whether these molecules activate a different form of Smo is not yet known.
Smo antagonists are intensely studied for Hh-dependent tumors. MRT-92 and its derivatives, including [3H]MRT-92, have increased the understanding of how small molecules interact at Smo 7TM and the development of novel antagonists interacting with both sites 1 and 2. This, we believe, will open new avenues for cancer therapy and novel therapeutics.
Supplementary Material
Acknowledgments
This work has benefited from the facilities of the TEFOR Molecular Biology Unit, IMAGIF, Gif Research Center. This work was supported by grants from La Ligue contre le Cancer (Comité des Yvelines) and Fondation Association pour la Recherche sur le Cancer (ARC) to M.R. from l’Institut National du Cancer (INCa R10067LS), Marie Curie Integration Grant FP7-PEOPLE-2011-CIG 294010, Centre National de la Recherche Scientifique/Institut National de la Santé et de la Recherche Médicale (CNRS/INSERM) and Institut Curie (to O.A.). L.H. is the recipient of a doctoral fellowship from La Ligue contre le Cancer and la Fondation Recherche Médicale (FDT20140930825) and L.B. from the Ministère de l'Enseignement Supérieur et de la Recherche.
Glossary
- 7TM
7-transmembrane
- AP
alkaline phosphatase
- BC
bodipy cyclopamine
- BCC
basal cell carcinoma
- ECD
extracellular domain
- ECL
extracellular loop
- GCP
granule cell precursor
- Hh
Hedgehog
- hSmo
human Smoothened
- IC50
half maximal inhibitory concentration
- PDB
Protein Data Bank
- Ptc
patched
- Smo
Smoothened
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Ruat M., Hoch L., Faure H., Rognan D. (2014) Targeting of Smoothened for therapeutic gain. Trends Pharmacol. Sci. 35, 237–246 [DOI] [PubMed] [Google Scholar]
- 2.Wang C., Wu H., Katritch V., Han G. W., Huang X. P., Liu W., Siu F. Y., Roth B. L., Cherezov V., Stevens R. C. (2013) Structure of the human smoothened receptor bound to an antitumour agent. Nature 497, 338–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang C., Wu H., Evron T., Vardy E., Han G. W., Huang X. P., Hufeisen S. J., Mangano T. J., Urban D. J., Katritch V., Cherezov V., Caron M. G., Roth B. L., Stevens R. C. (2014) Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 5, 4355–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruat M., Roudaut H., Ferent J., Traiffort E. (2012) Hedgehog trafficking, cilia and brain functions. Differentiation 83, S97–S104 [DOI] [PubMed] [Google Scholar]
- 5.Briscoe J., Thérond P. P. (2013) The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14, 416–429 [DOI] [PubMed] [Google Scholar]
- 6.Taipale J., Cooper M. K., Maiti T., Beachy P. A. (2002) Patched acts catalytically to suppress the activity of Smoothened. Nature 418, 892–897 [DOI] [PubMed] [Google Scholar]
- 7.Weierstall U., James D., Wang C., White T. A., Wang D., Liu W., Spence J. C., Bruce Doak R., Nelson G., Fromme P., Fromme R., Grotjohann I., Kupitz C., Zatsepin N. A., Liu H., Basu S., Wacker D., Han G. W., Katritch V., Boutet S., Messerschmidt M., Williams G. J., Koglin J. E., Marvin Seibert M., Klinker M., Gati C., Shoeman R. L., Barty A., Chapman H. N., Kirian R. A., Beyerlein K. R., Stevens R. C., Li D., Shah S. T., Howe N., Caffrey M., Cherezov V. (2014) Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 5, 3309–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J. K., Taipale J., Cooper M. K., Beachy P. A. (2002) Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 16, 2743–27489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amakye D., Jagani Z., Dorsch M (2013) Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 19, 1410–1422 [DOI] [PubMed] [Google Scholar]
- 10.Ng J. M., Curran T. (2011) The Hedgehog’s tale: developing strategies for targeting cancer. Nat. Rev. Cancer 11, 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hadden M. K. (2013) Hedgehog pathway inhibitors: a patent review (2009—present). Expert Opin Ther Pat 23, 345–361 [DOI] [PubMed] [Google Scholar]
- 12.Von Hoff D. D., LoRusso P. M., Rudin C. M., Reddy J. C., Yauch R. L., Tibes R., Weiss G. J., Borad M. J., Hann C. L., Brahmer J. R., Mackey H. M., Lum B. L., Darbonne W. C., Marsters J. C. Jr., de Sauvage F. J., Low J. A. (2009) Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 361, 1164–1172 [DOI] [PubMed] [Google Scholar]
- 13.LoRusso P. M., Rudin C. M., Reddy J. C., Tibes R., Weiss G. J., Borad M. J., Hann C. L., Brahmer J. R., Chang I., Darbonne W. C., Graham R. A., Zerivitz K. L., Low J. A., Von Hoff D. D. (2011) Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 17, 2502–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nachtergaele S., Whalen D. M., Mydock L. K., Zhao Z., Malinauskas T., Krishnan K., Ingham P. W., Covey D. F., Siebold C., Rohatgi R. (2013) Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. eLife 2, e01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rana R., Carroll C. E., Lee H. J., Bao J., Marada S., Grace C. R., Guibao C. D., Ogden S. K., Zheng J. J. (2013) Structural insights into the role of the Smoothened cysteine-rich domain in Hedgehog signalling. Nat. Commun. 4, 2965–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yauch R. L., Dijkgraaf G. J., Alicke B., Januario T., Ahn C. P., Holcomb T., Pujara K., Stinson J., Callahan C. A., Tang T., Bazan J. F., Kan Z., Seshagiri S., Hann C. L., Gould S. E., Low J. A., Rudin C. M., de Sauvage F. J. (2009) Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 326, 572–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buonamici S., Williams J., Morrissey M., Wang A., Guo R., Vattay A., Hsiao K., Yuan J., Green J., Ospina B., Yu Q., Ostrom L., Fordjour P., Anderson D. L., Monahan J. E., Kelleher J. F., Peukert S., Pan S., Wu X., Maira S. M., García-Echeverría C., Briggs K. J., Watkins D. N., Yao Y. M., Lengauer C., Warmuth M., Sellers W. R., Dorsch M. (2010) Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2, 51ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manetti F., Faure H., Roudaut H., Gorojankina T., Traiffort E., Schoenfelder A., Mann A., Solinas A., Taddei M., Ruat M. (2010) Virtual screening-based discovery and mechanistic characterization of the acylthiourea MRT-10 family as smoothened antagonists. Mol. Pharmacol. 78, 658–665 [DOI] [PubMed] [Google Scholar]
- 19.Roudaut H., Traiffort E., Gorojankina T., Vincent L., Faure H., Schoenfelder A., Mann A., Manetti F., Solinas A., Taddei M., Ruat M. (2011) Identification and mechanism of action of the acylguanidine MRT-83, a novel potent Smoothened antagonist. Mol. Pharmacol. 79, 453–460 [DOI] [PubMed] [Google Scholar]
- 20.Solinas A., Faure H., Roudaut H., Traiffort E., Schoenfelder A., Mann A., Manetti F., Taddei M., Ruat M. (2012) Acylthiourea, acylurea, and acylguanidine derivatives with potent hedgehog inhibiting activity. J. Med. Chem. 55, 1559–1571 [DOI] [PubMed] [Google Scholar]
- 21.Gorojankina T., Hoch L., Faure H., Roudaut H., Traiffort E., Schoenfelder A., Girard N., Mann A., Manetti F., Solinas A., Petricci E., Taddei M., Ruat M. (2013) Discovery, molecular and pharmacological characterization of GSA-10, a novel small-molecule positive modulator of Smoothened. Mol. Pharmacol. 83, 1020–1029 [DOI] [PubMed] [Google Scholar]
- 22.Hyman J. M., Firestone A. J., Heine V. M., Zhao Y., Ocasio C. A., Han K., Sun M., Rack P. G., Sinha S., Wu J. J., Solow-Cordero D. E., Jiang J., Rowitch D. H., Chen J. K. (2009) Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 106, 14132–14137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taipale J., Chen J. K., Cooper M. K., Wang B., Mann R. K., Milenkovic L., Scott M. P., Beachy P. A. (2000) Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 406, 1005–1009 [DOI] [PubMed] [Google Scholar]
- 24.Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y., Reiter J. F. (2005) Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021 [DOI] [PubMed] [Google Scholar]
- 25.Rohatgi R., Milenkovic L., Corcoran R. B., Scott M. P. (2009) Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. USA 106, 3196–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Amerongen R., Nusse R. (2009) Towards an integrated view of Wnt signaling in development. Development 136, 3205–3214 [DOI] [PubMed] [Google Scholar]
- 27.Chen B., Dodge M. E., Tang W., Lu J., Ma Z., Fan C. W., Wei S., Hao W., Kilgore J., Williams N. S., Roth M. G., Amatruda J. F., Chen C., Lum L. (2009) Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 5, 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodrich L. V., Milenković L., Higgins K. M., Scott M. P. (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113 [DOI] [PubMed] [Google Scholar]
- 29.Kim J., Aftab B. T., Tang J. Y., Kim D., Lee A. H., Rezaee M., Kim J., Chen B., King E. M., Borodovsky A., Riggins G. J., Epstein E. H. Jr., Beachy P. A., Rudin C. M. (2013) Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 23, 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinha S., Chen J. K. (2006) Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat. Chem. Biol. 2, 29–30 [DOI] [PubMed] [Google Scholar]
- 31.Jain A. N. (2003) Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 46, 499–511 [DOI] [PubMed] [Google Scholar]
- 32.Dijkgraaf G. J., Alicke B., Weinmann L., Januario T., West K., Modrusan Z., Burdick D., Goldsmith R., Robarge K., Sutherlin D., Scales S. J., Gould S. E., Yauch R. L., de Sauvage F. J. (2011) Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 71, 435–444 [DOI] [PubMed] [Google Scholar]
- 33.Rudin C. M., Hann C. L., Laterra J., Yauch R. L., Callahan C. A., Fu L., Holcomb T., Stinson J., Gould S. E., Coleman B., LoRusso P. M., Von Hoff D. D., de Sauvage F. J., Low J. A. (2009) Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 361, 1173–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scales S. J., de Sauvage F. J. (2009) Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 30, 303–312 [DOI] [PubMed] [Google Scholar]
- 35.McMillan R., Matsui W. (2012) Molecular pathways: the hedgehog signaling pathway in cancer. Clin. Cancer Res. 18, 4883–4888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin T. L., Matsui W. (2012) Hedgehog pathway as a drug target: Smoothened inhibitors in development. Onco Targets Ther 5, 47–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han Y. G., Kim H. J., Dlugosz A. A., Ellison D. W., Gilbertson R. J., Alvarez-Buylla A. (2009) Dual and opposing roles of primary cilia in medulloblastoma development. Nat. Med. 15, 1062–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao H., Jin Q., Koo D. I., Liao X., Englund N. P., Wang Y., Ramamurthy A., Schultz P. G., Dorsch M., Kelleher J., Wu X. (2011) Small molecule antagonists in distinct binding modes inhibit drug-resistant mutant of smoothened. Chem. Biol. 18, 432–437 [DOI] [PubMed] [Google Scholar]
- 39.Brinkhuizen T., Reinders M. G., van Geel M., Hendriksen A. J., Paulussen A. D., Winnepenninckx V. J., Keymeulen K. B., Soetekouw P. M., van Steensel M. A., Mosterd K. (2014) Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J. Am. Acad. Dermatol. 71, 1005–1008 [DOI] [PubMed] [Google Scholar]
- 40.Kim J., Tang J. Y., Gong R., Kim J., Lee J. J., Clemons K. V., Chong C. R., Chang K. S., Fereshteh M., Gardner D., Reya T., Liu J. O., Epstein E. H., Stevens D. A., Beachy P. A. (2010) Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 17, 388–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim D. J., Kim J., Spaunhurst K., Montoya J., Khodosh R., Chandra K., Fu T., Gilliam A., Molgo M., Beachy P. A., Tang J. Y. (2014) Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. 32, 745–751 [DOI] [PubMed] [Google Scholar]
- 42.Hadden M. K. (2014) Hedgehog pathway agonism: therapeutic potential and small-molecule development. ChemMedChem 9, 27–37 [DOI] [PubMed] [Google Scholar]
- 43.Teperino R., Amann S., Bayer M., McGee S. L., Loipetzberger A., Connor T., Jaeger C., Kammerer B., Winter L., Wiche G., Dalgaard K., Selvaraj M., Gaster M., Lee-Young R. S., Febbraio M. A., Knauf C., Cani P. D., Aberger F., Penninger J. M., Pospisilik J. A., Esterbauer H. (2012) Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell 151, 414–426 [DOI] [PubMed] [Google Scholar]
- 44.Masdeu C., Faure H., Coulombe J., Schoenfelder A., Mann A., Brabet I., Pin J. P., Traiffort E., Ruat M. (2006) Identification and characterization of Hedgehog modulator properties after functional coupling of Smoothened to G15. Biochem. Biophys. Res. Commun. 349, 471–479 [DOI] [PubMed] [Google Scholar]
- 45.Masdeu C., Bernard V., Faure H., Traiffort E., Ruat M. (2007) Distribution of Smoothened at hippocampal mossy fiber synapses. Neuroreport 18, 395–399 [DOI] [PubMed] [Google Scholar]
- 46.Coulombe J., Traiffort E., Loulier K., Faure H., Ruat M. (2004) Hedgehog interacting protein in the mature brain: membrane-associated and soluble forms. Mol. Cell. Neurosci. 25, 323–333 [DOI] [PubMed] [Google Scholar]
- 47.Charytoniuk D., Porcel B., Rodríguez Gomez J., Faure H., Ruat M., Traiffort E. (2002) Sonic Hedgehog signalling in the developing and adult brain. J. Physiol. Paris 96, 9–16 [DOI] [PubMed] [Google Scholar]
- 48.Zhao H., Ayrault O., Zindy F., Kim J. H., Roussel M. F. (2008) Post-transcriptional down-regulation of Atoh1/Math1 by bone morphogenic proteins suppresses medulloblastoma development. Genes Dev. 22, 722–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petrel C., Kessler A., Maslah F., Dauban P., Dodd R. H., Rognan D., Ruat M. (2003) Modeling and mutagenesis of the binding site of Calhex 231, a novel negative allosteric modulator of the extracellular Ca(2+)-sensing receptor. J. Biol. Chem. 278, 49487–49494 [DOI] [PubMed] [Google Scholar]
- 50.Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.