

Abstract
An interactive web server is developed for predicting the potential binding sites and its target operons for a given regulatory protein in prokaryotic genomes. The program allows users to submit known or experimentally determined binding sites of a regulatory protein as ungapped multiple sequence alignments. It analyses the upstream regions of all genes in a user-selected prokaryote genome and returns the potential binding sites along with the downstream co-regulated genes (operons). The known binding sites of a regulatory protein can also be used to identify its orthologue binding sites in phylogeneticaly related genomes where the trans-acting regulator protein and cognate cis-acting DNA sequences could be conserved. PredictRegulon can be freely accessed from a link on our world wide web server: http://www.cdfd.org.in/predictregulon/.
INTRODUCTION
With over 100 bacterial genomes sequenced, a key challenge of post-genomic research is to dissect the complex transcription regulatory network which controls the metabolic and physiological process of a cell. A first step towards this goal is to identify the genes within a genome that are controlled by a specific transcription regulatory protein. This paper describes a web server tool—PredictRegulon—for genome-wide prediction of potential binding sites and target operons of a regulatory protein for which few experimentally identified binding sites are known. This technique could utilize the available experimental data on binding sites of transcription regulatory proteins from various bacterial species (1–3) for identification of regulons in phylogenetically related species.
PREDICTREGULON METHOD
The program, PredictRegulon, first constructs the binding site recognition profile based on ungapped multiple sequence alignment of known binding sites. This profile is calculated using Shannon's positional relative entropy approach (4). The positional relative entropy Qi at position i in a binding site is defined as
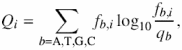
where b refers to each of the possible bases (A, T, G, C), fb,i is observed frequency of each base at position i and qb is the frequency of base b in the genome sequence. The contribution of each base to the positional Shannon relative entropy is calculated by multiplying each base frequency by positional relative entropy as follows:
![]()
where Wb,i refers to the weighted Shannon relative entropy of the base b (A, T, G, C) at position i. Finally, a 4 × L entropy matrix (L is the length of the binding site) is constructed representing the binding site recognition profile, where each matrix element is the weighted positional Shannon relative entropy of a base.
The profile, encoded as the matrix, is used to scan the upstream sequences of all the genes of the user-selected genome. The entropy score of each site is calculated as the sum of the respective positional nucleotide entropy (Wb,i). A maximally scoring site is selected from the upstream sequence of each gene. The score may represent the strength of interaction between regulatory protein and binding site (5). The lowest score among the input sites is considered as the cut-off score. The sites scoring higher than the the cut-off value are reported as potential binding sites conforming to the consensus profile.
Co-directionally transcribed genes downstream of the predicted binding site were selected as potential co-regulated genes (operons) according to one of the following criteria: (i) co-directionally transcribed orthologous gene pairs conserved in at least three genomes (6); (ii) genes belong to the same cluster of orthologous gene function category and the intergenic distance is <200 bp (7); (iii) the first three letters in gene names are identical (the gene names for all the bacterial species were assigned using the COG annotation); (iv) intergenic distance is <90 bp (8).
This method has two specific requirements: a few experimentally determined regulatory protein binding sites should be available for developing the binding site recognition profile, and the profile should be applicable to the genome where the regulator or its homologue is present. In the absence of any experimental information on the regulatory sites in a given genome one may look up the known regulatory motifs from other related species from one of the four online databases which host the information about known transcription regulatory protein binding sites in prokaryote genomes (1–3).
A limitation of this approach is that it may predict a few false positive sites as candidates. However, this limitation can be overcome by experimental validations, by either in vitro binding studies with double strand oligonucleotides containing the binding sites (designed based on prediction) and regulatory proteins or real-time PCR analysis of candidate co-regulated genes.
EXAMPLE: PREDICTION OF LEXA REGULON IN MYCOBACTERIUM TUBERCULOSIS
To demonstrate a typical usage of PredictRegulon, we predicted the LexA binding sites and LexA regulon of M.tuberculosis using the LexA binding sites of Bacillus subtilis. LexA regulators from B.subtilis and M.tuberculosis share a high sequence identity (45%) at protein level (data not shown). Table 1 lists the known LexA binding sites from B. subtilis given as input to the program (2) and Table 2 shows the output of predicted LexA binding sites in M.tuberculosis. The site column in Table 2 represents the predicted binding sites of LexA in M.tuberculosis. In a typical output the perfect match to the known binding sites and the downstream genes are highlighted with a yellow background, and the rest with score greater than cut-off is shown with a blue background (colours not shown in the table). Eighteen of these genes (indicated by ‘a’) belonging to the LexA regulon were also observed in data obtained by experimental means by others (9–12). The rest of the matches are potential novel regulatory sites which could be confirmed experimentaly.
Table 1. Known LexA binding sites of Bacillus subtilis from the PRODORIC database.
| Binding site | Gene |
|---|---|
| AGAACAAGTGTTCG | dinC |
| AGAACTCATGTTCG | dinB |
| CGAACTTTAGTTCG | dinA |
| CGAATATGCGTTCG | recA |
| CGAACGTATGTTTG | dinC |
| CGAACCTATGTTTG | dinR |
| CGAACAAACGTTTC | dinR |
| GGAATGTTTGTTCG | dinR |
Table 2. Output of PredictRegulon web server (predicted LexA binding sites).
| Score | Position | Site | Gene | Synonym | COG | Product |
|---|---|---|---|---|---|---|
| 5.37 | −8 | CGAACGTATGTTCG | — | Rv3776a | — | Hypothetical protein Rv3776 |
| 5.32 | −100 | CGAACATGTGTTCG | — | Rv3073ca | COG3189 | Uncharacterized conserved protein |
| 5.32 | −144 | CGAACATGTGTTCG | pyrR | Rv1379a | COG2065 | Pyrimidine operon attenuation protein |
| 5.22 | −8 | CGAACACATGTTCG | — | Rv3074a | — | Hypothetical protein Rv3074 |
| 5.2 | −142 | CGAACAATTGTTCG | — | Rv3371a | — | Hypothetical protein Rv3371 |
| 5.2 | −64 | CGAACAATTGTTCG | dnaE2 | Rv3370ca | COG0587 | DNA polymerase III |
| 5.19 | −36 | CGAACGATTGTTCG | ruvC | Rv2594ca | COG0817 | ruvC |
| 5.14 | −32 | CGAAAGTATGTTCG | — | Rv0336a | — | Hypothetical protein Rv0336 |
| 5.14 | −32 | CGAAAGTATGTTCG | — | Rv0515a | — | Hypothetical protein Rv0515 |
| 5.14 | −105 | CGAACACATGTTTG | lexA | Rv2720a | COG1974 | SOS-response transcriptional repressors |
| 5.11 | −122 | CGAACAGGTGTTCG | recA | Rv2737ca | COG1372 | recA |
| 5.08 | −87 | CGAACAATCGTTCG | — | Rv2595a | COG2002 | Hypothetical protein Rv2595 |
| 5.06 | −44 | CGAATATGCGTTCG | dnaB | Rv0058a | COG0305 | Replicative DNA helicase |
| 5.04 | −263 | GGAACTTGTGTTGG | ubiE | Rv3832c | COG2226 | Methylase involved in ubiquinone biosynthesis |
| 5.04 | −23 | AGAACGGTTGTTCG | splB | Rv2578ca | COG1533 | DNA repair photolyase |
| 5.02 | −6 | CGAATATGAGTTCG | — | Rv0071a | COG3344 | Retron-type reverse transcriptase |
| 5.01 | −255 | CGAACAAGTGTTGG | — | Rv1414 | COG3616 | Predicted amino acid aldolase or racemase |
| 4.99 | −181 | GGAACGCGTGTTTG | — | Rv0750 | — | Hypothetical protein Rv0750 |
| 4.98 | −105 | CGAACAACAGTTCG | baeS | Rv0600c | COG0642 | Signal transduction histidine kinase |
| 4.98 | −186 | CGAAGATGCGTTCG | rpsT | Rv2412 | COG0268 | Ribosomal protein S20 |
| 4.95 | −242 | TGAACGCAAGTTCG | fbpB | Rv1886c | COG0627 | fbpB |
| 4.95 | −192 | CGAACGGGAGTTCG | — | Rv1455 | — | Hypothetical protein Rv1455 |
| 4.94 | −270 | AGAACCACCGTTCG | phd | Rv3181c | COG4118 | Antitoxin of toxin–antitoxin stability system |
| 4.94 | −213 | CGAACGACGGTTCG | pe | Rv2099ca | — | PE |
| 4.92 | −118 | CGAACAGGTGTTGG | — | Rv0004 | COG5512 | Zn-ribbon-containing |
| 4.92 | −163 | CGAACTTGCGTTCA | — | Rv1887 | — | Hypothetical protein Rv1887 |
| 4.91 | −239 | GGAACGCGAGTTCG | fadB2 | Rv0468 | COG1250 | 3-hydroxyacyl-CoA dehydrogenase |
| 4.91 | −7 | TGAACGAATGTTCC | — | Rv0039c | — | Hypothetical protein Rv0039c |
| 4.9 | −237 | CGAAGCCTTGTTCG | dltE | Rv3174 | COG0300 | Short-chain dehydrogenase |
| 4.89 | −225 | GGAAGGTGCGTTCG | frnE | Rv2466c | COG2761 | Predicted dithiol-disulfide isomerase |
| 4.88 | −8 | GGAAGCCATGTTCG | — | Rv0769 | COG1028 | Hypothetical protein Rv0769 |
| 4.88 | −186 | CGAAGAGGTGTTCG | coxS | Rv0374c | COG2080 | Aerobic-type carbon monoxide dehydrogenase |
| 4.88 | −186 | CGAACCGCAGTTCG | leuA | Rv3534c | COG0119 | Isopropyl malate/citramalate synthases |
| 4.85 | −195 | CGAACGGCTGTTGG | — | Rv2061c | COG3576 | Hypothetical protein Rv2061c |
| 4.85 | −85 | AGAACGGTTGTTGG | accA1 | Rv2501c | COG4770 | COG4770 |
| 4.84 | −151 | CGAAATTGTGTTCC | nuoB | Rv3146 | COG0377 | NADH:ubiquinone oxidoreductase |
| 4.84 | −217 | CAAACATGTGTTCG | — | Rv2719ca | — | Hypothetical protein Rv2719c |
| 4.84 | −5 | CGAACATGTATTCG | — | Rv1702ca | — | Hypothetical protein Rv1702c |
| 4.84 | −199 | CGAAATCTTGTTTG | — | Rv1375 | COG1944 | Hypothetical protein Rv1375 |
Score: score of the binding sites, Position: position of the binding site relative to the translation start site, Site: binding site of a regulatory protein, Gene: gene downstream to the binding site, Synonym: synonym of the gene, COG: Cluster of Orthologous Gene code, Product: Gene product. arepresents the ORFs known to be regulated by the regulator. ‘a’ symbols are not part of the orginal output of the web server. Source of Genome: NCBI ftp site (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/Mycobacterium_tuberculosis_H37Rv/), Accession no. NC_000962.
The web output of PredictRegulon also contains the hyperlinked gene-synonym and COG number. A click on the former shows the predicted operon context of the regulatory motif while a click on the latter opens a new page showing a description of this gene in the NCBI Conserved Domain Database, which is in turn linked to Pubmed for published information on this gene. These additional links provides users a simple way to browse and understand the functional/physiological implication of the genes that are part of predicted regulon.
Acknowledgments
ACKNOWLEDGEMENTS
This work is partially supported by the Council of Scientific and Industrial Research (CSIR) NMITLI Grant to A.R. Y.S. and S.K. were recipients of Senior Research Fellowships from CSIR, Govt. of India.
REFERENCES
- 1.Salgado H., Santos-Zavaleta,A., Gama-Castro,S., Millan-Zarate,D., Diaz-Peredo,E., Sanchez-Solano,F., Perez-Rueda,E., Bonavides-Martinez,C. and Collado-Vides,J. (2001) RegulonDB (Version 3.2): transcriptional regulation and operon organization in Escherichia coli K-12. Nucleic Acids Res., 29, 72–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Munch R., Hiller,K., Barg,H., Heldt,D., Linz,S., Wingender,E. and Jahn,D. (2003) PRODORIC: prokaryotic database of gene regulation. Nucleic Acids Res., 31, 266–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishii T., Yoshida,K., Terai,G., Fujita,Y. and Nakai,K. (2001) DBTBS: a database of Bacillus subtilis promoters and transcription factors. Nucleic Acids Res., 29, 278–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shannon C.E. (1948) A mathematical theory of communication. Bell Sys. Tech. J., 379–423 and 623–656. [Google Scholar]
- 5.Benos P.V., Bulyk,M.L. and Stormo,G.D. (2002) Additivity in protein-DNA interactions: how good an approximation is it? Nucleic Acids Res., 30, 4442–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ermolaeva M.D., White,O. and Salzberg,S.L. (2001) Prediction of operons in microbial genomes. Nucleic Acids Res., 295, 1216–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salgado H., Moreno-Hagelsieb,G., Smith,T.F. and Collado-Vides,J. (2000) Operons in Escherichia coli: genomic analyses and predictions. Proc. Natl Acad. Sci., USA, 97, 6652–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strong M., Mallick P., Pellegrini,M., Thompson,M.J. and Eisenberg,D. (2003) Inference of protein function and protein linkages in Mycobacterium tuberculosis based on prokaryotic genome organization: a combined computational approach. Genome Biol., 4, R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durbach S.I., Andersen,S.J. and Mizrahi,V. (1997) SOS induction in mycobacteria: analysis of the DNA-binding activity of a LexA-like repressor and its role in DNA damage induction of the recA gene from Mycobacterium smegmatis. Mol. Microbiol., 26, 643–653. [DOI] [PubMed] [Google Scholar]
- 10.Brooks P.C., Movahedzadeh,F. and Davis,E.O. (2001) Identification of some DNA damage-inducible genes of Mycobacterium tuberculosis: apparent lack of correlation with LexA binding. J. Bacteriol., 183, 4459–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dullaghan E.M., Brooks,P.C. and Davis,E.O. (2002) The role of multiple SOS boxes upstream of the Mycobacterium tuberculosis lexA gene—identification of a novel DNA-damage-inducible gene. Microbiology, 148, 3609–3615. [DOI] [PubMed] [Google Scholar]
- 12.Boshoff H.I., Reed,M.B., Barry,C.E. and Mizrahi,V. (2003) DNAE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell, 113, 183–193. [DOI] [PubMed] [Google Scholar]