

Abstract
Genes that regulate osteoclast (OC) development and function in both physiologic and disease conditions remain incompletely understood. Shp2 (the Src homology-2 domain containing protein tyrosine phosphatase 2), a ubiquitously expressed cytoplasmic protein tyrosine phosphatase, is implicated in regulating M-CSF and receptor activator of nuclear factor-κB ligand (RANKL)–evoked signaling; its role in osteoclastogenesis and bone homeostasis, however, remains unknown. Using a tissue-specific gene knockout approach, we inactivated Shp2 expression in murine OCs. Shp2 mutant mice are phenotypically osteopetrotic, featuring a marked increase of bone volume (BV)/total volume (TV) (+42.8%), trabeculae number (Tb.N) (+84.1%), structure model index (+119%), and a decrease of trabecular thickness (Tb.Th) (−34.1%) and trabecular spacing (Tb.Sp) (−41.0%). Biochemical analyses demonstrate that Shp2 is required for RANKL-induced formation of giant multinucleated OCs by up-regulating the expression of nuclear factor of activated T cells, cytoplasmic 1 (Nfatc1), a master transcription factor that is indispensable for terminal OC differentiation. Shp2 deletion, however, has minimal effect on M-CSF–dependent survival and proliferation of OC precursors. Instead, its deficiency aborts the fusion of OC precursors and formation of multinucleated OCs and decreases bone matrix resorption. Moreover, pharmacological intervention of Shp2 is sufficient to prevent preosteoclast fusion in vitro. These findings uncover a novel mechanism through which Shp2 regulates osteoclastogenesis by promoting preosteoclast fusion. Shp2 or its signaling partners could potentially serve as pharmacological targets to regulate the population of OCs locally and/or systematically, and thus treat OC-related diseases, such as periprosthetic osteolysis and osteoporosis.—Zhou, Y., Mohan, A., Moore, D. C., Lin, L., Zhou, F. L., Cao, J., Wu, Q., Qin, Y.–X., Reginato, A. M., Ehrlich, M. G., Yang, W. SHP2 regulates osteoclastogenesis by promoting preosteoclast fusion.
Keywords: M-CSF, RANKL, Nfatc1
Osteoclasts (OCs), which are the exclusive bone-resorbing cells in mammals, play a central role in regulating skeletal development and bone mass (1). Originating from hematopoietic stem cells, they mature and differentiate through 3 general stages: mononuclear tartrate-resistant acid phosphatase (TRAP)-positive (TRAP+) preosteoclasts, nonfunctional polykaryons formed by the fusion of multiple (10–20) individual preosteoclasts, and, finally, fully activated functional OCs (2, 3). Osteoclast differentiation is regulated by 2 key growth factors, M-CSF and receptor activator of nuclear factor-κB ligand (RANKL) (4, 5). M-CSF, which signals through c-Fms, a transmembrane receptor tyrosine kinase, is crucial for myeloid cell survival and differentiation to preosteoclasts (6), and RANKL elicits a broad range of cellular responses, including preosteoclast fusion, polykaryon maturation, and OC activation (3). Many of the crucial RANKL-mediated changes, including preosteoclast fusion and OC maturation, are mediated by nuclear factor of activated T cells, cytoplasmic 1 (Nfatc1), a calcineurin- and calcium-regulated transcription factor (7).
A member of the nuclear factor of activated T cells family of transcription factors, NFATc1 is the transcription factor most strongly up-regulated following RANKL stimulation of bone marrow–derived macrophage (BMM) precursor cells. (7). Mouse monocyte/macrophage precursors with loss-of-function mutations in Nfatc1 (Nfatc1−/−) fail RANKL-stimulated differentiation into OCs (7), and with overexpression of Nfatc1 efficiently induce OC differentiation in the absence of RANKL (7). Mechanistically, NFATc1 induces the expression of osteoclastogenic genes, such as TRAP (8), cathepsin K (Ctsk) (9), the d2 isoform of vacuolar calcitonin receptor, and matrix metallopeptidase 9 (MMP9) (10). Importantly, NFATc1 is a pivotal regulator of differentiation as it modulates the expression of ATPase V0 domain (ATP6v0d2) and dendritic cell–specific transmembrane protein (DC-STAMP) (11), both of which are key mediators of preosteoclast fusion (12). Although multiple molecules and cellular signaling pathways are known to mediate the signal transduction from RANK to NFATc1, how they are regulated during the process of osteoclastogenesis remains incompletely understood.
Shp2, encoded by Ptpn11, is a widely expressed src-homology-2 domain containing protein tyrosine phosphatase (13, 14) that may play a role in the RANK/NFATc1 signaling pathway, and, by extension, OC development, skeletal remodeling, and bone mineral homeostasis. Shp2 is required for Ras/Erk activation by most, if not all, receptor tyrosine kinases and cytokine receptor signaling, influencing cell viability, proliferation, migration, and differentiation (13, 14). Previous studies using primary BMM and cell lines suggest a critical role for Shp2 in c-Fms (15) and RANK signaling (16). Shp2 gain-of-function mutations cause myeloproliferative disorders associated with skeletal abnormalities (17), and, interestingly, genetic deletion of Shp2 in skeletal muscle compromises myoblast fusion associated with impaired Nfatc1 expression and activation (18). However, relatively little is known about the role of Shp2 in osteoclastogenesis. What is known is that Shp2-null mice die early in gestation (19, 20), and mice with ubiquitous postnatal Shp2 deletion exhibit skeletal malformations and they fail to generate TRAP+ multinucleated OCs (21). To what extent these phenotypes emanate from Shp2 deficiency in OCs and/or OC precursors is impossible to determine, as Shp2 is deleted in all cell types, especially hematopoietic stem and progenitor cells, common myeloid progenitors, and granulocyte macrophage progenitors (21).
There is increasing evidence that Shp2 has a significant role in regulating hematopoiesis (13, 14). Tissue-specific Shp2 loss-of-function mutations lead to hematopoietic dysplasia, affecting multiple lineages of hematopoietic cells (22). As well, Shp2 gain-of-function mutations cause Noonan syndrome and other myeloid proliferative disorders in humans, many of which are associated with skeletal phenotypes, featuring craniofacial abnormalities, short stature, and cardiac defects (23, 24). These hematopoietic and skeletal manifestations in humans are recapitulated in mice carrying the same genetic mutations (17, 25). In an effort to understand the role of Shp2 in myeloid lineage cells, we have created mice lacking Shp2 specifically in lysozyme M-expressing cells, presumably macrophages and OCs (26). Shp2-deficient mice are born at the expected Mendelian ratios, have normal blood counts, differentials, and expression of myelomonocytic cell surface markers. However, they subsequently develop age-related osteopetrosis, strongly suggesting that Shp2 plays a role in regulation of OC development and OC bone resorptive activity.
To address these questions and understand the role of Shp2 in osteoclastogenesis and skeletal homeostasis, we took a tissue-specific gene knockout approach, generating a genetically modified mouse strain in which Shp2 is deleted only in Ctsk-expressing cells, predominantly OCs. The objective of this study was to evaluate the effect of Shp2 deficiency on OC development, skeletal remodeling, and bone mineral homeostasis. We found that Shp2 is essential for both in vivo and in vitro osteoclastogenesis, regulating giant multinucleated OC formation by promoting the fusion of preosteoclasts via the RANKL/NFATc1 signaling axis. Our study reveals a novel mechanism through which Shp2 regulates osteoclastogenesis and provides a potential alternative therapeutic target for the treatment of osteoporosis and osteolytic diseases.
MATERIALS AND METHODS
Transgenic animals
Mice carrying the Ptpn11 floxed allele (26), LysM-Cre (27), and Ctsk-Cre (28) were maintained in the pathogen-free animal facility at Rhode Island Hospital. All animal-related experiments were approved by the Institutional Animal Care and Use Committee. To generate mice lacking Shp2 in cells that express lysozyme or Ctsk, mice carrying the Ptpn11 floxed allele were bred to LysM-Cre and Ctsk-Cre mice respectively, yielding offspring with the following genotypes and nomenclature: Ptpn11fl/+;LysM-Cre (Shp2LysM-CTR), Ptpn11fl/fl:LysM-Cre (Shp2LysM-cKO), Ptpn11fl/+;Ctsk-Cre (Shp2Ctsk-CTR) and Ptpn11fl/fl;Ctsk-Cre (Shp2Ctsk-cKO). Shp2Ctsk-CTR and Shp2LysM-CTR were used as controls as there is no discernible phenotype between wild-type, Shp2LysM-CTR, and Shp2Ctsk-CTR mice. Both LysM-Cre and Ctsk-Cre are knock-in alleles and were maintained in a heterozygous state in this study.
Reagents
DMEM, fetal bovine serum, penicillin, and streptomycin were purchased from Invitrogen Inc. (Grand Island, NY, USA); soluble RANKL and murine M-CSF were purchased from Peprotech (Rocky Hill, NJ, USA). TRAP staining kits and cell viability/proliferation kits were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Roche (Indianapolis, IN, USA), respectively. Antibodies against Nfatc1 and c-Fos were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and against Shp2 and Erk2 from Upstate Inc. (Lake Placid, NY, USA). Shp2 inhibitor NSC-87877 (29) was purchased from TOCRIS Bioscience (Minneapolis, MN, USA).
Skeletal phenotype and bone quality evaluation
X-ray images of 8-week-old male mice were taken immediately after euthanasia using a digital radiography system (MX-20; Faxitron Bioptics, LLC, Tucson, AZ, USA). High-resolution 3-dimensional volume images were generated with a desktop micro-computed tomography system (µ-CT40; Scanco Medical, Bruttisellen, Switzerland) after fixation of bone specimens in 4% paraformaldehyde. To measure new bone mineralization rate, mice received intraperitoneal injections of calcein (10 mg/kg) and xylenol orange (90 mg/kg), respectively, at days 7 and 2 before euthanasia. After fixation, 8 µm thick sections of proximal tibia were mounted unstained for evaluation of calcein and xylenol orange fluorescence. Nanoindentation (TriboIndenter, Hysitron, Inc., Minneapolis, MN, USA) was performed to evaluate tissue-level mechanical properties. To do so, the proximal tibias were split longitudinally, cleaned, dried, and embedded in epoxy resin. Five intact trabeculae from the metaphyseal region were evaluated in each sample. Indentation was performed in a 3 × 3 grid pattern, resulting in a total of 90 indents for each sample. The distance between adjacent indentations was 1.5 µm. The indentation procedure involved loading to 1000 µN at a rate of 100 µN/second over 10 seconds, a 30-second dwell time, and then unloading at −100 µN/second, also over 10 seconds. The elastic modulus and hardness of the sample were calculated from 20 to 90% of the unloading curve (30).
Bone marrow cell isolation, culture, and osteoclastogenic differentiation
Marrow cells were flushed from explanted femurs and tibiae of mice and concentrated as described elsewhere (31). Pelletized cells were resuspended in red blood cell lysis buffer and cultured in macrophage medium (DMEM with 10% fetal bovine serum, 1% penicillin/streptomycin, and 10 ng/ml M-CSF) at 37°C and 5% CO2 for 3 days. The medium was changed every other day. To induce osteoclastic differentiation, the cells were incubated for an additional 5–7 days in OC differentiation medium (macrophage medium supplemented with 100 ng/ml RANKL). The resulting marrow-derived OCs were then used for various biologic and biochemical assays.
TRAP staining and pit assays
TRAP staining was conducted to identify multinucleated OCs in bone marrow cell cultures in vitro and tibia sections in vivo. Briefly, tibia collected from 8-week-old Shp2Ctsk-CTR and Shp2Ctsk-cKO mice were fixed in 4% paraformaldehyde overnight and then decalcified in EDTA solution. The specimens were then dehydrated following standard procedures and embedded in paraffin for sectioning. TRAP+ multinucleated OCs (>3 nuclei) were quantified using a Nikon (Tokyo, Japan) digital microscope. Osteoclast-mediated bone resorptive activity was quantified using a standard pit assay (32). Briefly, 5 × 104 preosteoclasts were plated directly onto a dentin slice (4.4 × 4.4 × 0.2 mm) in 48-well tissue culture plates and cultured for 7 days. After culture, dentin slices were treated with 1 M ammonia by sonication to remove all cellular components and then stained with toluidine blue to evaluate formation of pits microscopically.
Cell proliferation and viability assay
To measure the effect of Shp2 deletion on the proliferation and viability of preosteoclasts and OCs, bone marrow cells from Shp2Ctsk-CTR and Shp2Ctsk-cKO mice were cultured in DMEM supplemented with M-CSF, RANKL, or both with the indicated doses. Tetrazolium salt WST1 reagent was then added (1/10, v/v) 2 hours before measuring optical density on a spectrophotometer at 420 nm. The effects of Shp2 inhibition by NSC-87877 on OC precursor proliferation and viability were determined using the same method.
Quantitative RT-PCR assay
Total RNA was extracted from OC cultures using the RNeasy mini kit (Qiagen, Valencia, CA, USA). cDNA was synthesized using 1 µg total RNA with iScript DNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) and quantitative RT-PCR (qRT-PCR) was performed with iQ SYBR Green qPCR kit (Bio-Rad) on a CFX384 real-time PCR machine. Gene expression in OC was normalized to Gapdh level before fold increases or decreases were calculated between Shp2Ctsk-CTR and Shp2Ctsk-cKO mice. The stability of Gapdh expression was validated against Actin and Hprt. All primer sequences used for this study are listed Table 1.
TABLE 1.
Primer sequences used for qRT-PCR analysis of murine osteoclastogenic genes
| Gene | Forward | Reverse | Product (bp) | Gene Bank Access No. |
|---|---|---|---|---|
| Trap | 5′-TGGTCCAGGAGCTTAACTGC | 5′-GTCAGGAGTGGGAGCCATATG | 114 | AK016276 |
| Ctsk | 5′-GTGGGTGTTCAAGTTTCTGC | 5′-GGTGAGTCTTCTTCCATAGC | 98 | BC046320 |
| CltR | 5′-CTCCAACAAGGTGCTTGGGA | 5′-GAAGCAGTAGATAGTCGCCA | 91 | AK133983 |
| cFos | 5′-CCAAGCGGAGACAGATCAACTT | 5′-TCCAGTTTTTCCTTCTCTTTCAGCAGAT | 89 | BC029814 |
| GAPDH | 5′-CCCCCAATGTGTCCGTCG | 5′-CGGCATCGAAGGTGGAAGA | 183 | AK169742 |
| Mmp9 | 5′-GAGCTGTGCGTCTTCCCCTTC | 5′-GGAATGATCTAAGCCCAGTGC | 309 | D12712 |
| Atp6vOd2 | 5′-TTCAGTTGCTATCCAGGACTCGGA | 5′-GCATGTCATGTAGGTGAGAAATGTGCTCA | 350 | AK170578 |
| DC-STAMP | 5′-TTGCCGCTGTGGACTATCTG | 5′-GAATGCAGCTCGGTTCAAAC | 171 | AB109560 |
| Nfatc1 | 5′-CGGGAAGAAGATGGTGCTGT | 5′-TTGGACGGGGCTGGTTAT | 190 | EU887569 |
| Hprt | 5′-GTAATGATCAGTCAACGGGGGAC | 5′-CCAGCAAGCTTGCAACCTTAACCA | 177 | AK002286 |
| Actin | 5′-CGTGAAAAGATGACCCAGATCA | 5′-CACAGCCTGGATGGCTACGT | 72 | X03672 |
Cell signaling study
To examine the molecular mechanism of Shp2 in M-CSF and RANKL signaling, bone marrow–derived OCs from Shp2Ctsk-CTR and Shp2Ctsk-cKO mice were lysed into modified NP-40 lysis buffer [0.5% NP40, 150 mM NaCl, 1 mM EDTA, 50 mM Tris (pH 7.4)] supplemented with a protease inhibitor cocktail (1 mM PMSF, 10 mg/ml aprotinin, 0.5 mg/ml antipain, and 0.5 mg/ml pepstatin). Total cell lysates were separated from debris by centrifugation at 14,000 rpm for 10 minutes and protein concentrations in individual supernatants were determined by the BCA protein assay kit (Thermo Scientific/Pierce Products, Rockland, ME, USA) with a Nanodrop Spectrophotometer (Thermo Scientific). For immunoblotting, cell lysates (30–50 µg) were resolved by SDS-PAGE, transferred to PVDF membranes, and incubated with primary antibodies for 2 hours or overnight at 4°C. After a 1 hour wash in TBS-T buffer (1× TBS with 0.1% Tween-20), the membranes were incubated with horseradish peroxidase–conjugated secondary antibodies. Detection of the proteins of interest was performed by enhanced chemiluminescence (Amersham, Piscataway, NJ, USA).
Phosphatase activity assay
To measure the enzymatic activity of Shp2 in preosteoclasts in response to inhibitor (NSC-87877) treatment, cells were lysed into cold plain NP-40 lysis buffer. After removing cell debris, Shp2 in cell lysate (0.6 mg protein) was immunoprecipitated with an antibody to Shp2 coupled to Protein A-Sepharose beads (Invitrogen, Carlsbad, CA, USA) for 2 hours at 4°C. The immunoprecipitates were washed twice with NP-40 lysis buffer and protein tyrosine phosphatase reaction buffer (25 mM HEPES, pH 7.2, 50 mM sodium chloride, 5 mM dithiothreitol, 2.5 mM EDTA, and 5% glycerol) and the Shp2 immune complex was resuspended in 50 µl of reaction buffer containing 50 mM para-nitrophenyl phosphate (pNPP). The reaction is quenched after 10 minutes by the addition of 1 ml of 1 N NaOH. The amount of product, p-nitrophenol, was determined via the optical absorbance at 405 nm.
Bone marrow cell infection and Nfatc1 overexpression assay
pMX-IRES-GFP–based constructs harboring Nfatc1 were used, as previously described (7). This expression system allows the determination of Nfatc1 expression by monitoring GFP signal in the infected cells. To generate bone marrow cells expressing GFP or Nfatc1/iresGFP, Plat-E cells were transiently transfected with pMX constructs via Effectene (Qiagen) per the manufacturer’s instructions. Viruses were collected 48 hours post-transfection and used to infect bone marrow cells that were seeded the previous day in the presence of M-CSF (20 ng/ml), and polybrene (4 µg/ml) was added during the infection process. Infected cells were then expanded and used for osteoclastogenesis assays in vitro.
Statistical analysis
Statistical differences between groups were evaluated by Student’s t test. P values <0.05 were considered statistically significant. Analyses were performed using Excel (Microsoft, Redmond, WA, USA) and Prism 3.0 (GraphPad, San Diego, CA, USA). Data are presented as mean ± sd.
RESULTS
Mice lacking Shp2 in Ctsk-expressing cells are osteopetrotic
Shp2Ctsk-CTR and Shp2Ctsk-cKO mice were bred as described (Fig. 1A) and born at the expected Mendelian ratios. Western blot analysis demonstrated that Ctsk-Cre mediated efficient deletion of Shp2 in OCs derived from bone marrow cells of Shp2Ctsk-cKO mice compared with those of Shp2+/+ mice (Supplemental Fig. 1). Both Shp2Ctsk-CTR and Shp2Ctsk-cKO mice appeared normal for the first 3 weeks postbirth; however, Shp2Ctsk-cKO mice subsequently developed short stature (Fig. 1B i, ii), increased bone mineral density (BMD), and exostoses at the metaphyses of the tubular long bones (Fig. 1B ii, iv, vi, viii). The disease affected both genders; our subsequent in vivo experiments were focused on male mice to avoid potential interference of sex. Gross images of explanted bones revealed that Shp2Ctsk-cKO mice had short and widened femurs, tibiae, and humeri and that the vertebrae and scapulae were also affected (Fig.1B, Supplemental Fig. 1C). µ-CT image analysis revealed a marked increase in volumetric bone density (BV/TV, 16.2 vs. 23.1; +42.8%), structure model index (1.37 vs. 3.0; +119%), and Tb.N (3.35 vs. 6.17; +84.1%) in Shp2Ctsk-cKO mice compared with Shp2Ctsk-CTR controls, and Tb.Th (0.049 vs. 0.032; −34.1%) and Tb.Sp (0.225 vs. 0.133; −41.0%) decreased (Fig. 1B v–viii, C). Both Shp2Ctsk-CTR and Shp2Ctsk-cKO mice had normal tooth eruption (Supplemental Fig. 1B). Nanoindentation testing revealed that Shp2 deletion reduced bone quality, decreasing the elastic modulus (15.5 GPa vs. 11.6 GPa; −24.7%) and hardness (0.744 vs. 0.612; −17.6%) of trabeculae in Shp2Ctsk-cKO mice (Fig. 1D). Taken together our data demonstrate that OC-specific Shp2 deficiency significantly influences skeletal development and remodeling and that Shp2 mutant mice are osteopetrotic.
Figure 1.
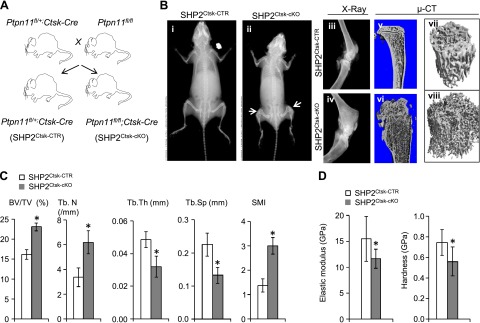
Mice lacking Shp2 in OC are osteopetrotic. A) Breeding schemes used to generate Shp2Ctsk-CTR and Shp2Ctsk-cKO mice. B) Radiographs (i–iv) and µ-CT imaging (v–viii) showed skeletal dwarfism and increases of BMD and trabecular numbers in Shp2Ctsk-cKO mice compared with Shp2Ctsk-CTR controls. C) Histomorphometric analysis demonstrated increases of volumetric bone density (BV/TV; +42.8%), trabeculae number (Tb.N; +84.1), structural modeling index (SMI; +119%), and decreases of Tb.Th (−34.1%) and Tb.Sp (−41.0%) in Shp2Ctsk-cKO mice. Data were collected from proximal tibiae from 8-week-old male Shp2Ctsk-CTR and Shp2Ctsk-cKO mice (n = 4). D) Nanoindentation analysis showing marked decreases in elastic modulus and hardness of trabecula from Shp2Ctsk-cKO mice compared with that of Shp2Ctsk-CTR (Shp2Ctsk-CTR vs. Shp2Ctsk-cKO: elastic modulus 15.5–11.6 GPa, −24.7%; hardness 0.744–0.612 GPa, −17.6%). *P < 0.05 (Student’s t test).
Shp2 deficiency impairs osteoclastogenesis in vivo and in vitro
To begin to understand how Shp2 influences skeletal phenotype, we examined the development of OC in vivo via TRAP staining of histologic sections (counterstained with hematoxylin). TRAP-positive multinucleated OCs were readily apparent on the surfaces of metaphyseal trabecular bone in Shp2Ctsk-CTR mice (Fig. 2A, arrows and pink staining), and they were absent from similar regions of the bones from Shp2Ctsk-cKO mice. Quantification revealed that the number of TRAP+ cells on the trabecular surfaces of Shp2Ctsk-cKO mice was significantly less than on the trabecular surfaces of Shp2Ctsk-CTR control (only ∼ 6.6%). Moreover, the few TRAP+ cells were present on the trabecular surfaces of Shp2Ctsk-cKO mice were mononuclear (Fig. 2A, red arrows).
Figure 2.
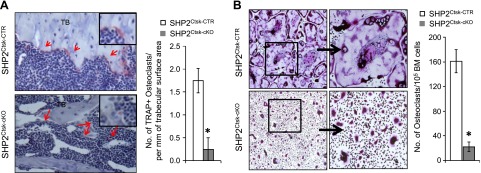
Shp2 deletion impairs osteoclastogenesis in vivo and in vitro. A) TRAP staining of tibia sections (left) from 8-week-old male mice revealed the presence of multinucleated OCs (red) on trabecular bone (TB) surface of Shp2Ctsk-CTR mice; these cells diminished in sex- and age-matched Shp2Ctsk-cKO mice. Slides were counterstained with hematoxylin (blue). Bar graph (right) depicting the average number of OC/mm on the trabecular bone surface (n = 3). B) TRAP staining showed the reduction of multinucleated OCs in an in vitro osteoclastogenesis assay using bone marrow cells from Shp2Ctsk-cKO mice compared with Shp2Ctsk-CTR controls. Bone marrow cells (1 × 105) were cultured in OC medium as described in the Materials and Methods section. Bar graph (right) depicting the quantification of TRAP+ OC (>3 nucleus) in this assay (n = 4). *P < 0.05 (Student’s t test).
Given the lack of TRAP+ cells, we postulated that the blockade of OC formation in Shp2 mutant mice might result from defective M-CSF and/or RANKL signaling. Because osteoclastogenesis in vivo can be recapitulated in vitro by culturing bone marrow cells in the presence of M-CSF and RANKL (4, 33), we adapted this assay to explore the role of Shp2 in osteoclastogenesis in further detail. Consistent with our previous observations, bone marrow cells from Shp2Ctsk-cKO mice largely failed to generate TRAP+ multinucleated OCs (only 16.6% of Shp2Ctsk-CTR) in response to M-CSF and RANKL treatment in vitro (Fig. 2B). Moreover, Shp2 deficiency in OC compromised new bone formation (reduced to 86% of Shp2Ctsk-CTR) as revealed by calcein and xylenol orange double labeling assays (Supplemental Fig. 2). This defective osteoclastogenesis both in vivo and in vitro indicates that Shp2 is essential for OC development and skeletal remodeling and that it functions downstream of c-Fms and RANK.
Shp2 regulates osteoclastogenesis by promoting preosteoclast fusion
Having established that Shp2 has a clear role in OC differentiation, we next addressed whether Shp2 deficiency influences the viability and/or proliferation of the OC lineage. Bone marrow cells from Shp2Ctsk-CTR and Shp2Ctsk-cKO mice were cultured in medium containing a fixed dose of M-CSF and varying doses of RANKL (Fig. 3A, B) or, conversely, a fixed dose of RANKL and varying doses of M-CSF (Fig. 3C, D). The viability and proliferation of OC precursors were evaluated using the WST1 test. Osteoclastogenic differentiation was assessed by TRAP staining of multinucleated giant cell formation in cultures.
Figure 3.
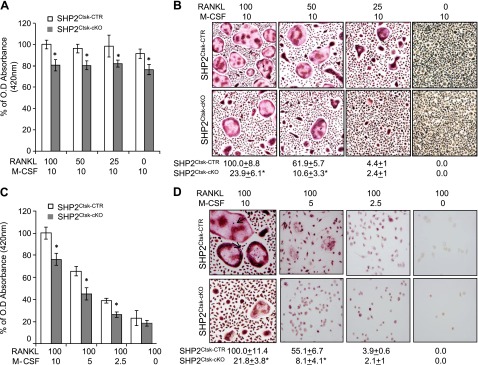
Shp2 is required for osteoclastogenesis by promoting preosteoclast fusion. A) Bar graphs showing the viability and proliferation of OC precursors from Shp2Ctsk-CTR and Shp2Ctsk-cKO mice cultured for 3 days in the presence of an optimal dose of M-CSF and variable doses of RANKL. Numbers of viable cells were determined by WST1 tests with a spectrophotometer at the 420 nm wavelength; the data represented the percentage of optical density relative to the optimal culture conditions (100 ng/ml RANKL and 10 ng/ml M-CSF). Shp2 deletion only slightly compromised the proliferation of OC precursors (n = 3) *P < 0.05 (Student’s t test). B) In vitro osteoclastogenesis and TRAP staining assays showed that Shp2 deficiency compromised M-CSF and RANKL-induced multinucleated OC formation. Bone marrow cells were cultured under the conditions indicated for 5 days. Note that an increase of RANKL dose promoted the formation of multinucleated OCs only in bone marrow cells from Shp2Ctsk-CTR but not Shp2Ctsk-cKO mice. The quantitative data represented the percentage of OC number relative to that of Shp2Ctsk-CTR marrow cells under the optimal culture conditions (100 ng/ml RANKL and 10 ng/ml M-CSF) is shown below the images (n = 3). *P < 0.05 (Student’s t test). C) Bar graph showing the viability and proliferation of OC precursors from Shp2Ctsk-CTR and Shp2Ctsk-cKO mice cultured for 3 days in the presence of an optimal dose of RANKL and variable doses of M-CSF. Numbers of viable cells were determined by WST1 tests as described above. Note that reduction of M-CSF dose markedly impaired the proliferation and viability of OC precursors from both Shp2Ctsk-CTR and Shp2Ctsk-cKO mice (n = 3). *P < 0.05 (Student’s t test). D) In vitro osteoclastogenesis and TRAP staining showed that M-CSF but not RANKL-evoked signaling was essential for the proliferation and survival of preosteoclasts under the same culture conditions as described in C. Shp2 deletion in these cells had minimal effect on their proliferation and survival. The data was presented as the percentage of OC numbers relative to that under the optimal culture conditions (100 ng/ml RANKL and 10 ng/ml M-CSF; n = 3). *P < 0.05 (Student’s t test).
The WST1 results demonstrated that with optimal M-CSF dosing, regardless of RANKL dosing, the proliferation and viability of osteoclastogenic cells were largely comparable in osteoclastogenic cultures of Shp2Ctsk-CTR and Shp2Ctsk-cKO mice (Fig. 3A, B). In contrast, the proliferation and viability of osteoclastogenic cells from both Shp2Ctsk-CTR and Shp2Ctsk-cKO mice declined when the M-CSF dose was reduced, even when an optimal dose of RANKL maintained (Fig. 3C, D). These data suggest that M-CSF-evoked signals primarily maintain the proliferation and viability of OC precursors.
Interestingly, when cultured in media supplemented with an optimal dose of M-CSF and increased doses of RANKL, bone marrow cells from Shp2Ctsk-CTR mice formed increased numbers of both TRAP+ preosteoclasts and giant multinucleated OCs, and bone marrow cells from Shp2Ctsk-cKO mice formed only TRAP+ preosteoclasts, with few if any fused giant multinucleated OCs (Fig. 3B). Conversely, when bone marrow cells were cultured in medium supplemented with optimal doses of RANKL and decreased M-CSF, both the proliferation of OC precursors and their osteoclastogenic differentiation were compromised in Shp2Ctsk-CTR and Shp2Ctsk-cKO bone marrow cultures (Fig. 3D). The above observations were corroborated with a fluorescent protein-based reporter study. Bone marrow cells from Shp2fl/fl and Shp2Ctsk-cKO mice carrying the R26mTmG reporter were cultured in OC differentiation medium. After induction of Ctsk-Cre activity, bone marrow–derived preosteoclasts from Shp2Ctsk-cKO mice changed from red (Tomato Red) to green (GFP+) (Supplemental Fig. 3). The Shp2-deficient green preosteoclasts failed to form giant multinucleated OC, which was consistent with our TRAP staining data. In contrast, the Shp2 competent red preosteoclasts from Shp2fl/fl;Rosa26mTmG mice formed giant multinucleated OC (Supplemental Fig. 3). These findings suggest that Shp2 regulates OC differentiation by modulating preosteoclast fusion via a RANK-dependent signaling pathway.
Shp2 regulates preosteoclast fusion by tilting NFATc1 expression
To explore the molecular mechanism by which Shp2 regulates osteoclastogenesis, we performed qRT-PCR analysis to examine the regulation of genes involved in OC fusion and terminal differentiation. Bone marrow cells from Shp2Ctsk-CTR and Shp2Ctsk-cKO mice were cultured in macrophage medium for 3 days and then switched to OC differentiation medium for an additional 5 days. The cells were then lysed and total RNA was extracted. qRT-PCR analysis with the primer sets listed in Table 1 revealed that the expression of OC marker genes, such as Ctsk, Mmp9, Trap, and Calcitonin receptor, were all down-regulated in OC cultures from Shp2Ctsk-cKO mice compared with Shp2Ctsk-CTR controls (Fig. 4A). These findings were consistent with our previous observations that OC terminal differentiation was impaired as a consequence of Shp2 deletion in vivo and in vitro (Fig. 2A, B). Importantly, we found that the expression of Nfatc1 and DC-STAM, 2 genes critical for OC fusion (11, 34), were markedly decreased in the OC cultures of bone marrow from Shp2Ctsk-cKO mice, to only 11.7 and 16.5% of the level of the Shp2Ctsk-CTR controls, respectively (Fig. 4A). To validate the above qRT-PCR results at a protein level, Western blotting analysis was conducted and Nfatc1 and DC-SAMP levels indeed decreased in the OC culture from Shp2Ctsk-cKO mice (Fig. 4B, Supplemental Fig. 4). These results strongly suggest that Shp2 regulates osteoclastogenesis by modulating preosteoclast fusion, possibly by up-regulating the expression of Nfatc1.
Figure 4.

RANK/Shp2/NFATc1 signaling axis regulates preosteoclast fusion. A) Bar graphs of qRT-PCR data showed the abundance of osteoclastogenic transcripts in OC cultures from Shp2Ctsk-CTR and Shp2Ctsk-cKO mice. Notice that Shp2 deletion compromised the expression of calcitonin receptor (CaltR), Ctsk, Mmp9, Trap, and the 2 key OC fusion-related genes, Nfatc1 and DC-STAMP (DC). B) Western blot analysis showed the reduced expression of Nfatc1 and DC-STAMP in the OC cultures from Shp2Ctsk-cKO mice; expression of c-Fos is comparable between Shp2Ctsk-CTR and Shp2Ctsk-cKO mice. Actin was used as a loading control. C) Restoration of Nfatc1 expression in SHP2CTSK-cKO bone marrow cells resume their ability to form multinucleated OCs. Bone marrow cells isolated from Shp2Ctsk-cKO mice were infected with retrovirus (see Materials and Methods for details) expressing either GFP or Nfatc1/GFP. The top panel showed the infection efficiency visualized by GFP expression at day 3 postinfection (blue dots are nuclei stained with DAPI). The bottom panel demonstrated restoration of multinucleated OC formation (arrows) of Shp2Ctsk-cKO bone marrow cells postretroviral Nfatc1 expression. *P < 0.05 (Student’s t test, compared with Shp2Ctsk-CTR group).
If the above hypothesis is correct, we reasoned that restoring the expression of Nfatc1 in Shp2-deficient bone marrow cells should rescue the fusion of Shp2-deficient preosteoclasts. To test this hypothesis, Nfatc1 was expressed in Shp2Ctsk-cKO bone marrow cells through retroviral infection. As expected, the fusion of Shp2-deficient preosteoclasts was restored by the overexpression of exogenous Nfatc1 (Fig. 4C). Therefore, we uncovered an important physiologic role for Shp2 in regulating preosteoclast fusion by modulating Nfatc1 expression in response to the RANK signaling.
Pharmacological inhibition of Shp2 blocks preosteoclast fusion in vitro
NSC-87877 is a commercially available inhibitor of Shp2 that functions in a variety of cells to specifically suppress its enzymatic activity. We used NSC-87877 to evaluate whether the pharmacological inhibition of Shp2 could mimic its genetic deletion to suppress preosteoclast fusion in vitro. First, we tested the efficacy of Shp2 inhibition by NSC-87877. Briefly, normal OC precursors were treated with NSC-87877 for 12 hours and Shp2 was immunoprecipitated from the cell lysates and subjected to pNPP-based phosphatase activity assay. NSC-87877 treatment resulted in a dose-dependent reduction in Shp2 activity of 62% (25 µM) or 79% (50 µM), respectively (Fig. 5A, left); though, there was no corresponding effect on the proliferation of preosteoclasts based on the WST1 test (Fig. 5A right). Importantly, the ability of preosteoclasts to form mature multinucleated OCs was substantially compromised by NSC-87877 treatments. As with the dose-related decreases in Shp2 activity, we also observed substantial dose-related reduction in the number of mature OCs formed from cells treated with NSC-87877 compared with the vehicle control, by ∼76% at 25 µM and 92% at 50 µM (Fig. 5B). With NSC-87877 treatment, transcripts of genes responsible for OC terminal differentiation, such as Ctsk, Mmp9, Trap, and Calcitonin receptor, particularly Nfatc1 and DC-STAMP, were all down-regulated as determined by qRT-PCR analysis (Fig. 5C). NSC-87877 also affects Shp1, another member of the 2 Src homology-2 domain–containing phosphatases. However, Shp1 normally represses OC formation, and mice lacking Shp1 have increased number of OCs (35, 36). Thus, blockade of OC formation by NSC-87877 cannot be attributed to Shp1 inhibition. Interestingly, withdrawal of NSC-87877 from the OC culture medium resulted in the progressive reappearance of giant multinucleated OCs (Fig. 5D), suggesting that the inhibition of OC formation by NSC-87877 is reversible.
Figure 5.
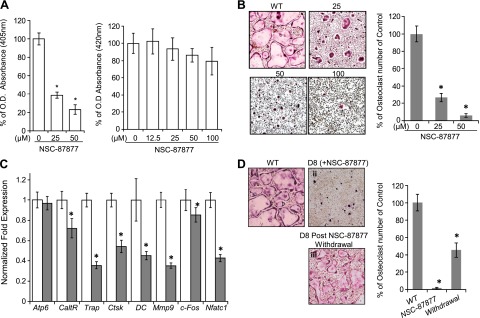
Pharmacological inhibition of Shp2 in vitro markedly compromised the formation of multinucleated OCs. A) Bar graph on the left depicts the enzymatic activity of Shp2 measured by pNPP tests in BMM pre- and post-NSC-87877 treatment. Data are presented as the percentage activity of Shp2 relative to that of untreated groups. Bar graph on the right depicts that NSC-87877 treatment of OC precursors had no apparent effect on their proliferation and viability as determined by WST1 tests. *P < 0.05 (Student’s t test). B) In vitro osteoclastogenesis and TRAP staining assays demonstrated that NSC-87877 treatment of OC precursors from wild-type mice (WT) impaired the formation of multinucleated OC but has no apparent effect on their proliferation and viability. Bar graphs on the right were quantitative data of the left presented as the percentage of OC numbers relative to NSC untreated group (n = 3). *P < 0.05 (Student’s t test). C) qRT-PCR data showed that NSC-87877 treatment (50 µM, solid bars) of OC precursors, compared to controls (open bars), cultured under osteoclastogenic condition impaired the expression of osteoclastogenic marker genes: calcitonin receptor (Caltr), Ctsk, Mmp9, Trap, c-Fos, Nfatc1, and DC-STAMP. *P < 0.05 (Student’s t test). D) In vitro osteoclastogenesis and TRAP staining assays demonstrated that NSC-87877 withdrawal in OC culture medium partially restored the formation of multinucleated OCs. Osteoclast precursors from WT mice were cultured in osteoclastogenic medium for 8 days without (i) or with NSC-87877 treatment (50 µM) (ii) and with NSC-87877 treatment for 8 days and withdrawal for 5 days (iii). Bar graph shows the same data as a percentage of OC numbers relative to NSC-87877 untreated group (n = 3). *P < 0.05 (Student’s t test).
Pharmacological inhibition of Shp2 blunts OC-mediated bone resorption
To evaluate the potential efficacy of pharmacologically blocking Shp2 to treat OC-mediated diseases, such as osteoporosis or osteolysis, we cultured bone marrow-derived preosteoclasts on dentin slices in the presence or absence of Shp2 or its inhibitor NSC-87877 (Fig. 6A, B). After culture, the cellular components were removed and the dentin slices were stained with toluidine blue. As expected, there were numerous large toluidine-stained pits on dentin slices exposed to OC with normal Shp2 activity, indicating that these OCs are capable of resorbing bone matrix. In contrast, only a few and smaller pits were formed when Shp2 was deleted or NSC-87877 was added to the culture medium. These findings strongly suggest that the formation of giant multinucleated OCs and the ability of these cells to resorb bone matrix are compromised by Shp2 deletion and inhibition.
Figure 6.
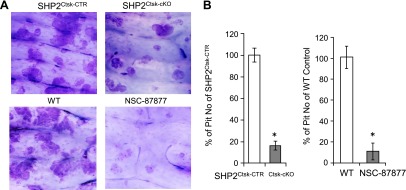
Pharmacological inhibition of Shp2 blunted OC-mediated bone resorption. A) Pit assays demonstrated that Shp2 deletion and inhibition with NSC-87877 (50 µM) markedly reduced OC-mediated bone resorption. Toluidine blue staining was used to visualize the pits formed on dentin slices after being cocultured with OC for 10 days. B) Bar graphs depicting the quantitative data corresponding to A. Data are presented as a percentage of pit numbers relative to the controls (n = 3). *P < 0.05 (Student’s t test; compared with the control group). WT, wild-type osteoclasts.
DISCUSSION
In this study, we explored the molecular origins of a serendipitously created osteopetrotic phenotype in macrophage-specific Shp2-deficient (Shp2LysM-cKO) mice. To do so, we ablated Shp2 in Ctsk-expressing cells—predominantly OCs—in mice via “Cre-loxP”-mediated gene deletion. Overall OC-specific Shp2-deficient mice (Shp2Ctsk-cKO) were smaller than corresponding control mice (Shp2Ctsk-CTR), with “stockier” long bones and vertebrae. At the microstructural level, the bones of the Shp2 mutant mice had a clear osteopetrotic phenotype, with increased volumetric density and alteration from platelike to rodlike architecture of trabeculae and a decrease in tissue-level bone material properties (hardness and elastic modulus). Importantly, Shp2 deficiency in Ctsk-expressing cells was associated with an absence of TRAP+ multinucleated OCs in vivo and in vitro, though a few TRAP+ mononuclear cells persisted. These findings indicate that Shp2 expression in OCs directly affects bone mass and microarchitecture, most likely by modulating osteoclastogenesis.
Upon establishing a genotype and phenotype relationship in OC-specific Shp2 mutant mice, our subsequent study revealed that Shp2 is a crucial mediator of preosteoclast fusion, rather than a supporter of its viability or proliferation. Shp2 has been identified in the c-Fms-associated signaling complex that forms upon M-CSF binding (37), suggesting a role for Shp2 in regulating the proliferation and survival of OC precursors and mature OCs. However, our data indicate that Shp2 deletion has minimal effect on preosteoclast proliferation or survival in the presence of sufficient M-CSF and RANKL. The number of TRAP-expressing preosteoclasts is comparable in bone marrow cultures from Shp2Ctsk-CTR and Shp2Ctsk-cKO mice in in vitro cell viability and proliferation assays. In contrast, Shp2 deletion markedly impaired the formation of giant multinucleated osteoclasts both in vivo and in vitro. Similar results were obtained in experiments on mice lacking Shp2 in more primitive OC precursors (myelomonocytic macrophages) due to lysozyme Cre-mediated Ptpn11 deletion (W. Yang and B. Neel, unpublished data). The results from experiments on these 2 mouse strains clearly indicate that Shp2 affects later OC differentiation, as opposed to viability or proliferation. These findings contrast with those of Bauler et al. who suggest that defective osteoclastogenesis is the result of impaired Akt activation and survival of OC precursors (21). At this point, the reason for this discrepancy is unclear, though it most likely reflects differences in the genetic approach of Ptpn11 deletion in the 2 mouse models. In our animals, the 2 loxP sites flank Ptpn11 exon 11, which encodes the enzymatic motif of “VHCSA/G” of Shp2. Excision of this floxed exon 11 results in a protein-null allele (26). Akt activation, upon M-CSF stimulation of BMM from our Shp2LysM-cKO mice (which had a normal total and differential blood cell counts), is pronounced (W. Yang and B. Neel, unpublished data).
The blockage of preosteoclast fusion by Shp2 deficiency was accompanied by decreased expression of several osteoclastogenic genes in Shp2-deficient preosteoclasts, including the calcitonin receptor, Ctsk, DC-STAMP, Mmp9, Trap, and Nfatc1. Published gene expression data have identified these genes as the primary downstream effectors in the RANK signaling cascade (7, 38, 39). Among them, Nfatc1 and its signaling partner DC-STAMP were reported to be critical for OC fusion (7, 11). Previous studies have shown that c-Fos functions upstream of Nfatc1 and that both genes are required for preosteoclast fusion in response to RANKL signaling (7, 34). Moreover, Nfatc1 directly controls ATP6v0d2 and DC-STAMP expression (11). Most importantly, although both Nfatc1 mRNA and protein levels markedly decreased in Shp2-deficient preosteoclasts, re-expression of Nfatc1 in these cells via virus infection restored their ability to form giant multinucleated OCs, which supports a model whereby Shp2 is an integral component of the RANK signaling pathway that regulates OC terminal differentiation, and the idea that Shp2 is essential for normal Nfatc1 expression and preosteoclast fusion at later stages of osteoclastogenesis. Interestingly, our data indicate that only Nfatc1 and DC-STAMP were significantly reduced in Shp2-deficient OCs in response to both M-CSF and RANKL stimulation, and c-Fos and ATP6v0d2 levels remained unchanged. Further studies will be required to elucidate this dichotomy.
Osteoclast-targeting drugs are the cornerstone of osteoporosis therapy and other conditions of OC-mediated bone fragility, including Paget disease, metastatic disease, and multiple myeloma. Currently, the most commonly used OC-targeting clinic drugs are bisphosphonates, which readily bind exposed hydroxyapatite and then inhibit bone resorption by promoting OC death. Free bisphosphonate can be rapidly cleared by the kidneys, but skeletally bound bisphosphonate persists indefinitely, effectively rendering treatment irreversible. Recently, bisphosphonate treatment has been associated with atypical bone fractures presumably due to lack of normal bone remodeling and repair (40), resulting in the accumulation of defective bone structures.
Our analysis of OC development in Shp2 mutant mice suggests a novel reversible approach for modulating osteoclastogenesis and OC-mediated bone resorptive dynamics. Shp2 enzymatic activity can be reversely inhibited by NSC-87877 (29), halting OC maturation, and that withdrawal of NSC-87877 restores the formation of giant multinucleated OCs. Although this provides strong additional evidence supporting Shp2’s key regulatory role in osteoclastogenesis (36), it also suggests that modulation of Shp2 activity, alone or in combination with other chemicals, could provide an attractive alternate approach for treating skeletal fragility. It may also be possible to develop novel alternative therapeutic options for the treatment of osteoporosis and osteolytic diseases (41, 42) by manipulating cellular signaling along the RANK/SHP2/NFATc1 axis. Identifying additional targets of Shp2 in OCs would be informative for this potential therapeutic application. However, the fact that Shp2 is widely expressed and influences various biologic functions will require additional examination of the risk/benefit ratio of Shp2 alone or in combination with other drugs to inhibit OC differentiation and serve as a treatment option for the management of osteoporosis and/or osteolytic diseases.
Supplementary Material
Acknowledgments
The authors thank Dr. Nhiem Tran for critical reading, Ms. Xiaohong Wang for histological section and staining, and Dr. T. Taniguchi for pMX-IRES-GFP and pMX-Nfatc1/IRES-GFP constructs. The authors are grateful to Mr. Howard Yang (Moses Brown High School) for assistance in performing experiments and data collection. This work was supported in part by the U.S. National Institutes of Health (NIH) National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant R21AR57156 (to W.Y.) and NIH National Institute of General Medical Sciences Grant P20 GM103468. This study was also aided by a grant from the Pediatric Orthopaedic Society of North America and the Orthopaedic Research and Education Foundation (to W.Y.), and the U.S. Department of Agriculture Research Service program Grant #5450-51000-046-00D (to J.C.).
Glossary
- BMD
bone mineral density
- BMM
bone marrow–derived macrophages
- BV
bone volume
- Ctsk
cathepsin K
- DC-STAMP
dendritic cell–specific transmembrane protein
- MMP
matrix metallopeptidase 9
- Nfatc1
nuclear factor of activated T cells, cytoplasmic 1
- OC
osteoclast
- pNPP
para-nitrophenyl phosphate
- Ptpn11
the gene encodes Shp2
- qRT-PCR
quantitative RT-PCR
- RANKL
receptor activator of nuclear factor-κB ligand
- Shp2
the Src homology-2 domain containing protein tyrosine phosphatase 2
- Tb.N
trabeculae number
- Tb.Sp
trabecular spacing
- Tb.Th
trabecular thickness
- TRAP
tartrate-resistant acid phosphatase
- TV
total volume
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Teitelbaum S. L. (2000) Bone resorption by osteoclasts. Science 289, 1504–1508 [DOI] [PubMed] [Google Scholar]
- 2.Boyle W. J., Simonet W. S., Lacey D. L. (2003) Osteoclast differentiation and activation. Nature 423, 337–342 [DOI] [PubMed] [Google Scholar]
- 3.Wada T., Nakashima T., Hiroshi N., Penninger J. M. (2006) RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 12, 17–25 [DOI] [PubMed] [Google Scholar]
- 4.Teitelbaum S. L., Ross F. P. (2003) Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 4, 638–649 [DOI] [PubMed] [Google Scholar]
- 5.Asagiri M., Takayanagi H. (2007) The molecular understanding of osteoclast differentiation. Bone 40, 251–264 [DOI] [PubMed] [Google Scholar]
- 6.Ross F. P. (2006) M-CSF, c-Fms, and signaling in osteoclasts and their precursors. Ann. N. Y. Acad. Sci. 1068, 110–116 [DOI] [PubMed] [Google Scholar]
- 7.Takayanagi H., Kim S., Koga T., Nishina H., Isshiki M., Yoshida H., Saiura A., Isobe M., Yokochi T., Inoue J., Wagner E. F., Mak T. W., Kodama T., Taniguchi T. (2002) Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 3, 889–901 [DOI] [PubMed] [Google Scholar]
- 8.Yu M., Moreno J. L., Stains J. P., Keegan A. D. (2009) Complex regulation of tartrate-resistant acid phosphatase (TRAP) expression by interleukin 4 (IL-4): IL-4 indirectly suppresses receptor activator of NF-kappaB ligand (RANKL)-mediated TRAP expression but modestly induces its expression directly. J. Biol. Chem. 284, 32968–32979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balkan W., Martinez A. F., Fernandez I., Rodriguez M. A., Pang M., Troen B. R. (2009) Identification of NFAT binding sites that mediate stimulation of cathepsin K promoter activity by RANK ligand. Gene 446, 90–98 [DOI] [PubMed] [Google Scholar]
- 10.Sundaram K., Nishimura R., Senn J., Youssef R. F., London S. D., Reddy S. V. (2007) RANK ligand signaling modulates the matrix metalloproteinase-9 gene expression during osteoclast differentiation. Exp. Cell Res. 313, 168–178 [DOI] [PubMed] [Google Scholar]
- 11.Kim K., Lee S.-H., Ha Kim J., Choi Y., Kim N. (2008) NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the dendritic cell-specific transmembrane protein (DC-STAMP). Mol. Endocrinol. 22, 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yagi M., Miyamoto T., Sawatani Y., Iwamoto K., Hosogane N., Fujita N., Morita K., Ninomiya K., Suzuki T., Miyamoto K., Oike Y., Takeya M., Toyama Y., Suda T. (2005) DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 202, 345–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neel, B. G., Chan, G., Dhanji, S. (2009) SH2 Domain-Containing Protein-Tyrosine Phosphatases. In Handbook of Cell Signaling (Dennis, S. A., ed.), pp. 771–809, Elsevier, Inc., New York [Google Scholar]
- 14.Grossmann K. S., Rosário M., Birchmeier C., Birchmeier W. (2010) The tyrosine phosphatase Shp2 in development and cancer. Adv. Cancer Res. 106, 53–89 [DOI] [PubMed] [Google Scholar]
- 15.Lee A. W., States D. J. (2006) Colony-stimulating factor-1 requires PI3-kinase-mediated metabolism for proliferation and survival in myeloid cells. Cell Death Differ. 13, 1900–1914 [DOI] [PubMed] [Google Scholar]
- 16.Wada T., Nakashima T., Oliveira-dos-Santos A. J., Gasser J., Hara H., Schett G., Penninger J. M. (2005) The molecular scaffold Gab2 is a crucial component of RANK signaling and osteoclastogenesis. Nat. Med. 11, 394–399 [DOI] [PubMed] [Google Scholar]
- 17.Araki T., Mohi M. G., Ismat F. A., Bronson R. T., Williams I. R., Kutok J. L., Yang W., Pao L. I., Gilliland D. G., Epstein J. A., Neel B. G. (2004) Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat. Med. 10, 849–857 [DOI] [PubMed] [Google Scholar]
- 18.Fornaro M., Burch P. M., Yang W., Zhang L., Hamilton C. E., Kim J. H., Neel B. G., Bennett A. M. (2006) SHP-2 activates signaling of the nuclear factor of activated T cells to promote skeletal muscle growth. J. Cell Biol. 175, 87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang W., Klaman L. D., Chen B., Araki T., Harada H., Thomas S. M., George E. L., Neel B. G. (2006) An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev. Cell 10, 317–327 [DOI] [PubMed] [Google Scholar]
- 20.Saxton T. M., Henkemeyer M., Gasca S., Shen R., Rossi D. J., Shalaby F., Feng G. S., Pawson T. (1997) Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J. 16, 2352–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauler T. J., Kamiya N., Lapinski P. E., Langewisch E., Mishina Y., Wilkinson J. E., Feng G.-S., King P. D. (2011) Development of severe skeletal defects in induced SHP-2-deficient adult mice: a model of skeletal malformation in humans with SHP-2 mutations. Dis. Model. Mech. 4, 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan G., Cheung L. S., Yang W., Milyavsky M., Sanders A. D., Gu S., Hong W. X., Liu A. X., Wang X., Barbara M., Sharma T., Gavin J., Kutok J. L., Iscove N. N., Shannon K. M., Dick J. E., Neel B. G., Braun B. S. (2011) Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood 117, 4253–4261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tartaglia M., Mehler E. L., Goldberg R., Zampino G., Brunner H. G., Kremer H., van der Burgt I., Crosby A. H., Ion A., Jeffery S., Kalidas K., Patton M. A., Kucherlapati R. S., Gelb B. D. (2001) Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 29, 465–468 [DOI] [PubMed] [Google Scholar]
- 24.Noonan J. A. (2006) Noonan syndrome and related disorders: alterations in growth and puberty. Rev. Endocr. Metab. Disord. 7, 251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan G., Kalaitzidis D., Usenko T., Kutok J. L., Yang W., Mohi M. G., Neel B. G. (2009) Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood 113, 4414–4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang W., Wang J., Moore D. C., Liang H., Dooner M., Wu Q., Terek R., Chen Q., Ehrlich M. G., Quesenberry P. J., Neel B. G. (2013) Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 499, 491–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clausen B. E., Burkhardt C., Reith W., Renkawitz R., Förster I. (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 [DOI] [PubMed] [Google Scholar]
- 28.Nakamura T., Imai Y., Matsumoto T., Sato S., Takeuchi K., Igarashi K., Harada Y., Azuma Y., Krust A., Yamamoto Y., Nishina H., Takeda S., Takayanagi H., Metzger D., Kanno J., Takaoka K., Martin T. J., Chambon P., Kato S. (2007) Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell 130, 811–823 [DOI] [PubMed] [Google Scholar]
- 29.Chen L., Sung S. S., Yip M. L., Lawrence H. R., Ren Y., Guida W. C., Sebti S. M., Lawrence N. J., Wu J. (2006) Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol. Pharmacol. 70, 562–570 [DOI] [PubMed] [Google Scholar]
- 30.Ozcivici E., Ferreri S., Qin Y. X., Judex S. (2008) Determination of bone’s mechanical matrix properties by nanoindentation. Methods Mol. Biol. 455, 323–334 [DOI] [PubMed] [Google Scholar]
- 31.Tanaka S., Takahashi N., Udagawa N., Tamura T., Akatsu T., Stanley E. R., Kurokawa T., Suda T. (1993) Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J. Clin. Invest. 91, 257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu D., Shi Z., McDonald J., Pan G., Cao X., Yu X., Feng X. (2004) Development of a chimaeric receptor approach to study signalling by tumour necrosis factor receptor family members. Biochem. J. 383, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tolar J., Teitelbaum S. L., Orchard P. J. (2004) Osteopetrosis. N. Engl. J. Med. 351, 2839–2849 [DOI] [PubMed] [Google Scholar]
- 34.Matsuo K., Galson D. L., Zhao C., Peng L., Laplace C., Wang K. Z. Q., Bachler M. A., Amano H., Aburatani H., Ishikawa H., Wagner E. F. (2004) Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J. Biol. Chem. 279, 26475–26480 [DOI] [PubMed] [Google Scholar]
- 35.Umeda S., Beamer W. G., Takagi K., Naito M., Hayashi S., Yonemitsu H., Yi T., Shultz L. D. (1999) Deficiency of SHP-1 protein-tyrosine phosphatase activity results in heightened osteoclast function and decreased bone density. Am. J. Pathol. 155, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoki K., Didomenico E., Sims N. A., Mukhopadhyay K., Neff L., Houghton A., Amling M., Levy J. B., Horne W. C., Baron R. (1999) The tyrosine phosphatase SHP-1 is a negative regulator of osteoclastogenesis and osteoclast resorbing activity: increased resorption and osteopenia in me(v)/me(v) mutant mice. Bone 25, 261–267 [DOI] [PubMed] [Google Scholar]
- 37.Lee A. W., States D. J. (2000) Both src-dependent and -independent mechanisms mediate phosphatidylinositol 3-kinase regulation of colony-stimulating factor 1-activated mitogen-activated protein kinases in myeloid progenitors. Mol. Cell. Biol. 20, 6779–6798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang H., Chang E.-J., Ryu J., Lee Z. H., Lee Y., Kim H.-H. (2006) Induction of c-Fos and NFATc1 during RANKL-stimulated osteoclast differentiation is mediated by the p38 signaling pathway. Biochem. Biophys. Res. Commun. 351, 99–105 [DOI] [PubMed] [Google Scholar]
- 39.Kuroda Y., Hisatsune C., Nakamura T., Matsuo K., Mikoshiba K. (2008) Osteoblasts induce Ca2+ oscillation-independent NFATc1 activation during osteoclastogenesis. Proc. Natl. Acad. Sci. USA 105, 8643–8648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shane E., Burr D., Ebeling P. R., Abrahamsen B., Adler R. A., Brown T. D., Cheung A. M., Cosman F., Curtis J. R., Dell R., Dempster D., Einhorn T. A., Genant H. K., Geusens P., Klaushofer K., Koval K., Lane J. M., McKiernan F., McKinney R., Ng A., Nieves J., O'Keefe R., Papapoulos S., Sen H. T., van der Meulen M. C. H., Weinstein R. S., Whyte M. (2010) Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J. Bone Miner. Res. 25, 2267–2294 [DOI] [PubMed] [Google Scholar]
- 41.Watts N. B., Diab D. L. (2010) Long-term use of bisphosphonates in osteoporosis. J. Clin. Endocrinol. Metab. 95, 1555–1565 [DOI] [PubMed] [Google Scholar]
- 42.Sweet M. G., Sweet J. M., Jeremiah M. P., Galazka S. S. (2009) Diagnosis and treatment of osteoporosis. Am. Fam. Physician 79, 193–200 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.