

Abstract
Several lines of evidence support immune response in brain as a mechanism of injury in Alzheimer disease (AD). Moreover, immune activation is heightened in apolipoprotein E (APOE) ε4 carriers; inhibitors of prostaglandin (PG) synthesis show a partially protective effect on AD risk from APOE ε4; and genetic variants in triggering receptor expressed on myeloid cells 2 (TREM2) are a rare but potent risk for AD. We tested the hypothesis that APOE ε4 inheritance modulates both the PGE2 pathway and TREM2 expression using primary murine microglia from targeted replacement (TR) APOE3/3 and APOE4/4 mice. Microglial cyclooxygenase-2, microsomal PGE synthase, and PGE2 expression were increased 2- to 25-fold in both genotypes by TLR activators; however, this induction was significantly (P < 0.01) greater in TR APOE4/4 microglia with TLR3 and TLR4 activators. Microglial TREM2 expression was reduced approximately 85% by all TLR activators; this reduction was approximately one-third greater in microglia from TR APOE4/4 mice. Importantly, both receptor-associated protein and a nuclear factor κ-light-chain-enhancer inhibitor blocked TR APOE4/4–dependent effects on the PGE2 pathway but not on TREM2 expression. These data demonstrate complementary, but mechanistically distinct, regulation of pro- and anti-inflammatory mediators in TR APOE4/4 murine microglia that yields a more proinflammatory state than with TR APOE3/3.—Li, X., Montine, K. S., Keene, C. D., Montine, T. J. Different mechanisms of apolipoprotein E isoform–dependent modulation of prostaglandin E2 production and triggering receptor expressed on myeloid cells 2 (TREM2) expression after innate immune activation of microglia.
Keywords: apolipoprotein E genotype, PGE2, Alzheimer disease, neuroinflammation, brain immunity
Multiple lines of evidence now support innate immune response as a mediator of neuron stress and damage in affected regions of brain in Alzheimer disease (AD). Current genomewide association studies and the search for rare variants that increase risk for AD have linked several genes categorized as immune response genes (1, 2). Perhaps the most compelling of these is the gene for triggering receptor expressed on myeloid cells 2 (TREM2) (3, 4). TREM2 encodes a transmembrane glycoprotein expressed in myeloid lineage cells, such as macrophages, dendritic cells, osteoclasts, and, in brain, microglia. The functions of TREM2 include regulation of phagocytosis and suppression of the innate immune response mediated through TLR activation (5). TREM2 loss-of-function mutations result in Nasu-Hakola disease, an autosomal-recessive form of early-onset dementia presenting with bone cysts (6–8). Reduced TREM2 expression is proinflammatory in macrophages or dendritic cells after TLR activation (9–11), and silencing of TREM2 mRNA in mouse brain enhances neuroinflammation and cognitive impairment (12).
Although TREM2 already has a clearly defined role in immune regulation both in the CNS and the periphery, other genetic variants linked with increased risk for AD also have significant immune regulatory roles, even those that do not have immune regulation as a canonical function; a prime example is isoforms of apolipoprotein E (apoE) (13). ApoE, first discovered because of its major role in cholesterol and lipid transport, is now known to be pleiotropic, including modulation of peripheral immune and inflammatory responses, among several others (14). The biologic functions of apoE in humans are further complicated by 3 common isoforms (apoE2, apoE3, and apoE4), which are encoded by 3 different alleles: ε2, ε3, and ε4. Isoform-specific differences in apoE functions are many and influence virtually all aspects of apoE biologic activity in humans. Because apoE is pleiotropic, and because apoE3 and apoE4 isoforms differ in many activities, it is entirely possible that there are multiple molecular mechanisms by which inheritance of APOE ε4 might increase the risk of AD relative to APOE ε3. Indeed, experimental evidence supports apoE4-specific effects on neurodevelopment, age-related cognitive decline, metabolic alterations in brain, trafficking of Aβ peptides, and regulation of innate immune response and its downstream damage in brain (15–20).
Despite numerous studies demonstrating a proinflammatory state with APOE ε4 (21–25), the mechanism or mechanisms by which apoE4 regulates innate immune response in brain are not clear. A clue might come from the epidemiologic literature that has consistently associated protracted use of nonsteroidal anti-inflammatory drug (NSAIDs) with reduced risk of subsequently developing AD dementia (26, 27). One of the largest studies to make this association, the Cardiovascular Health Study, also observed that the protective effect of NSAIDs resided overwhelmingly in individuals with APOE ε4 (28). Others have also have observed an apparent interaction of NSAIDs with APOE ε4 in a clinical trial to suppress cognitive decline in AD (29, 30). However, we are unaware of any mechanistic data demonstrating apoE isoform–specific regulation of the prostaglandin (PG) pathway. Finally, if products of TREM2 and APOE ε4 both act, at least in part, to increase risk for AD by regulating the immune response in brain, then it is unknown whether they work through shared or distinct pathways. Here we tested the hypothesis that inheritance of APOE ε4 modulates both the PGE2 pathway and TREM2 expression using a primary microglia culture system from targeted replacement (TR) APOE3/3 or APOE4/4 mice.
MATERIALS AND METHODS
Reagents
DMEM/F12 medium and fetal bovine serum were purchased from Hyclone Laboratories (Logan, UT, USA). Papain and DNase I were purchased from Worthington Biochemical (Lakewood, NJ, USA). Polyinosinic–polycytidylic acid (PIC, TLR3 ligand) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Pam3CSK4 (Pam3, TLR2 ligand), Loxoribine (TLR7 ligand), and CpG (TLR9 ligand) were purchased from InvivoGen (San Diego, CA, USA). LPS (TLR4 ligand) was purchased from Calbiochem (La Jolla, CA, USA). Receptor-associated protein (RAP) was purchased from Innovative Research Inc. (Novi, MI, USA), and nuclear factor κ-light-chain-enhancer (NF-κB) inhibitor BAY 117085 from Enzo Life Science (Farmingdale, NY, USA).
Animals
Wild-type (WT) C57BL/6 mice were purchased from The Jackson Laboratories (Bar Harbor, ME, USA). Homozygous APOE3/3 and APOE4/4 TR mice on a C57Bl/6 background were a gift from Dr. Nobuyo Maeda (31–33) and previously used by us (24, 25, 34–37). The animals were maintained in a specific pathogen-free environment. All procedures were approved by the University of Washington Institutional Animal Care and Use Committee.
Cell culture
Primary mixed glial cultures were generated from 1- to 3-day-old pups of WT or TR APO3/3 or TR APOE4/4 mice as described previously (38, 39). In brief, cerebral cortex was dissected from brains and remaining meninges were removed while in ice-cold Dulbecco PBS. The cortical tissues were digested by incubation in solution (DMEM/F12 medium, 15U /ml papain, 0.5 mM EDTA, 0.2 mg/ml l-cysteine, and 200 µg/ml DNase I) for 30 minutes at 37°C and dissociated mechanically. The resulting cell suspension was plated on poly-l-ornithine coated flasks and maintained at 37°C and 5% CO2 in DMEM/F12 supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. At 11 to 15 days, microglia were collected from the underlying astrocytic monolayer by gentle agitation and replated on poly-l-ornithine-coated plates for experimental treatments. After removing microglia, the underlying astrocytes were subcultured in plates coated with poly-l-ornithine. Cells reaching confluence at 3 days after plating were rinsed twice with 2.5 volumes of serum-free medium before treatment and then incubated with the indicated reagents in serum-free medium plus G5 glial supplement. Primary neuron cultures were generated from cerebral cortex of 1-day-old pups as described previously (40).
Quantitative real-time PCR
Total RNA was extracted from primary microglia using RNeasy extraction kit (Qiagen, Valencia, CA, USA). Reverse transcription was performed with Omniscript RT kit (Qiagen). TaqMan probes and primers of cyclooxygenase-2 (COX2), microsomal PGE synthase (mPGES), and TREM2 were purchased from Applied Biosystems (Carlsbad, CA, USA). PCR was performed on an Applied Biosystems ViiA 7 Real-Time PCR System using the standard curve method of relative quantitation (41) and normalization to glyceraldehyde phosphate dehydrogenase (GAPDH), as performed previously by us (38, 39).
Flow cytometric analysis of cell surface TREM2
After treatment with TLR3 activator PIC for 24 hours, microglia were preincubated with anti-mouse CD16/CD32 antibody (BD Biosciences) for 5 minutes on ice to block Fc receptor–mediated binding. The cells were then incubated with FITC-conjugated TREM2 or rat IgG control for 30 minutes on ice in the dark. Thereafter, microglia were washed twice with PBS and resuspended in 4% paraformaldehyde for fixation. Fluorescence intensity was measured using a FACScan flow cytometer. Flow data were analyzed using FlowJo software (TreeStar, Ashland, OR, USA).
PGE2 measurement
Microglia from TR APOE3/3 or TR APOE4/4 mice were plated at in 96-well plates. After 24 hours incubation, microglia were treated with 20 μg/ml TLR3 activator PIC in the presence or absence of 1 μM RAP for various times. The conditioned medium was collected after treatments, and PGE2 concentration was determined using a PGE2 ELISA kit (Cayman Chemical Company, Ann Arbor, MI, USA) according to the manufacturer’s protocol.
NF-κB ELISA
The activity of transcription factor NF-κB was assessed by phospho-p65 subunit ELISA. In brief, microglia from TR APOE3/3 or TR APOE4/4 mice were seeded in 6-well plates. One day later, the cells were treated with 20 μg/ml PIC in the presence or absence of 1 μM RAP for 1 hour. After removing the medium, the cells were rinsed with ice-cold PBS, and lysis buffer was added to each well. The plate was incubated on ice for 5 minutes. The lysates were collected and sonicated and then centrifuged for 10 minutes at 4°C. The supernatants were collected for phosphor-NF-κB p65 ELISA assay following the manufacturer’s protocol (Cell Signaling, Danvers, MA, USA).
Statistical analyses
Statistical analyses were performed using GraphPad Prism software with α set to 0.05. All P values presented in the text or figures have been corrected for multiple comparisons when appropriate by the Bonferroni method. Values of P < 0.05 were considered significant; levels of significance are indicated in the figures.
RESULTS
We determined whether specific TLR activators differentially induce expression of COX2 (Fig. 1A) or mPGES (Fig. 1B) in primary microglia from TR APOE3 or TR APOE4 mice. As expected, all TLR activators increased expression of COX2 and mPGES over vehicle-exposed microglia cultures. These experiments also showed that activators of TLR3 (PIC) and TLR4 (LPS), but not of TLR2, TLR7, or TLR9, significantly further induced expression of COX2 and mPGES by approximately 50–60% in TR APOE4/4 microglia compared to TR APOE3/3 microglia.
Figure 1.

COX2 and mPGES mRNA expression induced by TLR activators. Primary microglia cultures from TR APOE3/3 (solid) and TR APOE4/4 (hatched) mice were treated for 6 hours with vehicle (Veh) or with activators of TLR2 (Pam, 1 μg/ml), TLR3 (PIC, 20 μg/ml), TLR4 (LPS, 1 μg/ml), TLR7 (Lox, 1 mM), or TLR9 (CpG, 1 μM). RNA was extracted for quantitative real-time PCR. Levels of COX2 (A) or mPGES (B) mRNA in each sample were normalized to endogenous GAPDH mRNA levels. Data are presented as average relative units (±sem, n = 3 per group). Two-way ANOVA had P < 0.01 for APOE genotype, TLR activator, and interaction between these terms. ***P < 0.001 and **P < 0.01 for Bonferroni-corrected repeated pair comparisons for TR APOE3/3 vs. TR APOE4/4.
Focusing on the more robust PIC effect, we first compared TLR3 expression levels between TR APOE3/3 and TR APOE4/4 microglia under basal and PIC-activated conditions. Basal TLR3 mRNA levels for TR APOE3/3 microglia were set as 100%. Basal TLR3 mRNA levels for TR APOE4/4 microglia were 113 ± 14% (P > 0.05), while PIC exposure increased TLR3 expression in TR APOE3/3 and TR APOE4/4 microglia to a similar extent (823 ± 52% vs. 916 ± 78%, P > 0.05), similar to what has been reported previously by others (42). We next determined the time course and concentration–response of increased expression of COX2 and mPGES by TR APOE3/3 and TR APOE4/4 microglia cultures. COX2 and mPGES mRNA levels increased progressively with increasing concentration of PIC for 12 hours with significant differences between TR APOE3/3 and TR APOE4/4 at each concentration, but most robust at 20 μg/ml (Fig. 2A). We observed that after 6 hours exposure to 20 μg/ml PIC, both COX2 and mPGES mRNA levels were significantly greater in TR APOE4/4 microglia but that this difference was no longer present at 18 hours after exposure (Fig. 2B). As expected, increased medium PGE2 levels lagged behind mRNA expression of COX2 and mPGES and were significantly increased in TR APOE4/4 microglia at 12 hours compared with TR APOE3/3 microglia; this approximately 85% increase persisted to 18 hours. We next tested whether this TR APOE4/4–specific increase in COX2 and mPGES expression could be blocked by RAP, an antagonist of a subset of apoE receptors. We found that exposure to 1 μM RAP ablated the TR APOE4/4–dependent increase in COX2 expression (Fig. 2C) and largely suppressed the TR APOE4/4–dependent increase in mPGES expression (Fig. 2D). Finally, we determined that RAP also significantly suppressed the TR APOE4/4–dependent increase in medium concentration of PGE2 in primary microglia (Fig. 2E), so that the ratio of medium PGE2 from TR APOE4/4 and TR APOE3/3 microglia after 18 hours PIC exposure was not different from unity. These data demonstrate a RAP-sensitive mechanism underlying the TR APOE4/4–dependent increased expression of COX2, mPGES, and medium PGE2 after innate immune activation.
Figure 2.
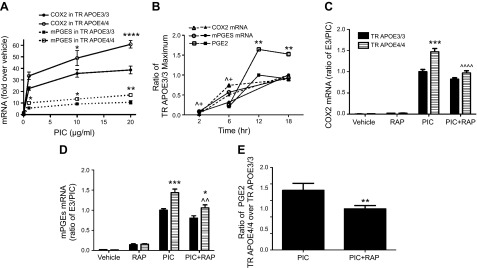
APOE genotype–specific effects on PGE2 pathway. Primary microglia cultures from TR APOE3/3 and TR APOE4/4 mice were exposed to indicated reagents and times and individual components of the PGE2 pathway were quantified. Data are average ± sem. A) Concentration–response relationship for PIC induction of COX2 and mPGES mRNA expression after 12 hours exposure (n = 3 per group). Two-way ANOVA was significant for concentration and time (P < 0.0001 for each) and Bonferroni-corrected posttests comparing TR APOE3/3 (solid symbols) with TR APOE4/4 (open symbols) were significant (*P < 0.05, **P < 0.01, ****P < 0.0001) at the concentrations indicated. B) Time course of PIC induction of COX2 mRNA and mPGES mRNA expression as measured by quantitative real-time PCR, as well as medium PGE2 concentration as measured by ELISA. Data are the ratio of TR APOE4/4 at each time point normalized to the maximum value for TR APOE3/3 cells activated with PIC. Two-way ANOVA for each endpoint followed by Bonferroni-corrected posttests comparing TR APOE3/3 (solid symbols) with TR APOE4/4 (open symbols) had P < 0.05 for ^COX2 mRNA and +mPGES mRNA at 2 and 6 hours, and **P < 0.01 for medium PGE2 concentration at both 12 and 18 hours. C, D) COX2 or mPGEs mRNA levels of primary microglia exposed to vehicle, RAP (1 μM), PIC (20 μg/ml), or PIC plus RAP for 6 hours. Data are the ratio normalized to the value of TR APOE3/3 cells activated with PIC. Two-way ANOVA had P < 0.0001 for TR APOE genotype, exposures, and interaction between these terms for both COX2 and mPGES mRNA. ***P < 0.001 and *P < 0.05 for Bonferroni-corrected posttests comparing TR APOE3/3 with TR APOE4/4. One-way ANOVA followed by Bonferroni-corrected posttests found that RAP significantly reduced both COX2 (C) and mPGES (D) mRNA levels in PIC-treated TR APOE4/4 cells (^^^^P < 0.0001 and ^^P < 0.01, respectively) but not in PIC-treated TR APOE3/3 cells. E) Data are the ratio of medium PGE2 concentration at 18 hours for TR APOE4/4 over TR APOE3/3 exposed to PIC or PIC plus RAP. **P < 0.01 for outcome of test.
To investigate signaling pathways that might underlie this RAP-sensitive effect, we focused on the transcription factor NF-κB, which is crucial to regulating gene expression in response to innate immune activation. As shown in Fig. 3A, basal NF-κB activity, as measured by phosphorylation of its subunit protein p65, increased markedly in both TR APOE3/3 and TR APOE4/4 microglia after PIC exposure. Significantly higher activity levels were observed in TR APOE4/4 microglia compared in TR APOE3/3 microglia; this effect was completely reversed by coincubation with RAP. PIC-stimulated expression of COX2 and mPGES in the presence of the NF-κB inhibitor BAY 117085 was dramatically reduced in both TR APOE3/3 and TR APOE4/4 microglia with ablation of the TR APOE–specific difference (Fig. 3B, C). Together, these data indicate that differential activation of NF-κB is, at least in part, responsible for TR APOE4/4–amplified PGE2 production, and that this effect is likely mediated by apoE4 at a RAP-sensitive receptor.
Figure 3.
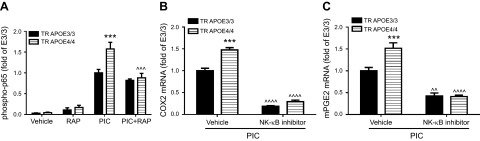
APOE genotype–specific effects on NF-κB–dependent expression of COX2 and mPGES. A) Primary microglia cultures from TR APOE3/3 and TR APOE4/4 mice were exposed to vehicle, RAP (1 μM), PIC (20 μg/ml), or PIC plus RAP for 1 hour, and phospho-p65 of NF-κB from cell lysates was quantified. Data are the average ratio (±sem) normalized to the value for TR APOE3/3 cells activated with PIC. Two-way ANOVA had P < 0.01 TR APOE, P < 0.001 for exposures, and P < 0.01 for interaction between these terms. ***P < 0.001 for Bonferroni-corrected posttests comparing TR APOE3/3 with TR APOE4/4. One-way ANOVA followed by Bonferroni-corrected posttests found that RAP significantly reduced activity in PIC-treated TR APOE4/4 cells (^^^P = 0.001) but not in PIC-treated TR APOE3/3 cells. B, C) Primary microglia cultures from TR APOE3/3 and TR APOE4/4 mice were exposed 20 μg/ml PIC plus vehicle or 1 μM NF-κB inhibitor BAY 117085 for 6 hours, and expression of COX2 (B) and mPGES (C) was determined by real-time PCR. Data are the average ratio (±sem) normalized to the value for TR APOE3/3 cells activated with PIC. Two-way ANOVA had P < 0.01 TR APOE, P < 0.001 for exposures, and P < 0.01 for interaction between these terms. ***P < 0.001 for Bonferroni-corrected posttests comparing TR APOE3/3 with TR APOE4/4. One-way ANOVA followed by Bonferroni-corrected posttests found that in the presence of PIC, BAY 117085 significantly reduced COX2 (B) or mPGES (C) levels in both genotypes (^^^^P < 0.0001 for COX2 in both genotypes; ^^P < 0.01 for mPGES in TR APOE3/3 cells and ^^^^P < 0.0001 in TR APOE4/4 cells).
Before TREM2 expression in TR APOE microglia, we first established whether the in vivo observation that cerebral cortical TREM2 expression is limited largely to microglia (43) extends to primary culture. We confirmed this by determining that TREM2 mRNA levels in primary cultures of WT cerebral microglia averaged 20- or 256-fold more than primary cultures of cerebral astrocytes or neurons, respectively (Fig. 4A, P < 0.0001). Next we examined the regulation of TREM2 expression in primary WT microglia incubated for 18 hours with the same TLR activators used to assess the PG pathway above (Fig. 4B). All TLR activators suppressed microglia expression of TREM2 by approximately 80% compared to vehicle control. Figure 4C is a time course for TREM2 expression by primary cerebral microglia cultures after activation of TLR3 with PIC. There was no change in TREM2 expression after 2 hours of PIC exposure (0.99 ± 0.06 TREM2 mRNA compared to time 0); however, after 4 hours exposure, TREM2 mRNA was reduced to 0.68 ± 0.04 and continued to decrease linearly to 0.23 ± 0.03 by 18 hours (P < 0.001). Previously we demonstrated that coincubation of primary microglia with PIC (20 μM) and an inhibitor of COX2 (NS398) for 18 hours completely blocks cytokine release (38). However, under these conditions, we observed only a small but significant 16% average protection from PIC-induced reduction of TREM2 expression (P < 0.01) and obtained similar results with a PGE2 receptor subtype 2 antagonist (not shown).
Figure 4.
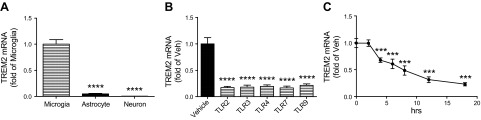
Down-regulation of TREM2 by TLR activators. A) Total RNA was extracted from primary cultures of WT microglia, astrocytes, or neurons. TREM2 mRNA levels were determined by real-time PCR at 1 (microglia) or 3 (astrocytes and neurons) days after plating. Data are the average ratio (±sem) normalized to the value for microglia. ****P < 0.0001 for Bonferroni-corrected repeated pair comparison with microglia. B) WT primary microglia were treated with vehicle (Veh) or the same TLR activators shown in Fig. 1—TLR2 (Pam, 1 μg/ml), TLR3 (PIC, 20 μg/ml), TLR4 (LPS, 1 μg/ml), TLR7 (Lox, 1 mM) or TLR9 (CpG, 1 μM)—for 18 hours. RNA was extracted for quantitative real-time PCR. TREM2 mRNA levels in each sample were normalized to endogenous GAPDH mRNA. Data are presented as average fold change (±sem) relative to cultures exposed to vehicle (n = 3 per group). ****P < 0.0001 for Bonferroni-corrected repeated pair comparison with vehicle. C) WT microglia cultures were exposed to TLR3 activator PIC (20 μg/ml) for the indicated periods, and expression of TREM2 was determined by real-time PCR. Data are average fold change (±sem) relative to cultures at 0 hours (CT). ***P < 0.001 for Bonferroni-corrected repeated pair comparisons with value at time 0.
We next determined TREM2 expression in TR APOE3/3 and TR APOE4/4 microglia after exposure to 1, 10, and 20 μg/ml PIC for 12 hours. Unlike PGE2 synthesis, PIC treatment suppressed TREM2 expression in microglia of both genotypes at all concentrations tested (Fig. 5A). Moreover, the concentration–response relationship was different from COX2 and mPGES, with the most robust difference between TR APOE microglia observed at 1 μg/ml. However, TREM2 mRNA levels were significantly greater in TR APOE3/3 than TR APOE4/4 after exposure to 1 or 10 μg/ml PIC, but not 20 μg/ml PIC. In contrast to COX2 or mPGES, TREM2 does not have enzymatic activity that can be assayed, so we measured cell surface TREM2 by flow cytometry. TREM2 antibody specificity was determined by comparing the resulting fluorescence intensity to that of cells incubated with rat IgG isotype control antibody or with no antibody. The fluorescence intensity of microglia incubated with TREM2 antibody was 3.6-fold greater than cells incubated with IgG control, and 4-fold greater than cells not incubated with antibody. Next, the regulation of TREM2 protein was analyzed by flow cytometry after 24 hours exposure to vehicle or 1 μg/ml PIC (Fig. 5B). Two-way ANOVA showed that PIC exposure significantly reduced TREM2 surface protein (P < 0.01), and there was a significant effect of TR APOE (P < 0.05). Bonferroni-corrected posttests showed that there was no significant TR APOE-dependent difference in microglia cell surface TREM2 with vehicle, but there was significantly less microglial cell surface TREM2 in TR APOE4/4 microglia after PIC exposure (P < 0.05). Finally, we determined changes in TREM2 mRNA levels in the presence of RAP (Fig. 5C) or NF-κB inhibitor BAY 117085 (Fig. 5D) to determine whether signaling pathways were similar to the TR APOE4/4-specific effect on PGE2 synthesis. RAP alone had no effect on TREM2 mRNA levels, nor did it alter the TR-APOE–specific effect of PIC. This contrasts our results with the PGE2 pathway, where RAP abolished the TR-APOE–specific PIC effect (Fig. 5C). In further contrast to the PGE2 pathway, BAY 117085 did not alter the APOE genotype-dependent effect on TREM2 expression after PIC treatment (Fig. 5D).
Figure 5.

Modulation of PIC-regulated TREM2 expression by TR APOE. Primary microglia cultures from TR APOE3/3 and TR APOE4/4 mice were exposed to indicated reagents and times and then TREM2 mRNA or surface protein levels were determined. A) Concentration–response relationship for PIC induction of TREM2 mRNA expression after 12 hours exposure. Data are average ± sem (n = 3 per group). Two-way ANOVA was significant for concentration and time (P < 0.0001 for each), and Bonferroni-corrected posttests comparing TR APOE3/3 (solid symbols) with TR APOE4/4 (open symbols) were significant (*P < 0.05, **P < 0.01, **P < 0.0001) at the concentrations indicated. B) Primary microglia from TR APOE3/3 and TR APOE4/4 mice were exposed to PIC (1 μg/ml) for 24 hours and then TREM2 protein on cell surface was determined by flow cytometry using with FITC-conjugated TREM2 antibody. Data are geometric means of fluorescence intensity (±sem, n = 4 per group). Two-way ANOVA had P < 0.05 TR APOE, exposures, and interaction between these terms for fluorescence intensity of TREM2. Bonferroni-corrected posttests comparing TR APOE3/3 with TR APOE4/4 had *P < 0.05. C) Primary microglia were exposed to vehicle, RAP (1 μM), PIC (1 μg/ml), or PIC plus RAP for 12 hours. Expression of TREM2 was determined by real-time PCR. Data are the average ratio (±sem) normalized to vehicle. Two-way ANOVA had P < 0.05 TR APOE, exposures, and interaction between these terms for TREM2 mRNA. Bonferroni-corrected posttests comparing TR APOE3/3 with TR APOE4/4 had *P < 0.05. One-way ANOVA followed by Bonferroni-corrected posttests found that RAP had no significant effect on TREM2 mRNA levels alone or in the presence of PIC in either genotype. D) Primary microglia were exposed to PIC (1 μg/ml) or to PIC plus vehicle or NF-κB inhibitor (1 μM BAY 117085) for 12 hours. Expression of TREM2 was determined by real-time PCR. Data are the average ratio (±sem) normalized to the value of TR APOE3/3. Two-way ANOVA had P < 0.01 TR APOE. Bonferroni-corrected posttests comparing TR APOE3/3 with TR APOE4/4 had *P < 0.05. One-way ANOVA followed by Bonferroni-corrected posttests found that in the presence of PIC, BAY 117085 had no significant effect on TREM2 mRNA levels in either genotype.
DISCUSSION
Although many approaches, including genomewide association studies, repeatedly confirm the association of APOE ε4 with AD, the pleiotropic functions of apoE plus the further complexity of isoform-specific activities have obscured a full understanding of the molecular mechanism or mechanisms of this common genetic risk for AD. Here we pursued 2 clues, one pharmacogenetic and another less common genetic risk for AD, to investigate potential mechanisms by which inheritance of APOE ε4 may increase risk for AD. Such mechanistic information is important because it will illuminate potential alternative therapeutic targets for preventing or treating AD.
AD has a complex cellular, molecular, and chemical biology. We and others already have shown in a variety of organotypic and in vivo models that selective activation of microglia innate immune response leads to paracrine neuron damage and degeneration, and that microglia activation and resulting neuron damage are greater in models that express apoE4. ApoE is synthesized by multiple cells in brain, including astrocytes and microglia; it is important to note that on a per-cell basis, murine primary microglia synthesize comparable amounts of apoE mRNA and protein as their astrocyte counterparts (23, 44). Innate immune activation in AD can occur by direct activation of some TLRs by Aβ peptides and indirectly by activation of other TLRs by products released from damaged neurons (21, 25, 34, 45). Moreover, microglia are more highly activated in affected regions of AD brain of individuals with APOE ε4 than with APOE ε3 (46). Here we focused on mechanisms of the enhanced innate immune response, so we used primary cultures of microglia from TR APOE3 or TR APOE4 mice and specific activators of TLRs to model selective innate immune responses. We did not include TR APOE2/2 microglia in these experiments because there are few data to suggest that the proposed protective effect of APOE ε2 for AD is mediated by innate immune regulation. Broadly, we demonstrated more robust induction of the proinflammatory PGE2 pathway in TR APOE4/4 than in TR APOE3/3 microglia after activation of TLR3 or TLR4. The innate immune suppressor, TREM2, showed decreased microglia expression with all TLR activators; however, its reduced expression was greater with TR APOE4/4 than with TR APOE3/3 microglia. Indeed, we found that doubling the RAP concentration or adding a 2 hour RAP pretreatment paradigm failed to elicit a greater effect in TR APOE4/4 microglia or any significant effect in TR APOE3/3 microglia (not shown). This suggests that apoE4 has a proinflammatory effect via RAP-dependent receptors which apoE3 does not. Together, these data demonstrate complementary regulation of pro- and anti-inflammatory mediators in TR APOE4/4 to yield a proinflammatory state compared to TR APOE3/3 microglia. Importantly, we showed that the signaling mechanisms underlying these TR APOE4/4–dependent effects were RAP and NF-κB dependent for the PGE2 pathway, and RAP and NF-κB independent for TREM2 expression.
Epidemiologic studies consistently observe that protracted use of NSAIDs prevent or delay onset of AD, and that the protective effects are more substantial in those with APOE ε4 (28, 47). Importantly, our results provide a molecular basis for these pharmacogenetic observations. Although epidemiologic data imply that NSAIDs may be effective in preventing AD, especially in individuals with APOE ε4, treatment trials with NSAIDs were not effective in groups with AD dementia or enriched for prodromal AD. Following recent advances in our knowledge of AD, it is now appreciated that different therapeutics may be needed for prevention vs. treatment of prodromal AD or AD dementia. Indeed, this reasoning is the basis for several ongoing prevention trials of anti-Aβ therapies in individuals at risk for AD despite failure of these same or closely related agents in treatment trials of AD dementia or prodromal AD. The same may be true for inhibiting the PG pathway; indeed, all experimental, epidemiologic, and clinical data for NSAIDs are consistent with them being effective in preventing AD but not in treating AD prodrome or dementia. We recognize that NSAIDs also have limitations due to toxicity; for this reason, we and others have pursued more selective targets and agents within the PG pathway. A major implication of our work is that any consideration of a future prevention trial with more selective PG pathway inhibitors should be organized to detect differential effects in participants with APOE ε4 vs. those without.
TREM2 is expressed on microglia in brain where it promotes phagocytosis of Aβ and apoptotic neuronal debris, which express its putative ligand (48–51). It is worth noting that TREM2 levels are increased in microglia surrounding amyloid plaques in autopsy brain from AD patients (52). Our contrasting findings from acute exposures of microglia suggest that a more complex relationship may exist in which early immune activation suppresses microglia TREM2 expression, whereas chronic activation and/or accumulation of fibrillar amyloid subsequently increases microglia expression of TREM2 in the vicinity of amyloid plaques. TREM2 also inhibits proinflammatory cytokine production in response to TLR activators in bone marrow macrophages and dendritic cells and thereby acts as an immune suppressor (9–11). We found that activation with each of the relatively selective TLR activators comparably decreased microglia TREM2 mRNA and surface protein, and that this reduction in a mediator of Aβ phagocytosis and an immune suppressor was more pronounced in TR APOE4/4 than TR APOE3/3. Importantly, and in contrast to the PG pathway, the effect of TR APOE4/4 on TREM2 expression was RAP and NF-κB independent, supporting at least 2 distinct mechanisms for a proinflammatory state in APOE ε4 brain: one enhancing the proinflammatory PGE2 pathway and another by suppressing the anti-inflammatory TREM2 pathway. We did not use models appropriate to investigate Aβ metabolism; however, given the results of others showing that inheritance of APOE ε4 is characterized by reduced Aβ clearance (53–56), we speculate that one mechanism for this observation may be greater reduction in TREM2 microglia expression in the context of immune activation such as occurs in transgenic models of AD.
In summary, we found that primary microglia from TR APOE4/4 mice had greater induction of PGE2 pathway including expression of COX2 and mPGES, as well as PGE2 production compared to TR APOE3/3 microglia in response to TLR3 or TLR4 activation. These results provide a mechanism to explain pharmacogenetic observations in the Cardiovascular Health Study, and one means by which expression of apoE4 produces a proinflammatory environment in brain. Unlike PGE2 signal, TREM2 expression was inhibited broadly by TLR activation and with a more pronounced effect in TR APOE4/4 than in TR APOE3/3 microglia, providing a second mechanism for the proinflammatory effect of APOE ε4. Signaling mechanisms for these apoE isoform–dependent immune modulatory effects differed in response to an inhibitor of NF-κB or RAP, suggesting involvement of apoE receptor activation in some inflammatory responses but not others. Our experimental studies provide guidance for the organization of future clinical trials that target immune suppression for the prevention of neuronal stress and damage in AD.
Acknowledgments
The authors thank Samantha Rice and Leanne Hellstern for assistance with animal husbandry, K. Arumuganathan and Jingjing Tang for assistance with flow cytometry, Eiron Cudaback for scientific input, and Aimee Schantz and Carol Arnold for administrative support. This work was funded by the U.S. National Institutes of Health (NIH) National Institute of Environmental Health Sciences (Grant R01-ES016754), NIH National Institute on Aging (Grant P50-AG05136), NIH National Institute of Neurological Disorders and Stroke (Grant P50-NS062684) and the Nancy and Buster Alvord Endowment.
Glossary
- AD
Alzheimer disease
- apo
apolipoprotein
- COX2
cyclooxygenase-2
- mPGES
microsomal PGE synthase
- NF-κB
nuclear factor κ-light-chain-enhancer
- NSAID
nonsteroidal anti-inflammatory drug
- PG
prostaglandin
- PIC
polycytidylic acid
- RAP
receptor-associated protein
- TR
targeted replacement
- TREM2
triggering receptor expressed on myeloid cells 2
- WT
wild-type
REFERENCES
- 1.Schellenberg G. D., Montine T. J. (2012) The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 124, 305–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert J. C., Ibrahim-Verbaas C. A., Harold D., Naj A. C., Sims R., Bellenguez C., DeStafano A. L., Bis J. C., Beecham G. W., Grenier-Boley B., Russo G., Thorton-Wells T. A., Jones N., Smith A. V., Chouraki V., Thomas C., Ikram M. A., Zelenika D., Vardarajan B. N., Kamatani Y., Lin C. F., Gerrish A., Schmidt H., Kunkle B., Dunstan M. L., Ruiz A., Bihoreau M. T., Choi S. H., Reitz C., Pasquier F., Cruchaga C., Craig D., Amin N., Berr C., Lopez O. L., De Jager P. L., Deramecourt V., Johnston J. A., Evans D., Lovestone S., Letenneur L., Morón F. J., Rubinsztein D. C., Eiriksdottir G., Sleegers K., Goate A. M., Fiévet N., Huentelman M. W., Gill M., Brown K., Kamboh M. I., Keller L., Barberger-Gateau P., McGuiness B., Larson E. B., Green R., Myers A. J., Dufouil C., Todd S., Wallon D., Love S., Rogaeva E., Gallacher J., St George-Hyslop P., Clarimon J., Lleo A., Bayer A., Tsuang D. W., Yu L., Tsolaki M., Bossù P., Spalletta G., Proitsi P., Collinge J., Sorbi S., Sanchez-Garcia F., Fox N. C., Hardy J., Deniz Naranjo M. C., Bosco P., Clarke R., Brayne C., Galimberti D., Mancuso M., Matthews F., Moebus S., Mecocci P., Del Zompo M., Maier W., Hampel H., Pilotto A., Bullido M., Panza F., Caffarra P., Nacmias B., Gilbert J. R., Mayhaus M., Lannefelt L., Hakonarson H., Pichler S., Carrasquillo M. M., Ingelsson M., Beekly D., Alvarez V., Zou F., Valladares O., Younkin S. G., Coto E., Hamilton-Nelson K. L., Gu W., Razquin C., Pastor P., Mateo I., Owen M. J., Faber K. M., Jonsson P. V., Combarros O., O’Donovan M. C., Cantwell L. B., Soininen H., Blacker D., Mead S., Mosley T. H. Jr., Bennett D. A., Harris T. B., Fratiglioni L., Holmes C., de Bruijn R. F., Passmore P., Montine T. J., Bettens K., Rotter J. I., Brice A., Morgan K., Foroud T. M., Kukull W. A., Hannequin D., Powell J. F., Nalls M. A., Ritchie K., Lunetta K. L., Kauwe J. S., Boerwinkle E., Riemenschneider M., Boada M., Hiltuenen M., Martin E. R., Schmidt R., Rujescu D., Wang L. S., Dartigues J. F., Mayeux R., Tzourio C., Hofman A., Nöthen M. M., Graff C., Psaty B. M., Jones L., Haines J. L., Holmans P. A., Lathrop M., Pericak-Vance M. A., Launer L. J., Farrer L. A., van Duijn C. M., Van Broeckhoven C., Moskvina V., Seshadri S., Williams J., Schellenberg G. D., Amouyel P.; European Alzheimer’s Disease Initiative (EADI) Genetic and Environmental Risk in Alzheimer’s Disease Alzheimer’s Disease Genetic Consortium Cohorts for Heart and Aging Research in Genomic Epidemiology (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J. S., Younkin S., Hazrati L., Collinge J., Pocock J., Lashley T., Williams J., Lambert J. C., Amouyel P., Goate A., Rademakers R., Morgan K., Powell J., St George-Hyslop P., Singleton A., Hardy J.; Alzheimer Genetic Analysis Group (2013) TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P. V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A. I., Lah J. J., Rujescu D., Hampel H., Giegling I., Andreassen O. A., Engedal K., Ulstein I., Djurovic S., Ibrahim-Verbaas C., Hofman A., Ikram M. A., van Duijn C. M., Thorsteinsdottir U., Kong A., Stefansson K. (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paradowska-Gorycka A., Jurkowska M. (2013) Structure, expression pattern and biological activity of molecular complex TREM-2/DAP12. Hum. Immunol. 74, 730–737 [DOI] [PubMed] [Google Scholar]
- 6.Paloneva J., Manninen T., Christman G., Hovanes K., Mandelin J., Adolfsson R., Bianchin M., Bird T., Miranda R., Salmaggi A., Tranebjaerg L., Konttinen Y., Peltonen L. (2002) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klünemann H. H., Ridha B. H., Magy L., Wherrett J. R., Hemelsoet D. M., Keen R. W., De Bleecker J. L., Rossor M. N., Marienhagen J., Klein H. E., Peltonen L., Paloneva J. (2005) The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 64, 1502–1507 [DOI] [PubMed] [Google Scholar]
- 8.Chouery E., Delague V., Bergougnoux A., Koussa S., Serre J. L., Mégarbané A. (2008) Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum. Mutat. 29, E194–E204 [DOI] [PubMed] [Google Scholar]
- 9.Turnbull I. R., Gilfillan S., Cella M., Aoshi T., Miller M., Piccio L., Hernandez M., Colonna M. (2006) Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 177, 3520–3524 [DOI] [PubMed] [Google Scholar]
- 10.Hamerman J. A., Jarjoura J. R., Humphrey M. B., Nakamura M. C., Seaman W. E., Lanier L. L. (2006) Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J. Immunol. 177, 2051–2055 [DOI] [PubMed] [Google Scholar]
- 11.Ito H., Hamerman J. A. (2012) TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur. J. Immunol. 42, 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang T., Yu J. T., Zhu X. C., Tan M. S., Gu L. Z., Zhang Y. D., Tan L. (2014) Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol. Aging 35, 1243–1251 [DOI] [PubMed] [Google Scholar]
- 13.Saunders A. M., Strittmatter W. J., Schmechel D., George-Hyslop P. H., Pericak-Vance M. A., Joo S. H., Rosi B. L., Gusella J. F., Crapper-MacLachlan D. R., Alberts M. J., et al. (1993) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472 [DOI] [PubMed] [Google Scholar]
- 14.Mahley R. W., Rall S. C. Jr (2000) Apolipoprotein E: far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet. 1, 507–537 [DOI] [PubMed] [Google Scholar]
- 15.Ng S., Lin C. C., Hwang Y. H., Hsieh W. S., Liao H. F., Chen P. C. (2013) Mercury, APOE, and children’s neurodevelopment. Neurotoxicology 37, 85–92 [DOI] [PubMed] [Google Scholar]
- 16.Wright R. O., Hu H., Silverman E. K., Tsaih S. W., Schwartz J., Bellinger D., Palazuelos E., Weiss S. T., Hernandez-Avila M. (2003) Apolipoprotein E genotype predicts 24-month Bayley Scales infant development score. Pediatr. Res. 54, 819–825 [DOI] [PubMed] [Google Scholar]
- 17.Kanekiyo T., Xu H., Bu G. (2014) ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron 81, 740–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keene C. D., Cudaback E., Li X., Montine K. S., Montine T. J. (2011) Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Curr. Opin. Neurobiol. 21, 920–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mosconi L., De Santi S., Brys M., Tsui W. H., Pirraglia E., Glodzik-Sobanska L., Rich K. E., Switalski R., Mehta P. D., Pratico D., Zinkowski R., Blennow K., de Leon M. J. (2008) Hypometabolism and altered cerebrospinal fluid markers in normal apolipoprotein E E4 carriers with subjective memory complaints. Biol. Psychiatry 63, 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valla J., Yaari R., Wolf A. B., Kusne Y., Beach T. G., Roher A. E., Corneveaux J. J., Huentelman M. J., Caselli R. J., Reiman E. M. (2010) Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE ε4 allele, the major late-onset Alzheimer’s susceptibility gene. J. Alzheimers Dis. 22, 307–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vitek M. P., Brown C. M., Colton C. A. (2009) APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 30, 1350–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez G. A., Tai L. M., LaDu M. J., Rebeck G. W. (2014) Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J. Neuroinflammation 11, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y., Cudaback E., Jorstad N. L., Hemingway J. F., Hagan C. E., Melief E. J., Li X., Yoo T., Khademi S. B., Montine K. S., Montine T. J., Keene C. D. (2013) APOE3, but not APOE4, bone marrow transplantation mitigates behavioral and pathological changes in a mouse model of Alzheimer disease. Am. J. Pathol. 183, 905–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cudaback E., Li X., Montine K. S., Montine T. J., Keene C. D. (2011) Apolipoprotein E isoform-dependent microglia migration. FASEB J. 25, 2082–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maezawa I., Nivison M., Montine K. S., Maeda N., Montine T. J. (2006) Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J. 20, 797–799 [DOI] [PubMed] [Google Scholar]
- 26.McGeer P. L., McGeer E. G. (2007) NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol. Aging 28, 639–647 [DOI] [PubMed] [Google Scholar]
- 27.Cudaback E., Jorstad N. L., Yang Y., Montine T. J., Keene C. D. (2014) Therapeutic implications of the prostaglandin pathway in Alzheimer’s disease. Biochem. Pharmacol. 88, 565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szekely C. A., Breitner J. C., Fitzpatrick A. L., Rea T. D., Psaty B. M., Kuller L. H., Zandi P. P. (2008) NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology 70, 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pasqualetti P., Bonomini C., Dal Forno G., Paulon L., Sinforiani E., Marra C., Zanetti O., Rossini P. M. (2009) A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer’s disease. Aging Clin. Exp. Res. 21, 102–110 [DOI] [PubMed] [Google Scholar]
- 30.Cole G. M., Frautschy S. A. (2010) Mechanisms of action of non-steroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 9, 140–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan P. M., Mezdour H., Quarfordt S. H., Maeda N. (1998) Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J. Clin. Invest. 102, 130–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sullivan P. M., Mezdour H., Aratani Y., Knouff C., Najib J., Reddick R. L., Quarfordt S. H., Maeda N. (1997) Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem. 272, 17972–17980 [DOI] [PubMed] [Google Scholar]
- 33.Knouff C., Hinsdale M. E., Mezdour H., Altenburg M. K., Watanabe M., Quarfordt S. H., Sullivan P. M., Maeda N. (1999) Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J. Clin. Invest. 103, 1579–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maezawa I., Zaja-Milatovic S., Milatovic D., Stephen C., Sokal I., Maeda N., Montine T. J., Montine K. S. (2006) Apolipoprotein E isoform-dependent dendritic recovery of hippocampal neurons following activation of innate immunity. J. Neuroinflammation 3, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maezawa I., Maeda N., Montine T. J., Montine K. S. (2006) Apolipoprotein E–specific innate immune response in astrocytes from targeted replacement mice. J. Neuroinflammation 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maezawa I., Jin L. W., Woltjer R. L., Maeda N., Martin G. M., Montine T. J., Montine K. S. (2004) Apolipoprotein E isoforms and apolipoprotein AI protect from amyloid precursor protein carboxy terminal fragment-associated cytotoxicity. J. Neurochem. 91, 1312–1321 [DOI] [PubMed] [Google Scholar]
- 37.Cudaback E., Li X., Yang Y., Yoo T., Montine K. S., Craft S., Montine T. J., Keene C. D. (2012) Apolipoprotein C-I is an APOE genotype-dependent suppressor of glial activation. J. Neuroinflammation 9, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X., Cudaback E., Keene C. D., Breyer R. M., Montine T. J. (2011) Suppressed microglial E prostanoid receptor 1 signaling selectively reduces tumor necrosis factor alpha and interleukin 6 secretion from toll-like receptor 3 activation. Glia 59, 569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X., Cudaback E., Breyer R. M., Montine K. S., Keene C. D., Montine T. J. (2012) Eicosanoid receptor subtype-mediated opposing regulation of TLR-stimulated expression of astrocyte glial-derived neurotrophic factor. FASEB J. 26, 3075–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X., Rose S. E., Montine K. S., Keene C. D., Montine T. J. (2013) Antagonism of neuronal prostaglandin E(2) receptor subtype 1 mitigates amyloid beta neurotoxicity in vitro. J. Neuroimmune Pharmacol. 8, 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larionov A., Krause A., Miller W. (2005) A standard curve based method for relative real time PCR data processing. BMC Bioinformatics 6, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Town T., Jeng D., Alexopoulou L., Tan J., Flavell R. A. (2006) Microglia recognize double-stranded RNA via TLR3. J. Immunol. 176, 3804–3812 [DOI] [PubMed] [Google Scholar]
- 43.Sessa G., Podini P., Mariani M., Meroni A., Spreafico R., Sinigaglia F., Colonna M., Panina P., Meldolesi J. (2004) Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur. J. Neurosci. 20, 2617–2628 [DOI] [PubMed] [Google Scholar]
- 44.Saura J., Petegnief V., Wu X., Liang Y., Paul S. M. (2003) Microglial apolipoprotein E and astroglial apolipoprotein J expression in vitro: opposite effects of lipopolysaccharide. J. Neurochem. 85, 1455–1467 [DOI] [PubMed] [Google Scholar]
- 45.Lynch J. R., Tang W., Wang H., Vitek M. P., Bennett E. R., Sullivan P. M., Warner D. S., Laskowitz D. T. (2003) APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J. Biol. Chem. 278, 48529–48533 [DOI] [PubMed] [Google Scholar]
- 46.Egensperger R., Kösel S., von Eitzen U., Graeber M. B. (1998) Microglial activation in Alzheimer disease: association with APOE genotype. Brain Pathol. 8, 439–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hayden K. M., Zandi P. P., Khachaturian A. S., Szekely C. A., Fotuhi M., Norton M. C., Tschanz J. T., Pieper C. F., Corcoran C., Lyketsos C. G., Breitner J. C., Welsh-Bohmer K. A.; Cache County Investigators (2007) Does NSAID use modify cognitive trajectories in the elderly? The Cache County study. Neurology 69, 275–282 [DOI] [PubMed] [Google Scholar]
- 48.Takahashi K., Rochford C. D., Neumann H. (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsieh C. L., Koike M., Spusta S. C., Niemi E. C., Yenari M., Nakamura M. C., Seaman W. E. (2009) A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 109, 1144–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Melchior B., Garcia A. E., Hsiung B. K., Lo K. M., Doose J. M., Thrash J. C., Stalder A. K., Staufenbiel M., Neumann H., Carson M. J. (2010) Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer’s disease. ASN Neuro 2, e00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones B. M., Bhattacharjee S., Dua P., Hill J. M., Zhao Y., Lukiw W. J. (2014) Regulating amyloidogenesis through the natural triggering receptor expressed in myeloid/microglial cells 2 (TREM2). Front. Cell. Neurosci. 8, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lue L. F., Schmitz C. T., Serrano G., Sue L. I., Beach T. G., Walker D. G. (2014) TREM2 protein expression changes correlate with Alzheimer’s disease neurodegenerative pathologies in post-mortem temporal cortices [Epub ahead of print]. Brain Pathol. doi: 10.1111/bpa.12190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castellano J. M., Kim J., Stewart F. R., Jiang H., DeMattos R. B., Patterson B. W., Fagan A. M., Morris J. C., Mawuenyega K. G., Cruchaga C., Goate A. M., Bales K. R., Paul S. M., Bateman R. J., Holtzman D. M. (2011) Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3, 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma J., Yee A., Brewer H. B. Jr., Das S., Potter H. (1994) Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 372, 92–94 [DOI] [PubMed] [Google Scholar]
- 55.Wisniewski T., Castaño E. M., Golabek A., Vogel T., Frangione B. (1994) Acceleration of Alzheimer’s fibril formation by apolipoprotein E in vitro. Am. J. Pathol. 145, 1030–1035 [PMC free article] [PubMed] [Google Scholar]
- 56.Moir R. D., Atwood C. S., Romano D. M., Laurans M. H., Huang X., Bush A. I., Smith J. D., Tanzi R. E. (1999) Differential effects of apolipoprotein E isoforms on metal-induced aggregation of A beta using physiological concentrations. Biochemistry 38, 4595–4603 [DOI] [PubMed] [Google Scholar]