

Abstract
Exposure to hyperoxia results in acute lung injury. A pathogenic consequence of hyperoxia is endothelial injury. Macrophage migration inhibitory factor (MIF) has a cytoprotective effect on lung endothelial cells; however, the mechanism is uncertain. We postulate that the MIF receptor CD74 mediates this protective effect. Using adult wild-type (WT), MIF-deficient (Mif−/−), CD74-deficient (Cd74−/−) mice and MIF receptor inhibitor treated mice, we report that MIF deficiency or inhibition of MIF receptor binding results in increased sensitivity to hyperoxia. Mif−/− and Cd74−/− mice demonstrated decreased median survival following hyperoxia compared to WT mice. Mif−/− mice demonstrated an increase in bronchoalveolar protein (48%) and lactate dehydrogenase (LDH) (68%) following 72 hours of hyperoxia. Similarly, treatment with MIF receptor antagonist resulted in a 59% and 91% increase in bronchoalveolar lavage protein and LDH, respectively. Inhibition of CD74 in primary murine lung endothelial cells (MLECs) abrogated the protective effect of MIF, including decreased hyperoxia-mediated AKT phosphorylation and a 20% reduction in the antiapoptotic effect of exogenous MIF. Treatment with MIF decreased hyperoxia-mediated H2AX phosphorylation in a CD74-dependent manner. These data suggest that therapeutic manipulation of the MIF-CD74 axis in lung endothelial cells may be a novel approach to protect against acute oxidative stress.—Sauler, M., Zhang, Y., Min, J.-N., Leng, L., Shan, P., Roberts, S., Jorgensen, W. L., Bucala, R., Lee, P. J. Endothelial CD74 mediates macrophage migration inhibitory factor protection in hyperoxic lung injury.
Keywords: apoptosis, endothelium, H2AX, MIF
Patients with adult respiratory distress syndrome (ARDS) often require high levels of supplemental oxygen. Although life sustaining, prolonged exposure to elevated partial pressures of oxygen can result in oxidative stress, cellular damage, and respiratory impairment (1, 2). Optimal treatment of individuals with severe respiratory distress therefore will rely on elucidating protective mechanisms against hyperoxic lung injury. Key pathogenic features of hyperoxic lung injury are endothelial injury and apoptosis, and molecular regulators of endothelial health may afford protection from hyperoxia (3).
Macrophage migration inhibitory factor (MIF) is an upstream regulator of the innate response, and MIF protects against oxidative stress–mediated apoptosis. In the lung, the mechanism for this protection remains poorly understood (4–7). MIF is a pleiotropic cytokine that exists in preformed cytoplasmic pools in alveolar epithelial, alveolar endothelial, and pulmonary macrophages. MIF is secreted by these diverse cell types in response to microbial products, proinflammatory cytokines, and oxidative stress (8, 9). Early investigations demonstrated that MIF may have a pivotal role in the pathogenesis of many forms of lung disease including ARDS (10). MIF is elevated in the bronchoalveolar lavage (BAL), and serum of individuals with ARDS and genetic deletion of the Mif gene is protective in certain models of murine lung injury (10–12). Multiple studies have linked functional polymorphisms in MIF to pulmonary disease, yet the effects of MIF polymorphisms in ARDS remain uncertain (13). Recently, investigations have suggested that MIF can protect against various forms of oxidative stress in the lung, including cigarette smoke and radiation (4–7, 14). Collectively, these observations suggest that whereas MIF secretion may correlate with disease severity, it may have a role in lung protection from oxidant injury. Exposure to hyperoxia is a validated model of ARDS that has led to successful translational studies (15, 16). Studying MIF in a murine hyperoxic injury model may inform our understanding of ARDS and suggest novel therapeutic targets in this disease.
MIF binds to its cell surface receptor CD74 to initiate signaling pathways that include the p44/p42 MAPK (ERK1/2) and PI3K/AKT pathways; these in turn lead to enhanced survival and a decrease in p53-dependent apoptosis (17–20). In the lung, CD74 expression has been reported in macrophages, type II pneumocytes, and upon oncogenic transformation (6, 21, 22). We hypothesized that constitutive or inducible CD74 expression is essential for MIF’s protective effect in the lung. Using a hyperoxia model of lung injury, we demonstrate that both Mif−/− and Cd74−/− mice are more sensitive to hyperoxic injury than wild-type (WT) mice, and pharmacologic antagonism of the MIF-CD74 interaction replicates the susceptibility of MIF-deficient mice to hyperoxic lung injury. Although CD74 is absent at baseline in endothelial cells, our data demonstrate that CD74 is induced by hyperoxia, suggesting a specific tissue-protective role for MIF-CD74 signal transduction during hyperoxic injury.
MATERIALS AND METHODS
Mice
Mif −/− mice, backcrossed for >10 generations onto a C57BL/6J background, were described previously (23). Cd74−/− mice on a C57BL/5J background and WT C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and bred in our facility. All mice were bred by homozygous mating under specific pathogen-free conditions at the animal facility of the Yale University School of Medicine, and animal protocols were reviewed and approved by the Animal Care and Use Committee at Yale University.
Hyperoxia exposure
Eight- to 10-week-old mice were bred and exposed to >95% oxygen in a Plexiglas-exposure chamber as previously described (24). For survival studies, animals were monitored, and time of death was noted. Mice were treated with the MIF antagonist 3-(3-hydroxybenzyl)-5-methylbenzooxazol-2-one, designated MIF098 [compound 5 (25)], which was delivered immediately prior to initiation of hyperoxia by i.p. injection and every 12 hours subsequently at a concentration of 40 mg/kg twice a day (26). MIF098 was dissolved in 125 μl PEG400 (Sigma-Aldrich, St. Louis, MO, USA) in a sonicating water bath; 1.125 ml Hp-P-cyclodextrin (Sigma-Aldrich) then was added to prepare a 4 mg/ml solution. Control mice were treated with vehicle alone.
Isolation of primary murine lung endothelial cells
The isolation of murine lung endothelial cells (MLECs) has been previously described (27). Briefly, microvascular endothelial cells were obtained from 4-week-old WT mice. Animals were euthanized via cervical dislocation, and lungs were collected in ice-cold DMEM. Lung tissue was minced and digested for 1 hour at 37°C in 0.1% collagenase-A. Digests then were filtered and resuspended in growth medium and plated in 0.1% gelatin-coated plates. Cells were subjected to sequential magnetic bead selection with CD31 and CD102 (BD Biosciences, Franklin Lakes, NJ, USA). For studies of nuclear damage, SV40-transformed MLECs were generated by expressing SV40 large T antigen (SV40) in early passage of primary MLECs as previously described (28).
Measurements of lung injury
BAL fluid was assessed at 72 hours after hyperoxia exposure. Mice were killed by intraperitoneal ketamine/xylazine injection, and the trachea was cannulated and perfused with two 0.9 ml aliquots of cold saline. The cellular contents and BAL fluid were separated by centrifugation. Cells were enumerated with a COULTER Counter (Beckman Coulter, Brea, CA, USA). Protein quantification was performed via a bicinchoninic acid protein assay (Thermo Fisher Scientific, Waltham, MA, USA). Lactate dehydrogenase (LDH) quantification was performed via a Cytotoxicity Detection Kit (Roche, Basel, Switzerland). H2O2 content of the BAL was measured using an Amplex Red hydrogen peroxide kit (Life Technologies, Carlsbad, CA, USA). BAL IL-6 was quantified using a commercially available ELISA kit (BD Biosciences).
Apoptosis assay
Fluorescence-activated cell sorting (FACS) analysis of MLECs for Annexin V and propidium iodide (PI) was performed per the manufacturer’s protocol (BD Biosciences). TUNEL staining per manufacturer protocol (Roche) was used on mouse tissue.
Immunofluorescence imaging
Formalin-fixed paraffin-embedded lung tissue samples were deparaffinized with xylene, rehydrated gradually with graded alcohol solutions, and then washed with deionized water. For immunofluorescent staining of CD74, antigen retrieval was performed by heating for 15 minutes with 10 mM citrate buffer (pH 6.0) in a microwave oven. Slides were blocked with serum-free blocking solution (Dako, Copenhagen, Denmark). Sections were incubated with a 1:50 dilution of antibody specific for the extracellular domain of CD74 (C-16; Santa Cruz Biotechnology, Dallas, TX, USA) and 1:50 dilution of anti-vWF (von Willebrand factor) antibody (Dako) overnight at 4°C in a humidified chamber. Secondary staining was accomplished with chicken anti-goat (Alexa Fluor 488) and goat anti-rabbit (Alexa Fluor 594) (Life Technologies), and samples were counterstained with DAPI (Life Technologies). Microscopy was performed with a Nikon Eclipse Ti-S microscope (Tokyo, Japan) equipped with an Andor Technology camera (Belfast, United Kingdom). Costaining for γ-H2AX and 53BP1 foci has been previously described (31). Briefly, cells were fixed with 2% paraformaldehyde (2% sucrose) for 10 minutes at room temperature. Cells were permeabilized with 0.5% Triton X-100 in PBS for 5 minutes on ice. To block nonspecific binding, cells were incubated in porphobilinogen for 1 hour (0.2% fish gelatin, 0.5% bovine serum albumin, and 1× PBS) and then incubated overnight at 4°C in a humidified chamber with antibodies against γ-H2AX (EMD Millipore, Billerica, MA, USA) or 53BP (Santa Cruz Biotechnology) followed by 488 goat anti-mouse (Alexa Fluor 488) and 594 goat anti-rabbit (Alexa Fluor 594). For colocalization studies, colocalized foci were enumerated visually and values confirmed with validated high-throughput imaging software (29, 30).
Western blot
Lung tissue was homogenized in a RIPA buffer (Thermo Fisher Scientific) supplemented with protease and phosphatase inhibitors (Roche). Western blotting was performed with an SDS-PAGE system (Bio-Rad, Hercules, CA, USA), and 20 μg protein sample was electrophoresed on a 4–20% Tris gel (Bio-Rad) in Tris running buffer, blotted to a PVDF membrane (Bio-Rad), and probed with primary antibodies against γ-H2AX, CD74, phosphorylated AKT (Ser-473), AKT, and β-actin (Cell Signaling Technology, Danvers, MA, USA).
Statistics
Data are expressed as the mean and/or median ± se. Survival studies were evaluated using log-rank analysis. Graphs and statistical comparisons were performed with GraphPad Prism version 6.00 (GraphPad Software, La Jolla, CA, USA).
RESULTS
Mif−/− and Cd74−/− mice are sensitive to hyperoxia
To determine if MIF protects against hyperoxic lung injury, Mif−/−, Cd74−/−, and WT adult mice were exposed to continuous hyperoxia by a standard protocol. WT mice had significantly improved survival when compared to Mif−/− and Cd74−/− mice (Fig. 1).
Figure 1.

Mif−/− and Cd74−/− mice are susceptible to hyperoxia. A) Survival proportions of Mif−/− and WT mice (n = 20–31 mice per group). *P < 0.01; log-rank analysis. B) Survival proportions of Cd74−/− and WT mice (n = 8). *P < 0.05; log-rank analysis.
Mif−/− mice are sensitive to hyperoxia-induced lung injury
We then investigated lung injury at 72 hours, a time point that we have previously shown to be associated with maximal lung injury (2). At 72 hours, Mif−/− mice demonstrated significantly increased lung injury as evidenced by increased BAL protein and LDH, increased apoptosis as evidenced by TUNEL staining, increased reactive oxygen species (ROS) production as suggested by measurements of BAL H2O2, and increased inflammation as suggested by measurements of IL-6 content (Fig. 2).
Figure 2.
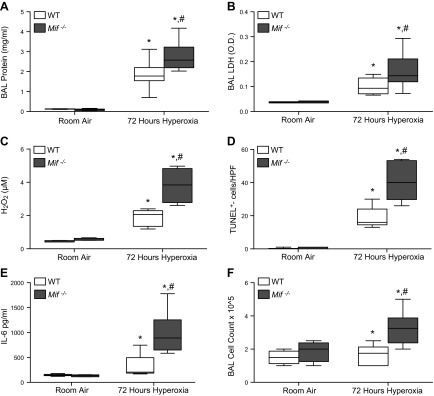
Mif−/− mice demonstrate increased sensitivity to 72 hours of hyperoxic lung injury. A) BAL protein concentration. B) BAL LDH activity. O.D., optic density. C) H2O2 as detected by Amplex Red from BAL. D) Median number of TUNEL+ cells per [high-powered field (HPF)]. E) BAL IL-6 concentration. F) BAL cell count (n = 8–12 mice per group). *P < 0.05 vs. room air WT and #P < 0.05 vs. WT mice exposed to hyperoxia; Student’s t test.
Inhibition of CD74 by MIF098 results in increased sensitivity to hyperoxic lung injury
MIF initiates multiple signaling pathways by engaging the cell surface receptor, CD74. Small molecule MIF receptor antagonists have been validated in different preclinical models of inflammation (31, 32), and recent work has led to the development of highly potent, orally bioavailable benzooxazolone-based inhibitors such as MIF098 (25, 26). This molecule functions as an allosteric inhibitor of MIF binding to the CD74 receptor. To test if MIF exerts its protective effect via the CD74 receptor, WT mice were treated with MIF098 or vehicle control and examined for their sensitivity to hyperoxia. Similar to the injury in the Mif−/− mice in Fig. 1, inhibition of MIF interaction with CD74 resulted in increased hyperoxic injury as evidenced by increased BAL protein and LDH, apoptosis, and ROS production (Fig. 3).
Figure 3.
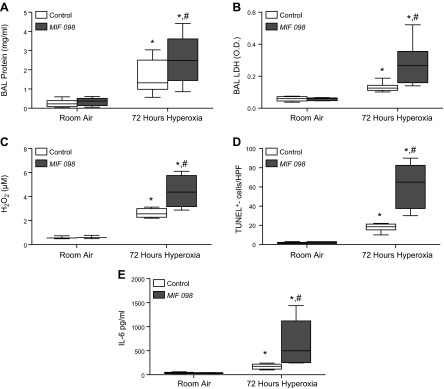
Treatment with MIF receptor antagonist (MIF098) results in increased susceptibility to hyperoxic lung injury. A) BAL protein concentration. B) BAL LDH activity. C) H2O2 as detected by Amplex Red from BAL. D) Median number of TUNEL+ cells per HPF. E) BAL IL-6 concentration (n = 8 mice per group). *P < 0.05 vs. room air WT and #P < 0.05 vs. WT mice exposed to hyperoxia; Student’s t test.
CD74 expression is increased in MLECs following exposure to hyperoxia
We next sought to determine the relevant cell type in the lung that mediates the protective effect of MIF-CD74 interaction. Previous reports from our lab have demonstrated a pivotal role for endothelial cells in hyperoxic lung injury. Using immunofluorescent staining and colocalization with vWF, we found only minimal CD74 staining on endothelial cells under baseline conditions. After exposure to hyperoxia, there was a marked increase in CD74 protein in isolated MLECs (Fig. 4A, B). In lung sections, CD74 colocalized with vWF, a marker for microvascular endothelial cells, and there was a marked induction of immunoreactive CD74 following hyperoxia exposure, similar to the Western blot data (Fig. 4C).
Figure 4.
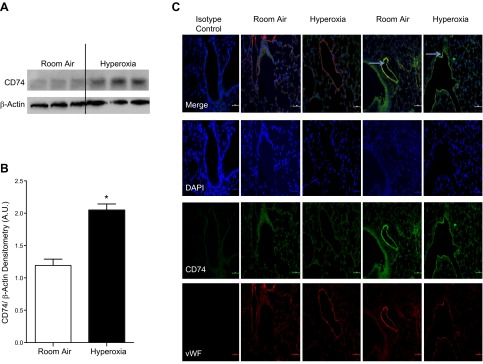
A) Western blot demonstrating up-regulation of the CD74 receptor in MLECs after 24 hours of hyperoxia. B) Densitometry of CD74 representing 3 independent experiments. A.U., arbitrary units. C) Immunofluorescence staining of CD74 (green) and vWF (red) demonstrating up-regulation of CD74 in endothelial cells. Arrows represent positive endothelial CD74 staining.
Inhibition of CD74 decreases AKT signaling
Phosphorylation of AKTSer473 is induced by oxidative stress in lung endothelial cells (33). Phospho-AKTSer473 in turn has been demonstrated to be protective in hyperoxic lung injury models as well as mediate protection from oxidative stress–induced apoptosis. Similarly, MIF has been demonstrated to mediate an antiapoptotic effect through CD74 and AKT signaling. The silencing of CD74 decreased AKT phosphorylation in MLECs in response to hyperoxia (Fig. 5).
Figure 5.

A) Silencing of CD74 RNA (SICD74) results in decreased phosphorylation of AKT, a downstream target of CD74 signaling. B) Densitometry of phosphorylated AKT from 3 independent experiments. All data represent the mean ± sem. *P < 0.05 vs. WT; Student’s t test.
CD74 is necessary for MIF’s antiapoptotic effect in MLECs
The antiapoptotic effect of MIF has been well characterized in a variety of cell types and toxic stressors; however, the precise role of MIF-CD74 signaling in the response of pulmonary endothelial cells has not been examined. Previously reported MLECs treated with 100 ng/ml MIF in vitro were protected from oxidative stress and apoptosis (Fig. 6A, B). Treatment with silencing RNA (siRNA) directed against MIF resulted in increased apoptosis when compared to control (scrambled) RNA, and this protective effect was reversed by treatment with MIF (Fig. 6C). However, treatment with a CD74 siRNA resulted in increased apoptosis, an effect that was not rescued by MIF addition (Fig. 6D). These data support the conclusion that CD74 is necessary for MIF-dependent protection against oxidative stress–induced apoptosis.
Figure 6.
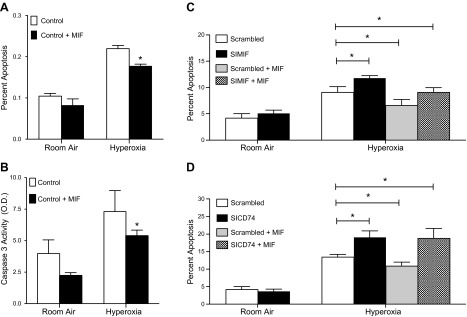
A) Treatment with MIF decreases hyperoxia-induced cell death in MLECs as detected by Annexin/PI staining and FACS analysis. *P < 0.05; Student's t test compared to control in hyperoxia. B) Treatment of MLECs with MIF decreases hyperoxia-induced caspase-3 activation. *P < 0.05; Student's t test compared to control in hyperoxia. C) Treatment with MIF rescues MLECs in which Mif gene expression is silenced, as detected by Annexin/PI staining and FACS analysis. D) Treatment with MIF cannot rescue MLECs in which Cd74 gene expression is silenced, as detected by Annexin/PI staining and FACS analysis. Data shown are representative of 3 independent experiments. All data represent the mean ± sem. *P < 0.05; Student’s t test compared to scrambled treated with hyperoxia (n = 3–7 per group).
MIF decreases oxidative stress–mediated γ-H2AX in a CD74-dependent manner
Hyperoxia results in the formation of ROS, which can promote DNA damage. This leads to the phosphorylation of serine 139 of Histone 2AX (γ-H2AX) as a consequence of both PI3K-related kinases ATM (ataxia-tenagiectasia mutated) and ATR (ataxia-and Rad3-related) (34). γ-H2AX is a sensitive marker of DNA damage and is frequently used to assess the degree of double-stranded DNA breaks. γ-H2AX recruits DNA repair proteins including 53BP1, and these complexes regulate both DNA repair and p53-dependent apoptosis (35). Our data suggest that MIF inhibits γ-H2AX and γ-H2AX/53BP1 complex formation in a CD74-dependent manner. In agreement with previous reports, 12 hours of hyperoxia resulted in phosphorylation of H2AX (36). Silencing of MIF resulted in increased γ-H2AX expression compared to control, an effect that was antagonized by treatment with recombinant MIF (Fig. 7A). Silencing of CD74 resulted in a similar increase in γ-H2AX, and as expected, this increase was unaffected by treatment with exogenous MIF (Fig. 7B, C). Similarly, whereas treatment with MIF reduced γ-H2AX/53BP1 foci formation as a consequence of hyperoxia exposure, pretreatment with SICD74 abrogated the protective effects of MIF treatment (Fig. 7D, E).
Figure 7.
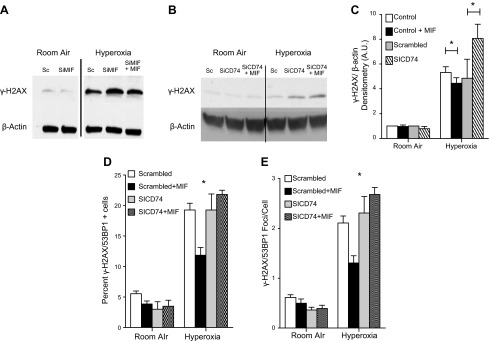
A) Western blot of γ-H2AX following MLEC exposure to hyperoxia for 12 hours. Cells treated with MIF silencing (SiMIF) showed increased γ-H2AX when compared to scrambled (Sc) siRNA, or SiMIF and recombinant MIF. B) Western blot of MLECs treated with CD74 silencing (SiCD74) and siCD74 plus recombinant MIF showed increased γ-H2AX compared to WT. C) Densitometry of γ-H2AX from 3 independent experiments of MLECs treated with MIF or SICD74. D, E) Treatment with MIF decreased the percentage of cells that demonstrated >4 γ-H2AX/53BP1 colocalized foci and the average number of colocalized foci per cell. Data shown are representative of 3 independent experiments. All data represent the mean ± sem. *P < 0.05; Student’s t test.
DISCUSSION
Acute lung injury and ARDS remain devastating respiratory syndromes that are associated with high mortality and few therapeutic options (37, 38). Recurrent failure to translate auspicious preclinical studies into successful therapeutics has been attributed to the complex nature of these diseases, which includes heterogeneity in the inciting stimuli as well as the determinants of clinical progression. The results of randomized controlled clinical studies have suggested that treatments may need to be disease and time point specific (38, 39). Because animal models of disease are limited, integrating multiple murine lung injury models is necessary to fully understand disease pathogenesis and develop new therapeutic targets. MIF is an innate immune protein whose expression has been inversely correlated with disease severity in ARDS and has been demonstrated to mediate deleterious cytokine production in certain lung injury models (10, 11, 40). Yet, studies have highlighted a protective effect of MIF, and recent evidence has suggested that MIF may exert a protective effect on lung endothelial cells (4–6). Therefore, we investigated the role of MIF and its receptor CD74 in a hyperoxic lung injury model of ARDS. We demonstrated that 1) MIF and CD74 mediate protection from hyperoxic lung injury, 2) CD74 expression is induced by hyperoxia in endothelial cells, and 3) MIF, via CD74, protects against oxidative stress–mediated apoptosis and DNA damage in isolated lung endothelial cells. The endothelial MIF-CD74 signaling pathway may represent a novel therapeutic target in oxidant-mediated lung disease.
Using an adult murine hyperoxic injury model, we demonstrate that genetic MIF deficiency is associated with increased lung injury, inflammation, and endothelial cell death. Similar sensitivity to hyperoxic lung injury was observed by use of a previously validated small molecule antagonist of MIF receptor binding. This molecule is an allosteric inhibitor of MIF binding to the CD74 receptor. These data suggest that MIF’s protective effect was, at least in part, mediated by its receptor CD74.
Endothelial cells constitute 30% of the lung surface, demonstrate early pathologic changes to acute hyperoxia, and have been shown to be a key mediator of hyperoxia-induced lung injury. Others have described that CD74 is induced on lung endothelial cells in response to stimuli (41). In the lung, CD74 has been demonstrated in type II cells, macrophages, and lymphocyte populations (2, 3, 42). Most recently, CD74 has been demonstrated in endothelial cells of patients with pulmonary HTN (43, 44). Although the protective effect of MIF in endothelial cells has been described previously, the involvement of CD74 and its inducible expression as a consequence of hyperoxia are novel observations (6, 22). An important limitation of this study is the lack of a mouse-specific monoclonal antibody that targets cell surface CD74. Additionally, there are multiple isoforms of the CD74 protein. However, other investigators have demonstrated that inflammation, possibly as a consequence of IFN-γ or cathepsin S in hyperoxic lung injury, can induce endothelial CD74 (45–48).
We demonstrate decreased AKT activation as a consequence of CD74 silencing. Phosphorylation of AKT induces a wide spectrum of proangiogenic, antiapoptotic, and antioxidant effects, including NF-E2–related factor 2 (NRF-2) (49–51). Activation of CD74 via AKT signaling has been described to mediate protection from oxidative stress (33). Given the pleiotropic nature of MIF, targeting endothelial CD74 may be the preferred therapeutic target. Our data suggest that a MIF-CD74 interaction may play an important role in protecting endothelial cells from hyperoxic lung injury. It remains to be determined if the protective effect of MIF is mediated solely by CD74 signaling through AKT or if alternative pathways also play a role, such as CD74-regulated intramembrane cleavage or MIF interaction with Jun-activating binding protein 1 (Jab1) (52–54). The mechanism of CD74 up-regulation remains uncertain and deserves further study.
Oxidative stress can result in genomic instability. Hyperoxia in particular can cause DNA damage, including single- and double-stranded DNA breaks that can induce cellular senescence and apoptosis (34, 45, 55–57). In order to preserve genetic integrity, organisms have adopted complex systems to counteract the effects of genotoxic stress. Cellular antioxidants mitigate DNA damage by neutralizing ROS to avoid damage. Elaborate checkpoint systems survey for DNA damage and facilitate activation of repair mechanisms while arresting progression through the cell cycle in order to prevent the propagation of genetically altered cells. Coordination of these systems is crucial for cellular defense and avoidance of malignant transformation (58, 59). Multiple studies have suggested that MIF plays an important role in cell cycle regulation and defense from genotoxic stress. One possibility is that this protection is mediated through increased antioxidant gene expression. Interestingly, a recent publication has suggested that silencing of MIF resulted in decreased NRF-2 activation, and hence, MIF would induce protection from oxidative stress–mediated damage (6). Multiple studies suggest that MIF provides protection from genotoxic stress and interferes with p53 and E2F-Rb signaling (19, 60, 61). Therefore, MIF may play a complex role in DNA stability in response to hyperoxia.
We showed herein that treatment of cells with MIF could suppress hyperoxia-induced DNA damage as evidenced by changes in the expression of γ-H2AX and 53BP1. γ-H2AX is a phosphorylated form of histone variant protein H2AX that serves to recruit 53BP1 and DNA repair enzymes, especially at sites of double-stranded DNA breaks (62, 63). The measurement of γ-H2AX is a validated surrogate for DNA damage in response to genotoxic stimuli. These data suggest that MIF may protect against double-stranded DNA breaks and may underlie in part the protective action of MIF in the lung (64). Further studies are necessary to determine if the increase in γ-H2AX is a consequence of decreased damage and/or increased repair as opposed to mechanisms that suppress γ-H2AX formation at the site of injury as a consequence of MIF. Elucidation of the interaction between MIF-CD74 and MIF-JAB1 may help to clarify the role of MIF-CD74 in the protection from oxidative stress. These studies will be crucial to determine the role of MIF in ARDS as well as other oxidative stress–mediated diseases, including cancer and aging.
Further studies are required to delineate the specific mechanisms by which MIF, through CD74, mediates its protection and how this pathway may be best approached pharmacologically, conceivably by small molecule antagonists or by anti-MIF, which is in phase I clinical testing (ClinicalTrials.gov, NCT01765790). d-Dopachrome tautomerase, an orthologous MIF protein that binds CD74, may have therapeutic potential as well (65–68). Finally, these data should prompt investigation into the potential protective role in ARDS of variant MIF alleles, which occur commonly in the human population and have been linked to the severity of other inflammatory or infectious conditions of the lung (e.g., asthma, pneumonia, and tuberculosis).
Acknowledgments
These studies were funded by the following grants from the U.S. National Institutes of Health: T32007778 (to M.S.), R01AI043210 (to R.B.), and R01HL090660, R01HL071595, and FAMRI Clinical Investigator Award (to P.J.L.).
Glossary
- ARDS
adult respiratory distress syndrome
- BAL
bronchoalveolar lavage
- FACS
fluorescence-activated cell sorting
- HTN
hypertension
- Jab1
Jun-activating binding protein 1
- LDH
lactate dehydrogenase
- MIF
macrophage migration inhibitory factor
- MLEC
murine lung endothelial cell
- NRF-2
NF-E2–related factor 2
- PI
propidium iodide
- ROS
reactive oxygen species
- siRNA
silencing RNA
- vWF
von Willebrand factor
- WT
wild-type
REFERENCES
- 1.Fisher A. B. (1980) Oxygen therapy. Side effects and toxicity. Am. Rev. Respir. Dis. 122, 61–69 [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y., Sauler M., Shinn A. S., Gong H., Haslip M., Shan P., Mannam P., Lee P. J. (2014) Endothelial PINK1 mediates the protective effects of NLRP3 deficiency during lethal oxidant injury. J. Immunol. 192, 5296–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y., Jiang G., Sauler M., Lee P. J. (2013) Lung endothelial HO-1 targeting in vivo using lentiviral miRNA regulates apoptosis and autophagy during oxidant injury. FASEB J. 27, 4041–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sauler M., Leng L., Trentalange M., Haslip M., Shan P., Piecychna M., Zhang Y., Andrews N., Mannam P., Allore H., Fried T., Bucala R., Lee P. J. (2014) Macrophage migration inhibitory factor deficiency in chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 306, L487–L496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun H., Choo-Wing R., Sureshbabu A., Fan J., Leng L., Yu S., Jiang D., Noble P., Homer R. J., Bucala R., Bhandari V. (2013) A critical regulatory role for macrophage migration inhibitory factor in hyperoxia-induced injury in the developing murine lung. PLoS One 8, e60560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathew B., Jacobson J. R., Siegler J. H., Moitra J., Blasco M., Xie L., Unzueta C., Zhou T., Evenoski C., Al-Sakka M., Sharma R., Huey B., Bulent A., Smith B., Jayaraman S., Reddy N. M., Reddy S. P., Fingerle-Rowson G., Bucala R., Dudek S. M., Natarajan V., Weichselbaum R. R., Garcia J. G. (2013) Role of migratory inhibition factor in age-related susceptibility to radiation lung injury via NF-E2-related factor-2 and antioxidant regulation. Am. J. Respir. Cell Mol. Biol. 49, 269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen M. T., Lue H., Kleemann R., Thiele M., Tolle G., Finkelmeier D., Wagner E., Braun A., Bernhagen J. (2003) The cytokine macrophage migration inhibitory factor reduces pro-oxidative stress-induced apoptosis. J. Immunol. 170, 3337–3347 [DOI] [PubMed] [Google Scholar]
- 8.Calandra T., Roger T. (2003) Macrophage migration inhibitory factor: a regulator of innate immunity. Nat. Rev. Immunol. 3, 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calandra T., Bernhagen J., Mitchell R. A., Bucala R. (1994) The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J. Exp. Med. 179, 1895–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donnelly S. C., Haslett C., Reid P. T., Grant I. S., Wallace W. A., Metz C. N., Bruce L. J., Bucala R. (1997) Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat. Med. 3, 320–323 [DOI] [PubMed] [Google Scholar]
- 11.Guo Y., Xie C. (2002) [The pathogenic role of macrophage migration inhibitory factor in acute respiratory distress syndrome]. Zhonghua Jie He He Hu Xi Za Zhi 25, 337–340 [PubMed] [Google Scholar]
- 12.Calandra T., Echtenacher B., Roy D. L., Pugin J., Metz C. N., Hültner L., Heumann D., Männel D., Bucala R., Glauser M. P. (2000) Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat. Med. 6, 164–170 [DOI] [PubMed] [Google Scholar]
- 13.Gao L., Flores C., Fan-Ma S., Miller E. J., Moitra J., Moreno L., Wadgaonkar R., Simon B., Brower R., Sevransky J., Tuder R. M., Maloney J. P., Moss M., Shanholtz C., Yates C. R., Meduri G. U., Ye S. Q., Barnes K. C., Garcia J. G. (2007) Macrophage migration inhibitory factor in acute lung injury: expression, biomarker, and associations. Transl. Res. 150, 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fallica J., Boyer L., Kim B., Serebreni L., Varela L., Hamdan O., Wang L., Simms T., Damarla M., Kolb T. M., Bucala R., Mitzner W., Hassoun P. M., Damico R. (2014) Macrophage migration inhibitory factor is a novel determinant of cigarette smoke-induced lung damage. Am. J. Respir. Cell Mol. Biol. 51, 94–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryter S. W., Alam J., Choi A. M. (2006) Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 86, 583–650 [DOI] [PubMed] [Google Scholar]
- 16.Paine R. III, Standiford T. J., Dechert R. E., Moss M., Martin G. S., Rosenberg A. L., Thannickal V. J., Burnham E. L., Brown M. B., Hyzy R. C. (2012) A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit. Care Med. 40, 90–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lue H., Thiele M., Franz J., Dahl E., Speckgens S., Leng L., Fingerle-Rowson G., Bucala R., Lüscher B., Bernhagen J. (2007) Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene 26, 5046–5059 [DOI] [PubMed] [Google Scholar]
- 18.Leng L., Metz C. N., Fang Y., Xu J., Donnelly S., Baugh J., Delohery T., Chen Y., Mitchell R. A., Bucala R. (2003) MIF signal transduction initiated by binding to CD74. J. Exp. Med. 197, 1467–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fingerle-Rowson G., Petrenko O., Metz C. N., Forsthuber T. G., Mitchell R., Huss R., Moll U., Müller W., Bucala R. (2003) The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc. Natl. Acad. Sci. USA 100, 9354–9359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell R. A., Liao H., Chesney J., Fingerle-Rowson G., Baugh J., David J., Bucala R. (2002) Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc. Natl. Acad. Sci. USA 99, 345–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marsh L. M., Cakarova L., Kwapiszewska G., von Wulffen W., Herold S., Seeger W., Lohmeyer J. (2009) Surface expression of CD74 by type II alveolar epithelial cells: a potential mechanism for macrophage migration inhibitory factor-induced epithelial repair. Am. J. Physiol. Lung Cell. Mol. Physiol. 296, L442–L452 [DOI] [PubMed] [Google Scholar]
- 22.Damico R. L., Chesley A., Johnston L., Bind E. P., Amaro E., Nijmeh J., Karakas B., Welsh L., Pearse D. B., Garcia J. G., Crow M. T. (2008) Macrophage migration inhibitory factor governs endothelial cell sensitivity to LPS-induced apoptosis. Am. J. Respir. Cell Mol. Biol. 39, 77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bozza M., Satoskar A. R., Lin G., Lu B., Humbles A. A., Gerard C., David J. R. (1999) Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J. Exp. Med. 189, 341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y., Zhang X., Shan P., Hunt C. R., Pandita T. K., Lee P. J. (2013) A protective Hsp70-TLR4 pathway in lethal oxidant lung injury. J. Immunol. 191, 1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hare A. A., Leng L., Gandavadi S., Du X., Cournia Z., Bucala R., Jorgensen W. L. (2010) Optimization of N-benzyl-benzoxazol-2-ones as receptor antagonists of macrophage migration inhibitory factor (MIF). Bioorg. Med. Chem. Lett. 20, 5811–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun H., Choo-Wing R., Fan J., Leng L., Syed M. A., Hare A. A., Jorgensen W. L., Bucala R., Bhandari V. (2013) Small molecular modulation of macrophage migration inhibitory factor in the hyperoxia-induced mouse model of bronchopulmonary dysplasia. Respir. Res. 14, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X., Shan P., Qureshi S., Homer R., Medzhitov R., Noble P. W., Lee P. J. (2005) Cutting edge: TLR4 deficiency confers susceptibility to lethal oxidant lung injury. J. Immunol. 175, 4834–4838 [DOI] [PubMed] [Google Scholar]
- 28.Min J. N., Tian Y., Xiao Y., Wu L., Li L., Chang S. (2013) The mINO80 chromatin remodeling complex is required for efficient telomere replication and maintenance of genome stability. Cell Res. 23, 1396–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.González J. E., Lee M., Barquinero J. F., Valente M., Roch-Lefère S., García O. (2012) Quantitative image analysis of gamma-H2AX foci induced by ionizing radiation applying open source programs. Anal. Quant. Cytol. Histol. 34, 66–71 [PubMed] [Google Scholar]
- 30.Kamentsky L., Jones T. R., Fraser A., Bray M. A., Logan D. J., Madden K. L., Ljosa V., Rueden C., Eliceiri K. W., Carpenter A. E. (2011) Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27, 1179–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arjona A., Foellmer H. G., Town T., Leng L., McDonald C., Wang T., Wong S. J., Montgomery R. R., Fikrig E., Bucala R. (2007) Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J. Clin. Invest. 117, 3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leng L., Chen L., Fan J., Greven D., Arjona A., Du X., Austin D., Kashgarian M., Yin Z., Huang X. R., Lan H. Y., Lolis E., Nikolic-Paterson D., Bucala R. (2011) A small-molecule macrophage migration inhibitory factor antagonist protects against glomerulonephritis in lupus-prone NZB/NZW F1 and MRL/lpr mice. J. Immunol. 186, 527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmad A., Ahmad S., Chang L. Y., Schaack J., White C. W. (2006) Endothelial Akt activation by hyperoxia: role in cell survival. Free Radic. Biol. Med. 40, 1108–1118 [DOI] [PubMed] [Google Scholar]
- 34.O’Reilly M. A. (2001) DNA damage and cell cycle checkpoints in hyperoxic lung injury: braking to facilitate repair. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L291–L305 [DOI] [PubMed] [Google Scholar]
- 35.Kinner A., Wu W., Staudt C., Iliakis G. (2008) Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 36, 5678–5694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kulkarni A., Das K. C. (2008) Differential roles of ATR and ATM in p53, Chk1, and histone H2AX phosphorylation in response to hyperoxia: ATR-dependent ATM activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L998–L1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ware L. B., Matthay M. A. (2000) The acute respiratory distress syndrome. N. Engl. J. Med. 342, 1334–1349 [DOI] [PubMed] [Google Scholar]
- 38.Matthay M. A., Ware L. B., Zimmerman G. A. (2012) The acute respiratory distress syndrome. J. Clin. Invest. 122, 2731–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meduri G. U., Golden E., Freire A. X., Taylor E., Zaman M., Carson S. J., Gibson M., Umberger R. (2007) Methylprednisolone infusion in early severe ARDS: results of a randomized controlled trial. Chest 131, 954–963 [DOI] [PubMed] [Google Scholar]
- 40.Lai K. N., Leung J. C., Metz C. N., Lai F. M., Bucala R., Lan H. Y. (2003) Role for macrophage migration inhibitory factor in acute respiratory distress syndrome. J. Pathol. 199, 496–508 [DOI] [PubMed] [Google Scholar]
- 41.Collins T., Korman A. J., Wake C. T., Boss J. M., Kappes D. J., Fiers W., Ault K. A., Gimbrone M. A. Jr, Strominger J. L., Pober J. S. (1984) Immune interferon activates multiple class II major histocompatibility complex genes and the associated invariant chain gene in human endothelial cells and dermal fibroblasts. Proc. Natl. Acad. Sci. USA 81, 4917–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crapo J. D., Barry B. E., Gehr P., Bachofen M., Weibel E. R. (1982) Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 126, 332–337 [DOI] [PubMed] [Google Scholar]
- 43.Montani D., Chaumais M. C., Guignabert C., Günther S., Girerd B., Jaïs X., Algalarrondo V., Price L. C., Savale L., Sitbon O., Simonneau G., Humbert M. (2014) Targeted therapies in pulmonary arterial hypertension. Pharmacol. Ther. 141, 172–191 [DOI] [PubMed] [Google Scholar]
- 44.Rabinovitch M., Guignabert C., Humbert M., Nicolls M. R. (2014) Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 115, 165–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beswick E. J., Bland D. A., Suarez G., Barrera C. A., Fan X., Reyes V. E. (2005) Helicobacter pylori binds to CD74 on gastric epithelial cells and stimulates interleukin-8 production. Infect. Immun. 73, 2736–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faure-André G., Vargas P., Yuseff M. I., Heuzé M., Diaz J., Lankar D., Steri V., Manry J., Hugues S., Vascotto F., Boulanger J., Raposo G., Bono M. R., Rosemblatt M., Piel M., Lennon-Duménil A. M. (2008) Regulation of dendritic cell migration by CD74, the MHC class II-associated invariant chain. Science 322, 1705–1710 [DOI] [PubMed] [Google Scholar]
- 47.Wang Z., Zheng T., Zhu Z., Homer R. J., Riese R. J., Chapman H. A. Jr, Shapiro S. D., Elias J. A. (2000) Interferon gamma induction of pulmonary emphysema in the adult murine lung. J. Exp. Med. 192, 1587–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hirakawa H., Pierce R. A., Bingol-Karakoc G., Karaaslan C., Weng M., Shi G. P., Saad A., Weber E., Mariani T. J., Starcher B., Shapiro S. D., Cataltepe S. (2007) Cathepsin S deficiency confers protection from neonatal hyperoxia-induced lung injury. Am. J. Respir. Crit. Care Med. 176, 778–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dai G., Vaughn S., Zhang Y., Wang E. T., Garcia-Cardena G., Gimbrone M. A. Jr, (2007) Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ. Res. 101, 723–733 [DOI] [PubMed] [Google Scholar]
- 50.Shiojima I., Walsh K. (2002) Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 90, 1243–1250 [DOI] [PubMed] [Google Scholar]
- 51.Franke T. F., Kaplan D. R., Cantley L. C. (1997) PI3K: downstream AKTion blocks apoptosis. Cell 88, 435–437 [DOI] [PubMed] [Google Scholar]
- 52.Becker-Herman S., Arie G., Medvedovsky H., Kerem A., Shachar I. (2005) CD74 is a member of the regulated intramembrane proteolysis-processed protein family. Mol. Biol. Cell 16, 5061–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kleemann R., Hausser A., Geiger G., Mischke R., Burger-Kentischer A., Flieger O., Johannes F. J., Roger T., Calandra T., Kapurniotu A., Grell M., Finkelmeier D., Brunner H., Bernhagen J. (2000) Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature 408, 211–216 [DOI] [PubMed] [Google Scholar]
- 54.Schwartz V., Krüttgen A., Weis J., Weber C., Ostendorf T., Lue H., Bernhagen J. (2012) Role for CD74 and CXCR4 in clathrin-dependent endocytosis of the cytokine MIF. Eur. J. Cell Biol. 91, 435–449 [DOI] [PubMed] [Google Scholar]
- 55.Barzilai A., Yamamoto K. (2004) DNA damage responses to oxidative stress. DNA Repair (Amst.) 3, 1109–1115 [DOI] [PubMed] [Google Scholar]
- 56.Pero R. W., Anderson M. W., Doyle G. A., Anna C. H., Romagna F., Markowitz M., Bryngelsson C. (1990) Oxidative stress induces DNA damage and inhibits the repair of DNA lesions induced by N-acetoxy-2-acetylaminofluorene in human peripheral mononuclear leukocytes. Cancer Res. 50, 4619–4625 [PubMed] [Google Scholar]
- 57.Martins E. A., Chubatsu L. S., Meneghini R. (1991) Role of antioxidants in protecting cellular DNA from damage by oxidative stress. Mutat. Res. 250, 95–101 [DOI] [PubMed] [Google Scholar]
- 58.Sancar A., Lindsey-Boltz L. A., Unsal-Kaçmaz K., Linn S. (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 [DOI] [PubMed] [Google Scholar]
- 59.Kanaar R., Hoeijmakers J. H., van Gent D. C. (1998) Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 8, 483–489 [DOI] [PubMed] [Google Scholar]
- 60.Fingerle-Rowson G., Petrenko O. (2007) MIF coordinates the cell cycle with DNA damage checkpoints. Lessons from knockout mouse models. Cell Div. 2, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petrenko O., Moll U. M. (2005) Macrophage migration inhibitory factor MIF interferes with the Rb-E2F pathway. Mol. Cell 17, 225–236 [DOI] [PubMed] [Google Scholar]
- 62.Paull T. T., Rogakou E. P., Yamazaki V., Kirchgessner C. U., Gellert M., Bonner W. M. (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10, 886–895 [DOI] [PubMed] [Google Scholar]
- 63.Ward I. M., Minn K., Jorda K. G., Chen J. (2003) Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J. Biol. Chem. 278, 19579–19582 [DOI] [PubMed] [Google Scholar]
- 64.Ivashkevich A., Redon C. E., Nakamura A. J., Martin R. F., Martin O. A. (2012) Use of the γ-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 327, 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Merk M., Mitchell R. A., Endres S., Bucala R. (2012) D-dopachrome tautomerase (D-DT or MIF-2): doubling the MIF cytokine family. Cytokine 59, 10–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qi D., Atsina K., Qu L., Hu X., Wu X., Xu B., Piecychna M., Leng L., Fingerle-Rowson G., Zhang J., Bucala R., Young L. H. (2014) The vestigial enzyme D-dopachrome tautomerase protects the heart against ischemic injury. J. Clin. Invest. 124, 3540–3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pasupuleti V., Du W., Gupta Y., Yeh I. J., Montano M., Magi-Galuzzi C., Welford S. M. (2014) Dysregulated D-dopachrome tautomerase, a hypoxia-inducible factor-dependent gene, cooperates with macrophage migration inhibitory factor in renal tumorigenesis. J. Biol. Chem. 289, 3713–3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xin D., Rendon B. E., Zhao M., Winner M., McGhee Coleman A., Mitchell R. A. (2010) The MIF homologue D-dopachrome tautomerase promotes COX-2 expression through β-catenin-dependent and -independent mechanisms. Mol. Cancer Res. 8, 1601–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]