

Abstract
Attention-deficit hyperactivity disorder (ADHD) is estimated to affect 8–12% of school-age children worldwide. ADHD is a complex disorder with significant genetic contributions. However, no single gene has been linked to a significant percentage of cases, suggesting that environmental factors may contribute to ADHD. Here, we used behavioral, molecular, and neurochemical techniques to characterize the effects of developmental exposure to the pyrethroid pesticide deltamethrin. We also used epidemiologic methods to determine whether there is an association between pyrethroid exposure and diagnosis of ADHD. Mice exposed to the pyrethroid pesticide deltamethrin during development exhibit several features reminiscent of ADHD, including elevated dopamine transporter (DAT) levels, hyperactivity, working memory and attention deficits, and impulsive-like behavior. Increased DAT and D1 dopamine receptor levels appear to be responsible for the behavioral deficits. Epidemiologic data reveal that children aged 6–15 with detectable levels of pyrethroid metabolites in their urine were more than twice as likely to be diagnosed with ADHD. Our epidemiologic finding, combined with the recapitulation of ADHD behavior in pesticide-treated mice, provides a mechanistic basis to suggest that developmental pyrethroid exposure is a risk factor for ADHD.—Richardson, J. R., Taylor, M. M., Shalat, S. L., Guillot III, T. S., Caudle, W. M., Hossain, M. M., Mathews, T. A., Jones, S. R., Cory-Slechta, D. A., Miller, G. W. Developmental pesticide exposure reproduces features of attention deficit hyperactivity disorder.
Keywords: ADHD, dopamine transporter, pyrethyroid, impulsivity, dopamine receptor
ADHD is the most common neurobehavioral disorder of childhood. However, the etiology and pathogenesis of this disorder are poorly understood. A recent review of 20 twin studies from Australia, Sweden, the United Kingdom and the United States found that ADHD has a heritability of 0.76, which is comparable to that of schizophrenia and bipolar disorder (1). Although the results from these studies are supportive of a genetic etiology of ADHD, candidate gene studies and genomewide scans (2–5) have failed to identify specific genes that can account for more than a fraction of cases. However, several candidate genes that are small, but significant, contributors to ADHD have been identified. These include several genes involved in catecholaminergic function, including DAT and the D1, D2, D4, and D5 dopamine receptors (6).
The difficulty in finding strong single gene effects suggests that ADHD is either a multigenic disorder or that environmental factors or gene-environment interactions may contribute to ADHD. Recently, evidence has been accumulating that environmental influences during development can lead to the occurrence of disease or dysfunction later in life (7). With regard to ADHD, epidemiologic studies have found that maternal nicotine use, neonatal hypoxia, lead exposure, and low birth weight increase risk for ADHD diagnosis (8–11). Although the mechanism by which seemingly disparate exposures associate with ADHD is not well established, it likely involves disruption of catecholaminergic systems, particularly the dopamine system. Because ADHD is known to involve disruption of catecholaminergic systems (12–14), developmental exposure to environmental factors that alter the proper development of these systems could be considered risk factors for ADHD.
In the past 2 decades, it has been recognized that children are more susceptible to the adverse effects of pesticides than adults (15). Pyrethroid pesticides, such as deltamethrin, are commonly used, particularly for household pest eradication and are more toxic to developing animals (16). However, little research has focused on the neurotoxic consequences of developmental exposure to pyrethroids (16). Previously, we and others have reported that exposure of adult mice to the pyrethroid pesticide deltamethrin alters the dopamine system and causes increased DAT levels (17, 18). This finding may be particularly relevant to ADHD, as DAT polymorphisms and elevated expression of the DAT are found in some ADHD patients (19). Here, we report that developmental deltamethrin exposure produces neurochemical and behavioral alterations similar to those observed in ADHD and that elevated urinary metabolites of pyrethroid pesticides in children are associated with increased likelihood of ADHD diagnosis.
MATERIALS AND METHODS
Animals and experimental design
Eight-week-old female and male C57BL/6J mice purchased from Jackson Laboratory (Bar Harbor, ME, USA) were used. Mice were maintained on a 12:12 light-dark cycle with food (Purina Rodent Chow #5001; Research Diets, New Brunswick, NJ, USA) and water available ad libitum. All procedures were conducted in accordance with the U.S. National Institutes of Health (NIH) Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Emory University and Rutgers–Robert Wood Johnson Medical School.
Based on the documented exposure of pregnant women to pyrethroids (20–22) and to mimic likely exposures in the human population, female mice were orally administered either 0.3, 1, or 3 mg/kg deltamethrin (ChemService, West Chester, PA, USA) dissolved in corn oil vehicle and mixed with peanut butter every 3 d during gestation and lactation (23). These doses are lower than the developmental no observable adverse effect level (12 mg/kg) (24) and span the no observable effect level (1 mg/kg), which is used by the Environmental Protection Agency to set allowable limits of pesticide exposure of humans. Dosing began the day a positive vaginal plug was observed and continued throughout gestation and lactation on the same schedule, ending at weaning of the pups on postnatal day 22. This administration method reduces handling stress associated with injections during pregnancy and most closely mimics human oral exposure conditions, which are by food residues or nondietary ingestion (25). Control mice received an equivalent amount of corn oil vehicle in peanut butter. The experiments reported here were derived from 3 separate cohorts of animals, representing 4–9 litters per treatment group, with the litter used as the individual unit of analysis and no more than 1 mouse of each sex per litter used for any behavioral test.
Locomotor activity
For locomotor activity assessment, mice were either placed in polycarbonate activity monitoring chambers (25.4 × 50.8 × 25.4 cm) and horizontal distance traveled quantified using Noldus Ethovision 3.0 (Wageningen, The Netherlands) or in sound-attenuated chambers equipped with photo beams (Med Associates, St. Albans, VT, USA). Mice were observed over 1–2 hours, with the first 30 minutes considered a habituation period, and locomotor activity quantified in 5-minute blocks. For experiments with methylphenidate and SKF82958, mice were injected with vehicle (saline) or drug after the 30-minute habituation period, and locomotor activity monitored for an additional 60 minutes. For the study with apomorphine, mice were depleted of dopamine by administration of reserpine (5 mg/kg) 24 hours prior to initiation of the experiment and α-methyl-ρ-tyrosine (250 mg/kg) 1 hour prior to injection with apomorphine. This combined treatment reduced striatal dopamine levels to less than 5% of control (data not shown). For the dose-response studies with drugs targeting dopamine receptors, mice were administered drugs in a counterbalanced design, with each dose preceded by a day with no injection and a day with saline injected. Thus, each dose was separated by 2 intervening days and no apparent sensitization was observed.
Operant assessment of impulsive behavior
The fixed ratio waiting for reward (FR-WAIT) procedures were measured in operant chambers (Med Associates), each of which was housed in a sound-attenuated box. Each chamber was equipped with 2 response levers. Only the right lever was active in these experiments. Behavioral responses were programmed and recorded using the MED-PC Research Control and Data Acquisition systems (Med Associates).
Prior to initiation of behavioral testing, mice were food-restricted in order to maintain a body weight equivalent to 85% of free feeding levels. Following stabilization of weight, lever press responding was shaped using an automated variable time and continuous reinforcement schedule with both components operating simultaneously. On the variable time schedule, food pellets were delivered independently of lever press responses, with deliveries separated by a variable time interval. The mean value of these intervals averaged 80 seconds. Any lever press response that occurred during the continuous reinforcement schedule session resulted in food delivery and the session terminated after 4 hours or the delivery of 100 reinforcers, whichever occurred first. A total of 3 such sessions were conducted.
FR-WAIT paradigm was utilized as described previously (26). Male and female offspring of mice exposed to 0 or 3mg/kg deltamethrin were trained to press 1 of 2 levers in an operant chamber for 45 minutes a day until the lever press response had been acquired. Once responding reliably, the mice were transitioned to a fixed ration (FR)1 schedule of reinforcement where every lever press resulted in pellet delivery. The response requirement was gradually incremented according to daily performance until satisfactory performance on an FR25 was observed, after which free rewards (food pellets) were delivered, with the time between each such food deliver increasing systematically. The starting time interval was 5 seconds, and this wait period increased by an additional 5 seconds following each free food delivery. Any intervening lever press reset the FR component. Sessions of 30 minutes duration were carried out 5–6 days per week between 10:00 am and 3:00 pm for a total of 14 sessions after stable baseline FR-WAIT performance was established. Outcome measures included waiting time (mean and longest) and number of FR resets during the waiting component. Data are presented from the final session.
Y-maze
Mice were allowed to explore the maze freely in an 8-minute test session. An arm entry was defined as the entry of all 4 paws into 1 arm. The sequence of arm entries was recorded with a digital video camera and tracked with Noldus Ethovision. Alternation behavior (actual alternations) was defined as the consecutive entry into 3 arms (i.e., the combination of 3 different arms) with stepwise combinations in the sequence. The maximum number of alternations was the total number of arms entered minus 2, and the percentage of alternation behavior was calculated as (actual alternations/maximum alternations) × 100. A same arm entry was recorded when mice exited an arm and returned to the same arm without visiting another arm.
Biochemical and molecular assays
Western blots, striatal dopamine uptake, and binding of 3H-mazindol were performed as described previously by our laboratories (27–31). For Western blots, samples (5–10 μg protein) were subjected to polyacrylamide gel electrophoresis on 10% precast NuPage gels (Invitrogen, Carlsbad, CA, USA) and transferred to a PVDF membrane. Membranes were then incubated overnight in a monoclonal antibody to the N terminus of DAT. DAT antibody binding was detected using a goat anti-rat secondary antibody coupled to horseradish peroxidase and enhanced chemiluminescence. The luminescence signal was captured on an Alpha Innotech Fluorochem imaging system (Protein Simple, San Jose, CA, USA) and stored as a digital image. Densitometric analysis was performed and calibrated to coblotted dilutional standards of pooled striata from all control samples. Membranes were stripped for 15 minutes at room temperature with Pierce stripping buffer and sequentially reprobed with additional antibodies. α-Tubulin was used to ensure equal protein loading across samples.
For dopamine uptake, striatal synaptosomes were prepared from fresh tissue and incubated in assay buffer (4 mM Tris, 6.25 mM HEPES, 120 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 0.6 mM ascorbic acid, 5.5 mM glucose, 10 µM pargyline; pH 7.4) containing a saturating concentration of dopamine (1 µM final concentration) and a tracer amount of [3H] dopamine (20 nM). Uptake was allowed to proceed for 3 min at 37°C, and then terminated by the addition of ice-cold buffer and rapid vacuum filtration over glass microfiber filter paper using a Brandel (Gaithersburg, MD, USA) harvester. Filters were washed, allowed to air dry, and placed in scintillation vials containing 8 ml of Econoscint (Fisher Scientific, Pittsburgh, PA, USA) for scintillation counting. Uptake rates were calculated as specific uptake (total uptake − nonspecific uptake), with nonspecific uptake defined by the inclusion of 10 µM nomifensine.
Receptor binding studies were conducted as described previously (32), with modifications to reduce the assay volume to 200 μl. For radioligand binding to the DAT, serotonin transporter, and norepinephrine transporter, striatal or cortical tissue was homogenized in 50 mM Tris-HCL containing 300 mM NaCl and 5 mM KCl and centrifuged at 48,000 g for 10 minutes. The pellet was resuspended in the same buffer and washed twice more by centrifugation. Striatal membranes were incubated with 10 nM 3H mazindol for 1 hour at 4°C in 96-well plates. Non-specific binding was determined by the inclusion of 10µM nomifensine. For paroxetine binding to the serotonin transporter, striatal membranes were incubated with 1nM 3H paroxetine for 1 hour at 25°C and nonspecific binding was determined by the inclusion of 10uM fluoxetine. For [3H] nisoxetine binding to the norepinephrine transporter, cortical membranes were incubated with 5 nM nisoxetine for 3 hours at 4°C and nonspecific binding was determined by inclusion of 10 µM desipramine. Incubations were terminated by rapid vacuum filtration onto glass microfiber filter plates, and radioactivity was determined by liquid scintillation counting.
HPLC, microdialysis, and fast-scan cyclic voltammetry
HPLC for catecholamines in tissue extracts was performed as described previously (33, 34). For microdialysis, mice were anesthetized with ketamine and xylazine and the scalp was shaved and disinfected with betadine-iodine solution. Mice were placed in a stereotaxic apparatus (Kopf Instruments, Tjunga, CA, USA) and a microdialysis guide cannula (MBR-5; BASi, West Lafayette, IN, USA) was implanted into the nucleus accumbens with following coordinates: anteroposterior + 1.5 mm (from bregma), mediolateral – 0.8 mm, and dorsoventral − 3.5 mm (from skull). Animals were allowed to recover for at least 72 hours before the beginning of the microdialysis experiment.
Following recovery, a microdialysis probe (MD-2211, BASi) was inserted carefully into the nucleus accumbens through the guide cannula. Sterile isotonic CNS perfusion fluid (CMA Microdialysis, North Chelmsford, MA, USA) was infused at a constant flow of 1 μl/min using a microperfusion pump devised to allow the mice to move freely in a round-bottomed Raturn bowl (Bioanalytical Systems, West Lafayette, IN, USA). The dialysate samples were collected every 30 minutes with a refrigerated fraction collector into microtubes containing 3 μl of 2.2 N per chloric acid, 2.89 mM sodium meta-bisulfite, and 1.48 mM Na2 EDTA. A volume of 20 µl from each collection was then injected into a HPLC system consisting of a Waters 2695 separation module, a cation exchange column (MD-150 × 3.2 mm, ESA Biosciences, Chelmsford, MA, USA) equipped with an Antec Leyden electrochemical detector set (Zoeterwoude, The Netherlands) at +0.8 dorsoventral. Quantification was determined based on a standard curve for dopamine. At the end of the experiments, animals were killed with CO2. Brains were removed and kept in 4% paraformaldehyde at 4°C for 2 days and then stored in 30% sucrose until histologic examination. Brains were sliced into 20 µm coronal sections using a Microm HM 450 microtome (MICROM International GmbH, Walldorf, Germany) and fixed on microscopic slides. Sections were stained with cresyl violet and locations of the probes were examined under a microscope.
Fast-scan cyclic voltammetry (FSCV) was used to assess dopamine release and reuptake kinetics in slices from the dorsal striatum. Dopamine release was evoked by a single, rectangular, electrical pulse (300 µA, 2 ms per phase, biphasic), applied every 15 minutes and detected by using FSCV as described previously (35).
Epidemiologic study
A nested case-control study of the association between urinary pyrethroid metabolite levels and diagnosis of ADHD was carried out on U.S. children between the ages of 6 and 15 years (n = 2123). Data for this analysis were obtained from the online resource maintained by the Centers for Disease Control, National Center for Health Statistics. Data from the 1999–2002 rounds of the National Health and Nutrition Examination Survey (NHANES) was utilized for a subset of 2123 children who had completed the health questionnaire and who had samples collected and analyzed for urinary metabolites of pyrethroid pesticides. The NHANES data were collected by a cross-sectional survey of noninstitutionalized individuals in the general population. It employed a complex, multistage probability sampling design that allows for oversampling of designated minorities and age groups (36–38). The 1999–2002 NHANES Protocol #98-12 was reviewed and approved by the National Center for Health Statistics (NCHS) Institutional Review Board (IRB). In 2003, the IRB changed its name to the NCHS Research Ethics Review Board. The NCHS Ethics Review Board reviewed and approved Protocol #2005-06 NHANES on September 20, 2006. IRB approval for the use of these data was also obtained from the Rutgers Robert Wood Johnson Medical School IRB (Protocol # 0220070187).
Exposure to pyrethroid pesticides, including deltamethrin, was assessed by measurement of urinary levels of 3-phenoxybenzoic acid (3-PBA). All reported values for 3-PBA dichotomized, with those below the limit of detection as the reference group. The ADHD status of subjects in the data set was evaluated in two ways. First the response to the question: “Has a doctor or health professional ever told (you) that (you/she or he) had attention deficit disorder?” This was further cross-referenced with the history of prescription drug usage of any of the following medications: amphetamine aspartate/ amphetamine sulfate, dextroamphetamine aspartate/dextroamphetamine sulfate, methylphenidate hydrochloride, which corresponded to the National Drug Codes 03700, 17900, 39500, and 82000, respectively. Calculated odds ratios for ADHD based on sex and race/ethnicity observed in this population were comparable to those previously reported in other NHANES data sets (39).
Statistical analysis
For the mouse studies, an individual litter was considered the smallest unit of analysis, with each litter representing an independent replication (n = 4–9 litters/treatment group). Body weight gain for dams and pups were analyzed by repeated-measures ANOVA (RMANOVA). All other neurochemical data were analyzed using 1-way or 2-way ANOVA where appropriate. Unpaired Student’s t tests were performed for comparisons involving only 2 groups and following 2-way ANOVA with the autoradiography data. FR-WAIT data were analyzed using RMANOVA with treatment as a between-group factor and sessions as the within-group factor. These analyses were carried out separately for males and females. When a significant F was determined, post hoc comparisons were performed using Dunnett’s test.
For the epidemiologic study, statistical analysis was carried out using STATA/SE statistical software package, version 8.2 (StataCorp, College Station, TX, USA) . Both univariate and multivariate analyses were carried out. Statistical significance was evaluated at the P = 0.05 level. For the multivariate analysis, a logistic regression model manual, step-down regression procedures were employed. These analyses took into account sample weights using Stata Survey Data procedures, strata, and sampling unit (interviewed and sample persons’ weights) of the NHANES data subset containing urinary pesticide analyses. Potential confounders and effect modifiers included in the model were age, sex, race/ethnicity, and total family income and whether there was health insurance coverage. Interaction terms for sex and other factors were not possible due to the constraints of the sample size available for analysis.
Results
Developmental deltamethrin exposure increases dopamine transporter levels
At 6 weeks of age, striatal DAT protein levels were significantly increased by deltamethrin in a dose-related manner. (F = 27.43; df = 3.29; P < 0.0001). There was also a significant deltamethrin × sex interaction (F = 3.69; df = 3.29; P = 0.023), with the male increases (70%; P < 0.001) more than twice that of the female (31%; P < 0.05; Fig. 1A). The magnitude of the increase in DAT is similar to that previously observed in some patients with ADHD (40). The elevated DAT levels at the highest dose in the males were confirmed by radioligand binding (t = 3.108; df = 6; P = 0.02; Fig. 1B) and functional uptake experiments in synaptosomes (t = 3.131; df = 6; P = 0.02; Fig. 1C). Autoradiography studies found that although DAT was significantly increased in the nucleus accumbens (t = 4.03; df = 6; P = 0.007), but not the dorsal striatum (Fig. 1D). To determine the in vivo consequence of increased DAT levels, we administered the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which requires the DAT for toxicity (41), and we found that MPTP toxicity, as determined by striatal dopamine loss, was potentiated in male mice developmentally exposed to 3 mg/kg deltamethrin (Fig. 1E). Deltamethrin’s effects on neurotransmitter transporters appears to be relatively specific for the DAT, as levels of the serotonin transporter and norepinephrine transporter were not significantly different from controls in the striatum and frontal cortex, respectively (Supplemental Fig. 1).
Figure 1.

Developmental pesticide exposure increases DAT levels. Developmental exposure to deltamethrin (DM) at 0, 0.3, 1, or 3 mg/kg increased dopamine transporter levels in the striatum of male offspring greater than female offspring at 6 weeks of age as determined by (A, B) Western blot. Data in male offspring developmentally exposed to 3 mg/kg deltamethrin were confirmed by (C) radioligand binding measurements and (D) functional uptake assays. Representative Western blots of the DAT and tubulin are provided above and below the graph, respectively. D, E) Autoradiographic determinations of DAT levels in the striatum (n = 4). ***P = 0.002. F) Increased MPTP toxicity, as assessed by striatal dopamine levels, is observed in male offspring following developmental deltamethrin exposure. (n = 5–6) for all endpoints. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Developmental deltamethrin exposure decreases extracellular dopamine and increases D1 dopamine receptor levels
The increased DAT levels observed in the males could suggest that deltamethrin exposure may produce a hypodopaminergic state through enhanced clearance of dopamine, which has been hypothesized to contribute to ADHD (42). Using HPLC, we observed no significant alteration of total tissue levels of dopamine or its metabolites in the striatum (Fig. 2A). However, in vivo microdialysis revealed significantly decreased (25%) extracellular dopamine levels in the nucleus accumbens of male mice developmentally exposed to deltamethrin (F = 4.413; df = 1.17; P = 0.05), with no significant effect in the females (Fig. 2B). This effect appeared to be specific for the mesocorticolimbic pathway, as cyclic voltammetry demonstrated no significant change in dopamine release or reuptake in the dorsal striatum (data not shown). Because synaptic levels of dopamine may influence dopamine receptor levels, we measured D1 and D2 dopamine receptor levels in the striatum by radioligand binding. Data revealed a significant effect of sex (F = 102.6; df 1.16; P < 0.001) and a sex by treatment interaction (F = 27.05; df = 1.16; P < 0.001), with male mice having significantly higher D1 receptor binding. Developmental deltamethrin exposure resulted in a significant increase (40%) of striatal D1 receptor levels in the deltamethrin male offspring (Fig. 2C; F = 31.21; df = 1.16; P < 0.001), with no significant change observed in the females. D2 receptor density was not significantly altered by deltamethrin in either sex (Supplemental Fig. 1). However, there was a significant effect of sex (F = 20.46; df = 1.16; P = 0.0003), with females having significantly higher D2 receptor binding. Autoradiographic studies confirmed that the nucleus accumbens demonstrated increased D1 receptor levels (Fig. 2D), consistent with the microdialysis data demonstrating significantly reduced synaptic dopamine levels in the accumbens.
Figure 2.
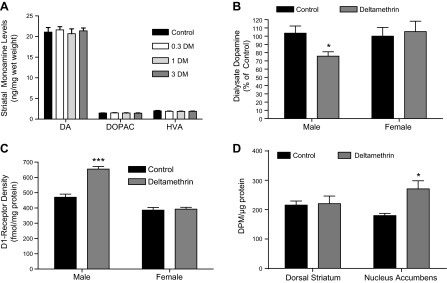
Developmental pesticide exposure reduces extracellular dopamine levels and increases D1 receptor levels. A) Total striatal levels of dopamine (DA) and its metabolites dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) as measured by HPLC with electrochemical detection were unchanged in male mice developmentally exposed to deltamethrin (DM) (n = 4–5). Similarly, no changes were observed in female mice developmentally exposed to deltamethrin (data not shown). B) Male but not female mice developmentally exposed to 3 mg/kg deltamethrin exhibit decreased extracellular dopamine levels in the nucleus accumbens as measured by microdialysis (n = 4–8). P < 0.05 by 2-way ANOVA. C) Male but not female mice developmentally exposed to 3 mg/kg deltamethrin exhibit increased striatal D1 receptor levels (n = 5). ***P < 0.001 by 2-way ANOVA. D) D1 receptor levels are increased specifically in the nucleus accumbens of male mice developmentally exposed to deltamethrin (n = 4). *P = 0.038.
Developmental deltamethrin exposure causes male-specific hyperactivity
At 6 weeks of age, a time roughly equivalent to early adolescence in humans, the male offspring of deltamethrin treated mice displayed significant dose-related increases in locomotor activity (F = 17.35; df = 3.21; P < 0.001; Fig. 3A). In contrast to the males, females exhibited much smaller increases in locomotor activity that did not reach statistical significance (F = 1.74; df = 3.18; P = 0.20). Examination of the time course of locomotor activity showed that the hyperactivity in the male mice developmentally exposed to deltamethrin did not appear until after a 30 min habituation period (Supplemental Fig. 2), analogous to that observed in patients with ADHD, who typically become hyperactive only after an extended period of time in a particular setting (43).
Figure 3.
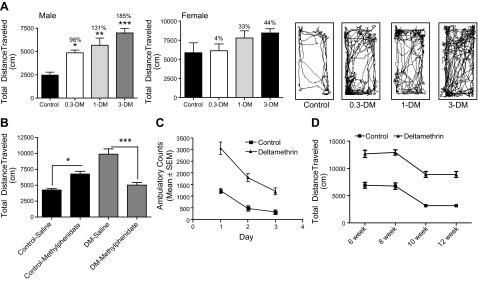
Developmental pesticide exposure causes hyperactivity that is ameliorated by methylphenidate. A) Following a 30-minute habituation period in a novel open field, locomotor activity was significantly elevated in male but not female offspring of deltamethrin (DM)-treated mice over the next 90 minutes (n = 4–7). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 as determined by ANOVA followed by Dunnett’s test. Representative track tracings of male mice movements are shown at the right. B) At 8 weeks of age, offspring of control mice or mice developmentally exposed to 3 mg/kg deltamethrin were placed in the open field for 30 minutes and then administered 1 mg/kg methylphenidate (intraperitoneally).) and locomotor activity monitored an additional 60 minutes. Methylphenidate significantly reduced locomotor activity in deltamethrin treated offspring to the levels of control offspring (n = 6–7). ***P ≤ 0.001 as determined by ANOVA followed by the Student-Newman-Keuls post hoc test. C) To determine whether the increased locomotor activity was a deficit in habituation, locomotor activity of male mice developmentally exposed to 3 mg/kg deltamethrin was measured in an open field for 60 minutes on 3 consecutive days. D) Locomotor hyperactivity persists through 12 weeks of age in male mice developmentally exposed to 3 mg/kg deltamethrin.
We next sought to determine whether administration of a therapeutically relevant dose of methylphenidate (1 mg/kg), one of the most widely used drugs for ADHD to male mice developmentally exposed to 3 mg/kg deltamethrin would ameliorate the increased locomotor activity. Statistical analysis revealed a main effect of methylphenidate (2-way ANOVA; F = 4.55; df = 1.22; P = 0.044) and a methylphenidate × deltamethrin interaction (F = 44.82; df = 1.22; P < 0.001). Administration of methylphenidate totally abolished the increased locomotor effect of developmental deltamethrin exposure (Fig. 3B). To determine whether there were deficits in habituation, we monitored locomotor activity in control and deltamethrin-exposed male offspring over 3 consecutive days. Locomotor activity decreased over the 3 d of testing in both control and treated mice (F = 33.74; df = 2.21; P < 0.001). However, mice developmentally exposed to deltamethrin continued to exhibit significantly greater locomotor activity (F = 90.22; df = 1.21; P < 0.001; Fig. 3C). Finally, we determined whether the hyperactivity observed persisted into adulthood. Locomotor activity was assessed in control and deltamethrin-exposed mice every 2 weeks from 6 to 12 weeks of age. Although locomotor activity decreased in both groups as the mice reached adulthood (F = 41.97; df = 3.48; P < 0.001), mice developmentally exposed to deltamethrin demonstrated significantly higher locomotor activity at all time points assessed (F = 298.70; df = 1.48; P < 0.001; Fig. 3D).
Altered dopamine receptors contribute to hyperactivity produced by developmental deltamethrin exposure
To determine the functional consequences of the increased dopamine receptor levels observed in mice developmentally exposed to deltamethrin, we administered the D1 and D2 antagonists SCH23390 and eticlopride (D2) and the D2-autoreceptor agonist quinpirole and monitored locomotor activity. SCH23390 administration caused a dose-related decrease in locomotor activity in all mice (F = 14.25; df = 3.36; P < 0.0001), and there was a significant interaction effect with deltamethrin (F = 4.067; df = 3.36; P = 0.014). Importantly, all doses of SCH23390 reduced locomotor activity in deltamethrin mice to control levels (P > 0.05; Fig. 4A). Because D1 and D2 receptors coordinately regulate motor output and locomotor activity, we next determined whether the D2-specific antagonist eticlopride would ameliorate locomotor hyperactivity. Similarly to SCH23390, eticlopride caused a dose-related decrease in locomotor activity (F = 18.84; df = 3.36; P < 0.0001) with a significant interaction effect (F = 4.635; df = 3; P = 0.0077). Again, all doses reduced locomotor activity to control levels (P > 0.05; Fig. 4B). SCH23390 and eticlopride decreased locomotor activity in both control and deltamethrin exposed females (P = 0.0055 and P = 0.0002). However, there were no significant differences observed between control and exposed females (P = 0.843 and P = 0.416) and no interaction between treatment and concentration (P = 0.995; Supplemental Fig. 3).
Figure 4.
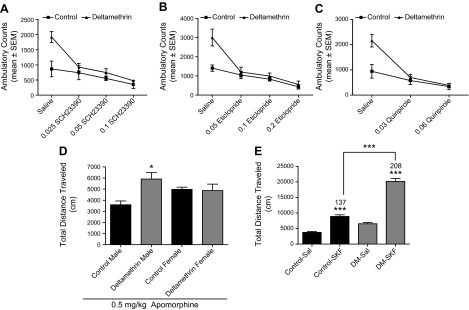
Dopamine receptor alterations mediate hyperactivity following developmental pesticide exposure. Administration of the dopamine receptor antagonists (A) SCH23390 and (B) eticlopride and the (C) D2-autoreceptor agonist quinpirole significantly reduced the hyperactivity observed in male mice developmentally exposed to 3 mg/kg deltamethrin (DM; n = 3). *P ≤ 0.05, ***P ≤ 0.001 as determined by ANOVA followed by means separation by the Student-Newman-Keuls test. D) Male but not female mice demonstrate enhanced locomotor response to apomorphine following developmental exposure to 3 mg/kg deltamethrin. Mice were depleted of endogenous dopamine by administration of reserpine and α-methyl-ρ-tyrosine and then administered apomorphine (n = 4). Locomotor activity was monitored over a 60-minute period. *P < 0.05 by 2-way ANOVA. E) Male mice developmentally exposed to 3 mg/kg deltamethrin exhibit enhanced locomotor response to the full D1-agonist SKF82958. Animals were allowed to habituate to the open field for 30 minutes and were administered SKF82958 (1 mg/kg, i.p.). Locomotor activity was monitored for an additional 60 minutes. ***P < 0.001 by 2-way ANOVA (n = 5–6).
Administration of low doses of the D2 receptor agonist quinpirole to target D2 autoreceptors dose-dependently decreased locomotor activity in both control and deltamethrin mice (F = 24.36; df = 2.27; P < 0.0001) with a significant interaction effect (F = 6.718; df = 2.27; P = 0.004). At both doses, quinpirole reduced the hyperactivity in deltamethrin mice to control levels (P > 0.05; Fig. 4C). Based on these findings, we also assessed D2 autoreceptor function in striatal slices using FSCV and found a slight, but nonsignificant increase in quinpirole (300 nM) response between control and deltamethrin treated mice (P = 0.06; Supplemental Fig. 3), suggesting that differences in autoreceptor responses likely do not play a major role in the differences observed in response to systemic quinpirole administration.
To further probe the role of dopamine receptors in the locomotor hyperactivity observed in male offspring, we first administered the general dopamine receptor agonist apomorphine to mice pretreated with reserpine and α-methyl-para-tyrosine to deplete dopamine. Apomorphine administration restored locomotor activity in all mice, but locomotor activation was potentiated in the male mice developmentally exposed to deltamethrin (F = 5.984; df = 1.8; P = 0.04; sex x treatment interaction; F = 7.178; df = 1.8; P = 0.028), with no corresponding potentiation in the females (Fig. 4D). In an additional experiment, we administered the full D1 agonist SKF82958 to the male mice developmentally exposed to deltamethrin to directly assess the contribution of the D1 receptor. SKF82958 caused significant increases in locomotor activity in control and deltamethrin mice (F = 221.3; df = 1.18; P < 0.0001). However, there was a significant deltamethrin × SKF interaction (F = 45.41; df = 1.18; P < 0.0001), and the deltamethrin-exposed male offspring had significantly higher locomotor activation following SKF treatment compared with controls (P < 0.001; Fig. 4E). Taken in concert, these findings identify a central role for altered dopamine receptor levels and response in the behavioral alterations observed in mice developmentally exposed to deltamethrin.
Developmental deltamethrin exposure causes impulsive-like behaviors and working memory deficits
Based on the previous data demonstrating that developmental deltamethrin exposure produces hyperactivity, we sought to assess impulsive-like behavior, inattention, and working memory deficits, all common features of ADHD. Given that the most robust effects were observed in mice developmentally exposed to 3 mg/kg deltamethrin, we focused our efforts on these mice.
To test impulsive-like behavior, we used FR-WAIT, in which mice received free pellets at increasing time intervals following 25 lever presses if press responding was withheld (44). Alternatively, mice could terminate the wait component and reset the FR, thereby having to perform an additional 25 lever presses to receive the next pellet. Male but not female mice developmentally exposed to 3 mg/kg deltamethrin had significantly reduced waiting times (F = 17.26; df = 1.22; P = 0.0004) and 3 times more FR resets (F = 26.17; df = 1.22; P < 0.001; Fig. 5A) compared with control mice.
Figure 5.
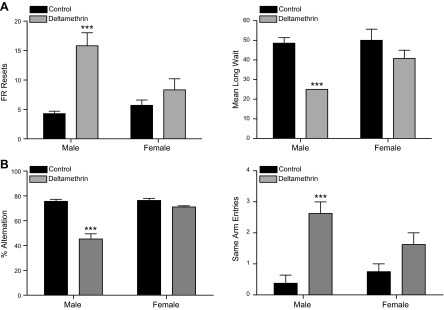
Developmental pesticide exposure causes impulsive-like behavior, working memory, and attention deficits. A) To test whether mice developmentally exposed to 3 mg/kg deltamethrin exhibited impulsive like behavior, mice (n = 6–8) were tested in operant chambers under an FR-WAIT paradigm over a 14-day period. Data were analyzed by RMANOVA and data from the final day of testing are presented. Deltamethrin exposure significantly increased the number of FR resets (left) and decreased mean long wait in males but not females. ***P ≤ 0.001. B) As a test of working memory and attention, mice were placed in a Y-maze and allowed freely explore for 8 min. Deltamethrin exposure significantly reduced alternation behavior (left) and increased the number of same arm entries (right) in male, but not female mice (n = 7–8). ***P ≤ 0.001 as determined by 2-way ANOVA.
To test the effects of deltamethrin exposure on working memory and attention, we used the Y-maze, which requires working memory (alternation behavior) and attention (same-arm entries) (45, 46). Male but not female offspring developmentally exposed to deltamethrin exhibited significantly decreased alternation behavior (F = 52.88; df = 1.28; P < 0.0001) and increased same-arm entries (F = 23.65; df = 1.28; P < 0.0001; Fig. 5B). These data may be interpreted as an indication of potential adverse effects on working memory and attention, respectively. Taken in concert, our behavioral data demonstrate that mice developmentally exposed to deltamethrin reproduce many of the core behavioral symptoms of ADHD, including hyperactivity, impulsive-like behavior, and deficits in working memory and attention, and that these effects are greater in the male offspring.
Urinary pyrethroid metabolites are associated with increased risk of ADHD diagnosis
To determine whether our laboratory findings were supported by observations in the human population, we carried out a cross-sectional study of pyrethroid pesticide exposure as a risk factor for ADHD. Data for this analysis were obtained from the 1999–2002 rounds of the NHANES using parent-reported diagnosis of ADHD and urinary levels of 3-PBA, a metabolite of pyrethroid pesticides including deltamethrin, as a measure of exposure. We also incorporated the use of prescription medications used to treat ADHD (i.e., various amphetamine formulations and methylphenidate) to improve specificity of the parent-reported diagnosis of ADHD. Of the 5489 children 6 to 15 years of age, 503 (9.2%) had parent-reported ADHD. The observed adjusted odds ratios for sex, age, and race/ethnicity were consistent with those previously reported for ADHD in other subsets of the NHANES data (39, 47). Urinary 3-PBA levels were available for 2123 of the 5489 children. The odds ratio of ADHD diagnosis for children with levels of 3-PBA in their urine above the limit of detection was 2.3 [95% confidence interval [CI] 1.4–3.9; P = 0.002; Table 1]. These data demonstrate that exposure to pyrethroid pesticides increases the risk of being diagnosed with ADHD.
TABLE 1.
Pyrethroid pesticide exposure in children results in increased risk of ADHD diagnosis
| Variable | Sample size (n) | Adjusted odds ratio (95% CI) | P value |
|---|---|---|---|
| Total | 2123 | ||
| Age (yr) | 2123 | 1.1 (1.0–1.2) | 0.162 |
| Sex | |||
| Female | 1121 | Referent | |
| Male | 1002 | 2.9 (1.5–5.7) | 0.003 |
| Race | |||
| Non-Hispanic white | 702 | Referent | |
| Other Hispanic | 98 | 0.4 (0.1–1.3) | 0.138 |
| Mexican American | 559 | 0.3 (0.1–0.5) | <0.001 |
| Non-Hispanic black | 106 | 0.6 (0.4–0.9) | 0.019 |
| Other, including multiracial | 658 | 0.6 (0.1–3.0) | 0.557 |
| Covered by health insurance | |||
| No | 394 | Referent | |
| Yes | 1729 | 1.3 (0.6–2.9) | 0.545 |
| 3-PBA above the limit of detection (0.1 μg/L) | |||
| No | 482 | Referent | |
| Yes | 1641 | 2.3 (1.4–3.9) | 0.002 |
Adjusted odds ratio for ADHD among U.S. children by urinary 3-PBA concentration. Logistic regression analysis for parent-reported ADHD diagnosis cross-referenced with prescription drug information for drugs used to treat ADHD in children aged 6–15 from NHANES 1999–2002. The model was adjusted for child’s age, sex, race/ethnicity, and insurance status.
DISCUSSION
Over the past decade, the search for etiological factors in ADHD has focused primarily on the identification of genetic contributors (1). The lack of success in identifying genes that contribute significantly to ADHD has led to the idea that environmental factors may also promote the development of ADHD. Our current work demonstrates that the offspring of mice exposed in utero to the pyrethroid pesticide deltamethrin exhibit behavioral abnormalities similar to those observed in children with ADHD, including hyperactivity, impulsive-like behaviors, and deficits in working memory and attention as well as a male sex preference of these effects. Supporting these experimental data, epidemiologic evidence demonstrates that exposure to pyrethroid pesticides in children is associated with an increased risk of ADHD diagnosis. From a mechanistic standpoint, these behavioral alterations appear to be driven by disruption of the dopamine system, including elevated DAT levels, lower synaptic dopamine, and increased D1 dopamine receptor levels. Taken together, these data provide a mechanistic basis to suggest that developmental pyrethroid exposure is a significant risk factor for ADHD.
There is considerable controversy over the potential role of the DAT in ADHD. Neuroimaging studies have found increased DAT levels in ADHD patients using a variety of imaging modalities (48). However, others have reported either no difference in or decreased DAT levels in ADHD patients (33). Similarly, the animal literature is inconsistent with regard to the DAT. The spontaneously hyperactive rat (SHR) is a widely used animal model of ADHD, exhibiting increased DAT levels that are normalized by methylphenidate (49). However, DAT knockout mice are hyperactive (50), and DAT-overexpressing mice demonstrate either no change (51) or increased locomotor activity (52). The data here demonstrate that the elevated DAT levels observed following developmental deltamethrin exposure are associated with hyperactivity, which appears only after habituation in a novel environment and persists from adolescence through adulthood, similar to that observed in the SHR model (53). This is in contrast to the DAT knockout mouse, which is hyperactive in a novel environment and has impaired habituation (50). With regard to additional behaviors, mice developmentally exposed to deltamethrin show deficits in Y-maze performance, indicative of working memory and attention deficits. However, no change in performance was observed in DAT knockout mice (54).
The discrepancies observed between different DAT knockout and overexpression models, the SHR, and our results may lie in the neuroanatomical specificity of the alterations in DAT levels. In the DAT knockout and BAC transgenic mouse (50, 51), DAT levels are uniformly increased throughout the brain, whereas developmental deltamethrin exposure and the SHR exhibit greater DAT increases in the nucleus accumbens (NAc). Additionally, increased impulsive-like behavior, as observed here in the male offspring of deltamethrin-exposed mice (Fig. 4A, B), is also found in rats with virally mediated DAT overexpression in the NAc (55). Moreover, virally mediated DAT knockdown in the NAc also increased impulsive-like behavior, demonstrating that either overexpression or knockdown of the DAT in the NAc results in increased impulsive behavior. These findings are also consistent with lesion studies demonstrating the role of the NAc in impulsive choice and hyperactivity (56). Thus, either overexpression or reduction of the DAT in the NAc appears to have potential to contribute significantly to behavioral dysfunction, particularly in the area of impulsive behavior. This is reminiscent of the classic inverted U-shaped dose-response of D1 receptor stimulation in the prefrontal cortex and regulation of working memory (57).
Although we found robust effects of developmental deltamethrin exposure on the DAT, our data demonstrate that elevated D1 dopamine receptors also contribute significantly to the increased locomotor activity of male mice following developmental deltamethrin exposure. This is supported by the findings that D1 agonist administration augments locomotor activity in deltamethrin offspring and that D1 antagonist treatment reduces locomotor activity to control levels in these mice. Although there have been no studies performed to directly determine D1 receptor density in ADHD patients, D1 dopamine receptor polymorphisms have been associated with ADHD (58, 59) and elevated D1 receptor levels are found in the SHR model of ADHD (60). The lack of change in D1 receptor levels in the female offspring of mice exposed to deltamethrin provides the most compelling support for the relative lack of behavioral alterations in these mice compared with male offspring developmentally exposed to deltamethrin. Indeed, sex differences in dopamine receptor levels, particularly the D1 receptor, and elimination of dopamine receptors during postnatal brain development in rodents have been hypothesized to play a role in ADHD (61). Most relevant to the current findings, D1 receptors remain higher in the NAc of males throughout adolescence and adulthood (61), which is also supported by our data. In DAT overexpressing mice, D1 (30%) and D2 receptor (60%) levels are significantly increased in striatal homogenates, suggesting that the D1 receptor changes found in deltamethrin exposed mice may be a result of up-regulation in response to increased DAT numbers and subsequently decreased extracellular DA levels.
Although there have been several reports on the potential developmental neurotoxicity of pyrethroids in animals, the data on neurobehavioral effects are highly variable, and many of the studies are hampered by inappropriate study design, the use of commercial formulations of pyrethroids, and inadequate statistical analysis (16). More recently, attention has focused on the potential for developmental pyrethroid exposure to contribute to neurobehavioral dysfunction in humans. In the U.S. general population, data from the 1999–2002 rounds of the NHANES found that children aged 6–11 yr had much higher urinary metabolites of pyrethroids compared with adults (62). Additionally, urinary pyrethroid concentrations in children have increased since the phase-out of residential uses of other pesticides, leading to concern over potential neurodevelopmental effects (63). Oulhote and Bouchard recently reported that postnatal pyrethroid exposure was associated with behavioral difficulties (64). After this manuscript was under review, Quirós-Alcalá and coworkers (65) examined association of pyrethroid exposure in a slightly different subset of NHANES participants. However, the association between 3-PBA levels and parent report of ADHD diagnosis did not quite reach statistical significance (odds ratio 1.26; 95% CI 0.96–1.65; P = 0.10). The differences between the 2 studies likely relate to differences in sample size, the use of imputed data for nondetects in the other study, and our use of dichotomized variables (detect or nondetect). Thus, additional epidemiologic studies with larger sample sizes and better measures of ADHD will be required to accurately determine the potential contribution of developmental pyrethroid exposure to ADHD.
Taken together with our animal data, the epidemiologic data provide strong support for developmental pyrethroid exposure as a risk factor for ADHD. Although there are several limitations to our epidemiologic data, including the cross-sectional nature of the data, potential recall bias, a one-time measurement of pesticide metabolites, and the inability to adjust for many important covariates and confounders, a previous study using the NHANES cohort found similar odds ratios for prenatal tobacco exposure and ADHD as reported in case-control studies (66). Prenatal tobacco exposure has also been demonstrated to interact with lead exposure and DAT and D4 receptor polymorphisms to produce behavioral dysfunction (67), suggesting the potential for gene environment in ADHD, which may include pyrethroid exposure. Thus, our work highlights the need to further explore environmental contributors to ADHD and provides mechanistic information on how environmental exposures during development alter the developing brain to produce behavioral dysfunction.
Supplementary Material
Acknowledgments
The authors thank Curtis Keller, Jason Mathew, Minzheng Wang, Chen Zhang, and Jessica Lam for excellent technical assistance, and also thank Dr. Eric Richfield for assistance with the autoradiography studies. This work was supported by U.S. National Institutes of Health Grants R21ES013828, R01ES015991,R01ES015991-04S1, and P30ES005022 (to J.R.R.), T32ES012870 (to W.M.C.), R01ES012702 (to D.C.S.), R01DA021325 and P50DA006634 (to S.R.J.), T32ES007148 (to M.M.T.), U54ES012068 and R21ES012315 (to G.W.M.); a U.S. Department of Defense Grant 00267036 (to G.W.M.); an Environmental Protection Agency STAR Fellowship (to T.S.G.); and R01ES020415 (to S.L.S.).
Glossary
- ADHD
attention-deficit hyperactivity disorder
- CI
confidence interval
- DAT
dopamine transporter
- FI
fixed interval
- FR-WAIT
the fixed ratio waiting for reward
- FSCV
fast-scan cyclic voltammetry
- IRB
Institutional Review Board
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NAc
nucleus accumbens
- NCHS
National Center for Health Statistics
- NHANES
National Health and Nutrition Examination Survey
- 3-PBA
3-phenoxybenzoic acid
- RMANOVA
repeated-measures ANOVA
- SHR
spontaneously hyperactive rat
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Faraone S. V., Mick E. (2010) Molecular genetics of attention deficit hyperactivity disorder. Psychiatr. Clin. North Am. 33, 159–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fisher S. E., Francks C., McCracken J. T., McGough J. J., Marlow A. J., MacPhie I. L., Newbury D. F., Crawford L. R., Palmer C. G., Woodward J. A., Del’Homme M., Cantwell D. P., Nelson S. F., Monaco A. P., Smalley S. L. (2002) A genomewide scan for loci involved in attention-deficit/hyperactivity disorder. Am. J. Hum. Genet. 70, 1183–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakker S. C., van der Meulen E. M., Buitelaar J. K., Sandkuijl L. A., Pauls D. L., Monsuur A. J., van ’t Slot R., Minderaa R. B., Gunning W. B., Pearson P. L., Sinke R. J. (2003) A whole-genome scan in 164 Dutch sib pairs with attention-deficit/hyperactivity disorder: suggestive evidence for linkage on chromosomes 7p and 15q. Am. J. Hum. Genet. 72, 1251–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shastry B. S. (2004) Molecular genetics of attention-deficit hyperactivity disorder (ADHD): an update. Neurochem. Int. 44, 469–474 [DOI] [PubMed] [Google Scholar]
- 5.Hebebrand J., Dempfle A., Saar K., Thiele H., Herpertz-Dahlmann B., Linder M., Kiefl H., Remschmidt H., Hemminger U., Warnke A., Knölker U., Heiser P., Friedel S., Hinney A., Schäfer H., Nürnberg P., Konrad K. (2006) A genome-wide scan for attention-deficit/hyperactivity disorder in 155 German sib-pairs. Mol. Psychiatry 11, 196–205 [DOI] [PubMed] [Google Scholar]
- 6.Gizer I. R., Ficks C., Waldman I. D. (2009) Candidate gene studies of ADHD: a meta-analytic review. Hum. Genet. 126, 51–90 [DOI] [PubMed] [Google Scholar]
- 7.Heindel J. J. (2007) Role of exposure to environmental chemicals in the developmental basis of disease and dysfunction. Reprod. Toxicol. 23, 257–259 [DOI] [PubMed] [Google Scholar]
- 8.Banerjee T. D., Middleton F., Faraone S. V. (2007) Environmental risk factors for attention-deficit hyperactivity disorder. Acta Paediatr. 96, 1269–1274 [DOI] [PubMed] [Google Scholar]
- 9.Froehlich T. E., Anixt J. S., Loe I. M., Chirdkiatgumchai V., Kuan L., Gilman R. C. (2011) Update on environmental risk factors for attention-deficit/hyperactivity disorder. Curr. Psychiatry Rep. 13, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eubig P. A., Aguiar A., Schantz S. L. (2010) Lead and PCBs as risk factors for attention deficit/hyperactivity disorder. Environ. Health Perspect. 118, 1654–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swanson J. M., Kinsbourne M., Nigg J., Lanphear B., Stefanatos G. A., Volkow N., Taylor E., Casey B. J., Castellanos F. X., Wadhwa P. D. (2007) Etiologic subtypes of attention-deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychol. Rev. 17, 39–59 [DOI] [PubMed] [Google Scholar]
- 12.Del Campo N., Chamberlain S. R., Sahakian B. J., Robbins T. W. (2011) The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol. Psychiatry 69, e145–e157 [DOI] [PubMed] [Google Scholar]
- 13.Arnsten A. F. (2006) Fundamentals of attention-deficit/hyperactivity disorder: circuits and pathways. J. Clin. Psychiatry 67(Suppl 8), 7–12 [PubMed] [Google Scholar]
- 14.Fischman A. J., Madras B. K. (2005) The neurobiology of attention-deficit/hyperactivity disorder. Biol. Psychiatry 57, 1374–1376 [DOI] [PubMed] [Google Scholar]
- 15.National Research Council (U.S.). (1993) Committee on Pesticides in the Diets of Infants and Children. In Pesticides in the Diets of Infants and Children, National Academy Press, Washington, DC [Google Scholar]
- 16.Shafer T. J., Meyer D. A., Crofton K. M. (2005) Developmental neurotoxicity of pyrethroid insecticides: critical review and future research needs. Environ. Health Perspect. 113, 123–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elwan M. A., Richardson J. R., Guillot T. S., Caudle W. M., Miller G. W. (2006) Pyrethroid pesticide-induced alterations in dopamine transporter function. Toxicol. Appl. Pharmacol. 211, 188–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bloomquist J. R., Barlow R. L., Gillette J. S., Li W., Kirby M. L. (2002) Selective effects of insecticides on nigrostriatal dopaminergic nerve pathways. Neurotoxicology 23, 537–544 [DOI] [PubMed] [Google Scholar]
- 19.Madras B. K., Miller G. M., Fischman A. J. (2005) The dopamine transporter and attention-deficit/hyperactivity disorder. Biol. Psychiatry 57, 1397–1409 [DOI] [PubMed] [Google Scholar]
- 20.Castorina R., Bradman A., Fenster L., Barr D. B., Bravo R., Vedar M. G., Harnly M. E., McKone T. E., Eisen E. A., Eskenazi B. (2010) Comparison of current-use pesticide and other toxicant urinary metabolite levels among pregnant women in the CHAMACOS cohort and NHANES. Environ. Health Perspect. 118, 856–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berkowitz G. S., Obel J., Deych E., Lapinski R., Godbold J., Liu Z., Landrigan P. J., Wolff M. S. (2003) Exposure to indoor pesticides during pregnancy in a multiethnic, urban cohort. Environ. Health Perspect. 111, 79–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams M. K., Barr D. B., Camann D. E., Cruz L. A., Carlton E. J., Borjas M., Reyes A., Evans D., Kinney P. L., Whitehead R. D. Jr., Perera F. P., Matsoanne S., Whyatt R. M. (2006) An intervention to reduce residential insecticide exposure during pregnancy among an inner-city cohort. Environ. Health Perspect. 114, 1684–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Armstrong L. E., Driscoll M. V., Donepudi A. C., Xu J., Baker A., Aleksunes L. M., Richardson J. R., Slitt A. L. (2013) Effects of developmental deltamethrin exposure on white adipose tissue gene expression. J. Biochem. Mol. Toxicol. 27, 165–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kavlock R., Chernoff N., Baron R., Linder R., Rogers E., Carver B., Dilley J., Simmon V. (1979) Toxicity studies with decamethrin, a synthetic pyrethroid insecticide. J. Environ. Pathol. Toxicol. 2, 751–765 [PubMed] [Google Scholar]
- 25.Caudle W. M., Richardson J. R., Wang M., Miller G. W. (2005) Perinatal heptachlor exposure increases expression of presynaptic dopaminergic markers in mouse striatum. Neurotoxicology 26, 721–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allen J. L., Conrad K., Oberdörster G., Johnston C. J., Sleezer B., Cory-Slechta D. A. (2013) Developmental exposure to concentrated ambient particles and preference for immediate reward in mice. Environ. Health Perspect. 121, 32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gatley S. J., Volkow N. D., Gifford A. N., Ding Y. S., Logan J., Wang G. J. (1997) Model for estimating dopamine transporter occupancy and subsequent increases in synaptic dopamine using positron emission tomography and carbon-11-labeled cocaine. Biochem. Pharmacol. 53, 43–52 [DOI] [PubMed] [Google Scholar]
- 28.Guillot T. S., Asress S. A., Richardson J. R., Glass J. D., Miller G. W. (2008) Treadmill gait analysis does not detect motor deficits in animal models of Parkinson’s disease or amyotrophic lateral sclerosis. J. Mot. Behav. 40, 568–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caudle W. M., Richardson J. R., Delea K. C., Guillot T. S., Wang M., Pennell K. D., Miller G. W. (2006) Polychlorinated biphenyl-induced reduction of dopamine transporter expression as a precursor to Parkinson's disease-associated dopamine toxicity. Toxicol. Sci. 92, 490–499 [DOI] [PubMed] [Google Scholar]
- 30.Richardson J. R., Caudle W. M., Wang M., Dean E. D., Pennell K. D., Miller G. W. (2006) Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of Parkinson's disease. FASEB J. 20, 1695-1697 [DOI] [PubMed] [Google Scholar]
- 31.Guillot T. S., Richardson J. R., Wang M. Z., Li Y. J., Taylor T. N., Ciliax B. J., Zachrisson O., Mercer A., Miller G. W. (2008) PACAP38 increases vesicular monoamine transporter 2 (VMAT2) expression and attenuates methamphetamine toxicity. Neuropeptides 42, 423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernagut P. O., Chalon S., Diguet E., Guilloteau D., Tison F., Jaber M. (2003) Motor behaviour deficits and their histopathological and functional correlates in the nigrostriatal system of dopamine transporter knockout mice. Neuroscience 116, 1123–1130 [DOI] [PubMed] [Google Scholar]
- 33.Volkow N. D., Wang G. J., Newcorn J., Fowler J. S., Telang F., Solanto M. V., Logan J., Wong C., Ma Y., Swanson J. M., Schulz K., Pradhan K. (2007) Brain dopamine transporter levels in treatment and drug naïve adults with ADHD. Neuroimage 34, 1182–1190 [DOI] [PubMed] [Google Scholar]
- 34.Schuh R. A., Richardson J. R., Gupta R. K., Flaws J. A., Fiskum G. (2009) Effects of the organochlorine pesticide methoxychlor on dopamine metabolites and transporters in the mouse brain. Neurotoxicology 30, 274–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Budygin E. A., John C. E., Mateo Y., Jones S. R. (2002) Lack of cocaine effect on dopamine clearance in the core and shell of the nucleus accumbens of dopamine transporter knock-out mice. J. Neurosci. 22, RC222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Centers for Disease Control and Prevention (2002) National Health and Nutrition Examination Survey Data. National Center for Health Statistics, Hyattsville, MD, USA. Available: http://www.cdc.gov/nchs/about/major/nhanes/datalink.htm
- 37.Centers for Disease Control and Prevention (2002) National Health and Nutrition Examination Survey Questionnaire (or Examination Protocol, or Laboratory Protocol). National Center for Health Statistics, Hyattsville, MD, USA. Available: http://www.cdc.gov/nchs/about/major/nhanes/nhanes01-02.htm
- 38.Centers for Disease Control and Prevention (2004) NHANES Analytic Guidelines: June 2004 Version. National Center for Health Statistics, Hyattsville, MD, USA. Available: http://www.cdc.gov/nchs/data/nhanes/nhanes_general_guidelines_june_04.pdf
- 39.Braun J. M., Kahn R. S., Froehlich T., Auinger P., Lanphear B. P. (2006) Exposures to environmental toxicants and attention deficit hyperactivity disorder in U.S. children. Environ. Health Perspect. 114, 1904–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spencer T. J., Biederman J., Madras B. K., Faraone S. V., Dougherty D. D., Bonab A. A., Fischman A. J. (2005) In vivo neuroreceptor imaging in attention-deficit/hyperactivity disorder: a focus on the dopamine transporter. Biol. Psychiatry 57, 1293–1300 [DOI] [PubMed] [Google Scholar]
- 41.Gainetdinov R. R., Fumagalli F., Jones S. R., Caron M. G. (1997) Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J. Neurochem. 69, 1322–1325 [DOI] [PubMed] [Google Scholar]
- 42.Johansen E. B., Killeen P. R., Russell V. A., Tripp G., Wickens J. R., Tannock R., Williams J., Sagvolden T. (2009) Origins of altered reinforcement effects in ADHD. Behav. Brain Funct. 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sagvolden T., Johansen E. B., Aase H., Russell V. A. (2005) A dynamic developmental theory of attention-deficit/hyperactivity disorder (ADHD) predominantly hyperactive/impulsive and combined subtypes. Behav. Brain Sci. 28, 397-419; discussion 419–368 [DOI] [PubMed] [Google Scholar]
- 44.Brockel B. J., Cory-Slechta D. A. (1998) Lead, attention, and impulsive behavior: changes in a fixed-ratio waiting-for-reward paradigm. Pharmacol. Biochem. Behav. 60, 545–552 [DOI] [PubMed] [Google Scholar]
- 45.Katz R. J., Schmaltz K. (1980) Dopaminergic involvement in attention. A novel animal model. Prog. Neuropsychopharmacol. 4, 585–590 [DOI] [PubMed] [Google Scholar]
- 46.Sarter M., Bodewitz G., Stephens D. N. (1988) Attenuation of scopolamine-induced impairment of spontaneous alteration behaviour by antagonist but not inverse agonist and agonist beta-carbolines. Psychopharmacology (Berl.) 94, 491–495 [DOI] [PubMed] [Google Scholar]
- 47.Bouchard M. F., Bellinger D. C., Wright R. O., Weisskopf M. G. (2010) Attention-deficit/hyperactivity disorder and urinary metabolites of organophosphate pesticides. Pediatrics 125, e1270–e1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krause J. (2008) SPECT and PET of the dopamine transporter in attention-deficit/hyperactivity disorder. Expert Rev. Neurother. 8, 611–625 [DOI] [PubMed] [Google Scholar]
- 49.Roessner V., Sagvolden T., Dasbanerjee T., Middleton F. A., Faraone S. V., Walaas S. I., Becker A., Rothenberger A., Bock N. (2010) Methylphenidate normalizes elevated dopamine transporter densities in an animal model of the attention-deficit/hyperactivity disorder combined type, but not to the same extent in one of the attention-deficit/hyperactivity disorder inattentive type. Neuroscience 167, 1183–1191 [DOI] [PubMed] [Google Scholar]
- 50.Giros B., Jaber M., Jones S. R., Wightman R. M., Caron M. G. (1996) Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379, 606–612 [DOI] [PubMed] [Google Scholar]
- 51.Salahpour A., Ramsey A. J., Medvedev I. O., Kile B., Sotnikova T. D., Holmstrand E., Ghisi V., Nicholls P. J., Wong L., Murphy K., Sesack S. R., Wightman R. M., Gainetdinov R. R., Caron M. G. (2008) Increased amphetamine-induced hyperactivity and reward in mice overexpressing the dopamine transporter. Proc. Natl. Acad. Sci. USA 105, 4405–4410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Donovan D. M., Miner L. L., Perry M. P., Revay R. S., Sharpe L. G., Przedborski S., Kostic V., Philpot R. M., Kirstein C. L., Rothman R. B., Schindler C. W., Uhl G. R. (1999) Cocaine reward and MPTP toxicity: alteration by regional variant dopamine transporter overexpression. Brain Res. Mol. Brain Res. 73, 37–49 [DOI] [PubMed] [Google Scholar]
- 53.Sagvolden T. (2000) Behavioral validation of the spontaneously hypertensive rat (SHR) as an animal model of attention-deficit/hyperactivity disorder (AD/HD). Neurosci. Biobehav. Rev. 24, 31–39 [DOI] [PubMed] [Google Scholar]
- 54.Carpenter A. C., Saborido T. P., Stanwood G. D. (2012) Development of hyperactivity and anxiety responses in dopamine transporter-deficient mice. Dev. Neurosci. 34, 250–257 [DOI] [PubMed] [Google Scholar]
- 55.Adriani W., Boyer F., Gioiosa L., Macrì S., Dreyer J. L., Laviola G. (2009) Increased impulsive behavior and risk proneness following lentivirus-mediated dopamine transporter over-expression in rats’ nucleus accumbens. Neuroscience 159, 47–58 [DOI] [PubMed] [Google Scholar]
- 56.Cardinal R. N., Howes N. J. (2005) Effects of lesions of the nucleus accumbens core on choice between small certain rewards and large uncertain rewards in rats. BMC Neurosci. 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vijayraghavan S., Wang M., Birnbaum S. G., Williams G. V., Arnsten A. F. (2007) Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat. Neurosci. 10, 376–384 [DOI] [PubMed] [Google Scholar]
- 58.Davis L. M., Michaelides M., Cheskin L. J., Moran T. H., Aja S., Watkins P. A., Pei Z., Contoreggi C., McCullough K., Hope B., Wang G. J., Volkow N. D., Thanos P. K. (2009) Bromocriptine administration reduces hyperphagia and adiposity and differentially affects dopamine D2 receptor and transporter binding in leptin-receptor-deficient Zucker rats and rats with diet-induced obesity. Neuroendocrinology 89, 152–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bobb A. J., Addington A. M., Sidransky E., Gornick M. C., Lerch J. P., Greenstein D. K., Clasen L. S., Sharp W. S., Inoff-Germain G., Wavrant-De Vrieze F., Arcos-Burgos M., Straub R. E., Hardy J. A., Castellanos F. X., Rapoport J. L. (2005) Support for association between ADHD and two candidate genes: NET1 and DRD1. Am. J. Med. Genet. B Neuropsychiatr. Genet. 134B, 67–72 [DOI] [PubMed] [Google Scholar]
- 60.Carey M. P., Diewald L. M., Esposito F. J., Pellicano M. P., Gironi Carnevale U. A., Sergeant J. A., Papa M., Sadile A. G. (1998) Differential distribution, affinity and plasticity of dopamine D-1 and D-2 receptors in the target sites of the mesolimbic system in an animal model of ADHD. Behav. Brain Res. 94, 173–185 [DOI] [PubMed] [Google Scholar]
- 61.Andersen S. L., Teicher M. H. (2000) Sex differences in dopamine receptors and their relevance to ADHD. Neurosci. Biobehav. Rev. 24, 137–141 [DOI] [PubMed] [Google Scholar]
- 62.Barr D. B., Olsson A. O., Wong L. Y., Udunka S., Baker S. E., Whitehead R. D., Magsumbol M. S., Williams B. L., Needham L. L. (2010) Urinary concentrations of metabolites of pyrethroid insecticides in the general U.S. population: National Health and Nutrition Examination Survey 1999-2002. Environ. Health Perspect. 118, 742–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morgan M. K. (2012) Children’s exposures to pyrethroid insecticides at home: a review of data collected in published exposure measurement studies conducted in the United States. Int. J. Environ. Res. Public Health 9, 2964–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oulhote Y., Bouchard M. F. (2013) Urinary metabolites of organophosphate and pyrethroid pesticides and behavioral problems in Canadian children. Environ. Health Perspect. 121, 1378–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quirós-Alcalá L., Mehta S., Eskenazi B. (2014) Pyrethroid Pesticide Exposure and Parental Report of Learning Disability and Attention Deficit/Hyperactivity Disorder in U.S. Children: NHANES 1999-2002. Environ. Health Perspect. 122, 1336–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Froehlich T. E., Lanphear B. P., Auinger P., Hornung R., Epstein J. N., Braun J., Kahn R. S. (2009) Association of tobacco and lead exposures with attention-deficit/hyperactivity disorder. Pediatrics 124, e1054–e1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Froehlich T. E., Lanphear B. P., Dietrich K. N., Cory-Slechta D. A., Wang N., Kahn R. S. (2007) Interactive effects of a DRD4 polymorphism, lead, and sex on executive functions in children. Biol. Psychiatry 62, 243–249 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.