

Abstract

Selective catalytic synthesis of Z-olefins has been challenging. Here we describe a method to produce 1,2-disubstituted olefins in high Z selectivity via reductive cross-coupling of alkyl halides with terminal arylalkynes. The method employs inexpensive and nontoxic catalyst (iron(II) bromide) and reductant (zinc). The substrate scope encompasses primary, secondary, and tertiary alkyl halides, and the reaction tolerates a large number of functional groups. The utility of the method is demonstrated in the synthesis of several pharmaceutically relevant molecules. Mechanistic study suggests that the reaction proceeds through an iron-catalyzed anti-selective carbozincation pathway.
Olefins are among the most important organic compounds in chemical, materials, and pharmaceutical industries.1,2 While the preparation of E-olefins is well established, the synthesis of Z-olefins is less straightforward.3 The most general method of Z-olefin synthesis is the Wittig-type reaction, which is noncatalytic and generates a stoichiometric amount of phosphine oxide waste.4 Recently, significant progress has been made in the development of catalytic methods, such as cross-coupling,5,6 semihydrogenation of alkynes,2,7,8 and olefin metathesis9,10 for Z-selective olefin synthesis. Moreover, catalytic carbomagnesiation and carbozincation of alkynes with Grignard and organozinc reagents have been developed to allow stereoselective synthesis of polysubstituted alkenes from readily available alkynes.5a,5c,6e,6f,11
Here, we report an alternative method for Z-selective synthesis of 1,2-disubstituted olefins via iron-catalyzed reductive cross-coupling of alkyl halides with terminal arylalkynes (Figure 1).12,13 This approach offers a valuable alternative compared to the above-mentioned methods: (i) the reaction employs inexpensive and nontoxic FeBr2 as catalyst without the need of sophisticated ligands; (ii) no sensitive organometallic reagents are used; (iii) the starting materials are readily available. Mechanistic study suggests that the reductive coupling occurs via an Fe-catalyzed anti-selective carbozincation of arylalkynes, which leads to high Z-selectivity (Figure 1).
Figure 1.

Z-Selective olefin synthesis by Fe-catalyzed reductive coupling of alkyl electrophiles with terminal arylalkynes.
We targeted Fe catalysis for this transformation. Fe catalysts are known to catalyze cross-coupling of alkyl halides with aryl Grignard reagents.14 While the mechanism of these coupling reactions are still under investigation, there is evidence that iron(I) species are involved for the activation of alkyl halides.15 We envisioned that an iron(I) intermediate might be generated by reduction of an iron(II) salt with a stable reductant such as zinc, thereby avoiding the use of Grignard reagents,13 which not only are inconvenient to handle but also may lead to undesired side reactions such as cross-coupling and reduction.
We commenced the study by examining the reaction of ethynylbenzene (1a) with iodocyclohexane (2a) (see Supporting Information, Tables S1–S6; Figure S1). After a screening of reaction parameters, we found that the optimized conditions involved the use of N,N-dimethylacetamide (DMA) as solvent, FeBr2 (10 mol %) as catalyst, Zn powder (1.5 equiv) as reductant, iodine (I2, 2 mol %) as Zn-activating reagent, and 2a in slight excess (1.5 equiv). The reaction took place at room temperature for 16 h. After aqueous workup, (2-cyclohexylvinyl)benzene (3a) was obtained in 91% GC yield and high Z-selectivity (Z:E > 13:1) (Table S1, entry 7). When Zn was replaced by another reductant such as Mn and Mg, the yields were only 5% and 7%, respectively (Table S1, entries 10 and 11). Among various iron salts, FeBr2 was the best catalyst (Table S2). When 99.99% pure FeBr2 was used, the yield was similar (Table S2, entry 4). When FeBr2 was replaced by catalysts based on Cu, Ni, Co, Mn, Cr, Ag, or Pd, the yield and/or selectivity was lower (Table S3). Both FeBr2 and Zn were essential for the reaction (Table S6).
The Fe-catalyzed olefination method proved to be general (Figure 2).16 The reaction is insensitive to the electronics of arylalkynes, as electron-neutral (3a), electron-rich (3b–3e), and electron-deficient arylalkynes (3f–3n) all reacted to yield the corresponding Z-olefins in good to excellent yields. Dimethylamino (3b), thiomethyl (3d), bromo (3f), chloro (3g), fluoro (3h), trifluoromethyl (3n), and thiophenyl (3q) groups are well tolerated. Carbonyl (3i–3l) groups are more sensitive. Still, amide (3i), ester (3j), and keto (3k) groups are compatible, and olefins containing these groups were produced with yields in the range of 50–60%17 and high Z to E ratios (Z:E ≥ 9). However, the presence of an aldehyde group reduced the yield to 32%17 and Z:E to 7.4. Alkynes bearing the nitrile (3m) and pyridine groups (3r) reacted properly only in the presence of 40 mol % N,N,N′,N′-tetramethylethylenediamine (TMEDA), presumably due to the coordination of nitrile and pyridine groups to the Fe ion that could be alleviated by TMEDA coordination. Alkynes with sterically congested o-tolyl (3o) and naphthyl (3p) groups are also suitable reaction partners. The reaction protocol allows the use of secondary alkyl iodides with a variety of cyclic and acyclic alkyl groups with different steric properties (3a, 3n, 3r, 4b, 4d–4f, 4i–4l) as well as functionality (4a, 4h, 4m). Gram-scale synthesis (20 mmol) could also be achieved to give synthetically useful yields of Z-olefin (3a). Furthermore, tertiary alkyl iodides reacted equally well to form adamantyl- (4c) and tert-butyl substituted Z-olefins (4g). Alkyl bromides, including bromocyclooctane (5f), 2-bromo-2-methylbutane (5g), and 3-bromo-3-ethylpentane (5h), reacted readily under similar conditions (Figure 3). For other alkyl bromides (Figure 3, 5a–5e, 4g), an increase in the loading of alkyl bromide (5 equiv) and reaction temperature (60 °C) was necessary. The Z:E selectivity in most cases is more than 12:1.
Figure 2.

Z-Olefin synthesis with secondary and tertiary alkyl iodides. The conditions were described in detail in the SI. (a) 4 d. (b) RI (2 equiv), Zn (2 equiv), I2 (3 mol %). (c) RI (3 equiv), Zn (3 equiv), I2 (5 mol %). (d) RI (2.5 equiv), Zn (2.5 equiv), I2 (5 mol %). (e) RI (5 equiv), Zn (5 equiv), I2 (10 mol %). (f) TMEDA (40 mol %) added. (g) FeBr2 (20 mol %), RI (6 equiv), Zn (6 equiv), I2 (10 mol %).
Figure 3.
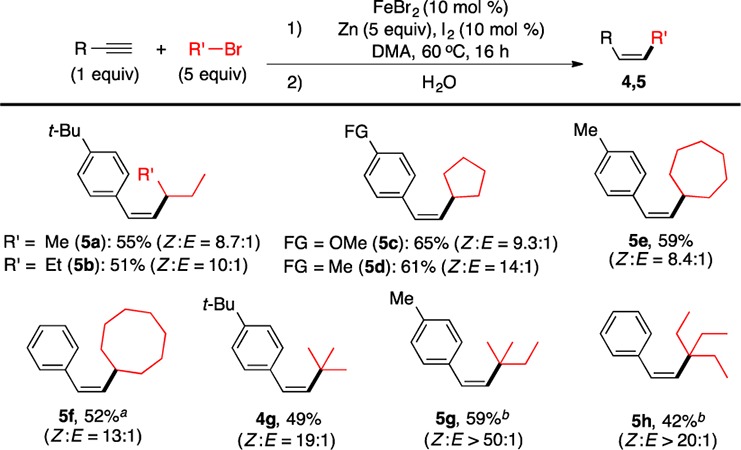
Z-Olefin synthesis with secondary and tertiary alkyl bromides. The conditions were described in detail in the SI. (a) RI (2 equiv), Zn (2 equiv), I2 (3 mol %), rt. (b) RI (3 equiv), Zn (3 equiv), I2 (5 mol %), rt.
The reaction conditions described above are less efficient for the coupling of primary alkyl iodides (Table S7, entries 1 and 2). A reoptimization showed that replacement of I2 by iodotrimethylsilane (TMSI), addition of CuBr2 (10 mol %) as cocatalyst, and higher concentrations of reactants were beneficial. Under these modified conditions, the coupling of 1a with 1-iodoheptane (2b) gave (Z)-non-1-en-1-ylbenzene in 53% yield (compared to about 30% under the protocol in Figure 2) (Table S7, entry 16). The modified protocol was applied to couple a wide range of primary alkyl iodides (Figure 4).18 Alkyl iodides with various chain lengths and isomeric structures did not significantly affect the product yields and Z-selectivity (6a–6c, 6e, 6f). High functional group compatibility was exhibited, tolerating alkyne (6d), ether (6g), chloro (6h), ester (6i), nitrile (6j, 6k), protected alcohol (6l), carbazole (6m), and olefin (6n) moieties. Alkyl tosylates (6q–6s) were also coupled at 60 °C when tetrabutylammonium iodide (1–2 equiv) was used as additive.19 The reaction could be run in gram scales (8–15 mmol) with similar yields (Figure 4, 6a, 6c).
Figure 4.

Z-Olefin synthesis with primary alkyl substrates. The conditions were described in detail in the SI. (a) RI (3 equiv), Zn (3.5 equiv), TMSI (30 mol %). (b) 2 d. (c) RI (5 equiv), Zn (5.5 equiv), TMSI (50 mol %). (d) ROTs (3 equiv), Zn (3.5 equiv), TMSI (30 mol %), TBAI (1 equiv). (e) 60 °C, 3 d. (f) ROTs (5 equiv), Zn (5.5 equiv), TMSI (50 mol %), TBAI (2 equiv).
The Fe-catalyzed olefination method was applied for the synthesis of Z-olefins containing bioactive moieties. Natural groups such as cholestanol, protected galactose, tocopherol (Figure 2, 4n–4p), and menthol (Figure 4, 6o) could be introduced on both the alkyne and alkyl coupling partners. 3,4,5-Trimethoxyphenyl ethylenyl group is a common motif found in the combretastatin family of natural products, which are promising antitumor agents.20Z-Stilbenoids containing the 3,4,5-trimethoxyphenyl group are widely studied. Their alkyl analogues are less encountered probably due to synthetic difficulty. The Fe-based olefination method was applied to prepare 3,4,5-trimethoxystyrenes substituted with a primary, secondary, or tertiary group with decent Z-selectivity (Figure 2, 4q, 4r; Figure 4, 6p). Finally, the Fe catalysis was used to prepare drug-like molecules and their key intermediates. With this method, Z-styrene 7a, an intermediate to a caffeic acid phenethyl ester analogue, which exhibits antiproliferative activity toward carcinoma,21 was synthesized in one step, 44% yield, and 7.6 Z:E selectivity (Figure 5).22 A previous method, using Wittig reaction, gave 25% yield and 3.3 Z:E selectivity. Likewise, the iron catalysis was applied for the synthesis of complex Z-styrenes that offer potential treatments for allergic diseases (Figure 5, 7b and 7c), achieving higher yields and/or Z-selectivity than the corresponding Wittig reactions.23
Figure 5.
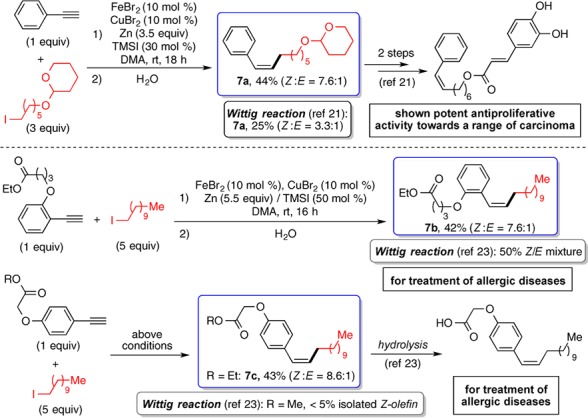
Synthesis of the bioactive molecules or their key intermediates for potential treatments of diseases.
To gain preliminary understanding of the reaction mechanism, several experiments were conducted. When 6-iodohept-1-ene and 6-bromohex-1-ene were used as radical clock substrates, the corresponding ring-cyclized Z-olefins (8a and 8b) were obtained (see Figure S3). (Z)-8-Iodooct-3-ene also reacted to give predominantly the cyclized product (8c) (Figure 6a). When a radical scavenger, 2,2,6,6-tetramethylpiperidineoxy (TEMPO), was added, an alkyl-TEMPO adduct (8d) was obtained while no Z-olefin was formed (Figure S4). These results suggest that an alkyl radical is generated in the reaction. The reaction was conducted in the presence of triethyl phosphite, an alkenyl radical trap24 (Figure 6b). Diethyl (E)-(2-cyclohexyl-1-phenylvinyl)phosphonate (8e) was formed, suggesting the formation of an alkenyl radical intermediate. We investigated the source of α-olefinic hydrogen in the final olefin products. The hydrogen might originate from the DMA solvent or water. Thus, a reaction was conducted in DMA-d9 followed by aqueous workup; a parallel reaction was conducted in DMA-H9 followed by workup with deuterium oxide (D2O) (Figure S7). α-Hydro-olefin and α-deuterioenriched olefin were formed from these two reactions, respectively, suggesting that water was the source of the olefinic hydrogen. We hypothesized that this proton was transferred by protonation of an alkenyl anion with water. To test this hypothesis, an electrophilic iodinating reagent, IBr, was added at the end of the reaction. The corresponding iodo-olefin (8f) was formed as the major product (Figure 6c), supporting the intermediacy of an alkenyl anion. The Fe-catalyzed reaction was also monitored by 1H NMR spectroscopy (Figure S9a). An alkenyl species was observed, which might be attributed to the alkenylzinc intermediate. A small amount of Z-olefin product was also observed before aqueous workup. The proton source of this olefin might be the trace amount of water contaminant in the reaction medium or the terminal alkyne itself.25 When the reaction was carried out under “extra-dry” conditions, a similar amount of olefin was produced before aqueous workup (Figure S9b), suggesting that water contaminant was not the main proton source. When the reaction was conducted using phenylacetylene-d1 (80% D) as the substrate, D-incorporation in the internal α-position of the olefin product was observed (Figure S9c,d). Thus, the terminal alkyne substrate itself was the proton source to partially protonate the alkenylzinc intermediate during the reactions. Since zinc can react with alkyl halides to give alkylzinc reagents,26 we used alkylzinc reagents in the reaction protocols (Figure S10). However, only trace amounts of Z-olefins were formed, excluding the intermediacy of in situ formed alkylzinc reagents. Iron-catalyzed isomerization of E-olefin to Z-olefin also did not occur under the reaction conditions (Figure S11).
Figure 6.

Mechanistic study. (a) Radical clock experiment. (b) Trapping of alkenyl radical. (c) Trapping of alkenyl anion. (d) Proposed mechanism.
Based on the above results, we propose a tentative catalytic cycle of the iron-catalyzed Z-olefin synthesis (Figure 6d). The reaction starts by reduction of the Fe(II) catalyst with Zn to form an Fe(I) intermediate (step i),15,27 which reacts with alkyl iodide to form an alkyl radical via single electron transfer (steps ii and iii).15 The alkyl radical attacks the sterically less hindered terminal carbon of arylalkyne to form a linearized alkenyl radical (I) due to the resonance stabilization of π-type radical by the aromatic ring (step iv).28 The linear geometry of (I) allows a Fe(I) ion to attack more favorably the sterically less hindered side of π-radical [bottom side of (I)], forming an Fe(II)-alkenyl complex (II) in which the aryl and alkyl groups are cis to one another (step v).28 The subsequent Fe–Zn exchange leads to an alkenylzinc species (III) and regenerates the Fe(II) catalyst (step vi). Protonation of the alkenylzinc species by water, and to a less degree, by terminal alkyne, furnishes the Z-olefin (step vii).29 For coupling of primary alkyl iodides, CuBr2 is proposed to stabilize the reactive primary alkyl radical via the formation of a Cu-alkyl species,30,31 thereby increasing the yields of reactions.
The main limitation of this Fe-catalyzed method is the scope of alkynes. For the moment, only terminal arylalkynes can react efficiently. The coupling of terminal alkylalkynes and internal arylalkynes only gave trace amounts of desired products. While a wide range of nonactivated alkyl halides were suitable substrates, activated alkyl halides, such as benzylic/allylic bromides and α-bromoketones, did not react to give the desired Z-olefins but underwent reductive dimerization under the catalytic conditions. Thus, the current method is not yet as general as Wittig reaction, cross-coupling, or semihydrogenation. However, the high Z-selectivity and the simple and practical conditions provided by this method make it a novel and valuable approach for the synthesis of Z-β-alkylstyrenes.
Acknowledgments
This work is supported by a European Research Council (ERC) starting grant (no. 257096) and by the Swiss State Secretariat of Education, Research, and Innovation (C13.0038).
Supporting Information Available
Experimental and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Williams J. M. J.Preparation of Alkenes: A Practical Approach; Oxford University Press: Oxford, U.K., 1996. [Google Scholar]
- Oger C.; Balas L.; Durand T. Chem. Rev. 2013, 113, 1313–1350. [DOI] [PubMed] [Google Scholar]
- Siau W.-Y.; Zhang Y.; Zhao Y.; Galano J.-M. Top. Curr. Chem. 2012, 327, 33–58. [DOI] [PubMed] [Google Scholar]
- For recent examples of Wittig-type reactions, see:; a Gu Y.; Tian S.-K. Top. Curr. Chem. 2012, 327, 197–238. [DOI] [PubMed] [Google Scholar]; b Dong D.-J.; Li H.-H.; Tian S.-K. J. Am. Chem. Soc. 2010, 132, 5018–5020. [DOI] [PubMed] [Google Scholar]
- a Hegedus L. S.; Söderberg B. C. G.. Transition Metals in the Synthesis of Complex Organic Molecules; University Science Books: Sausalito, CA, 2009. [Google Scholar]; b Beletskaya I. P.; Cheprakov A. V. Chem. Rev. 2000, 100, 3009–3066. [DOI] [PubMed] [Google Scholar]; c Negishi E.-I.; Huang Z.; Wang G.; Mohan S.; Wang C.; Hattori H. Acc. Chem. Res. 2008, 41, 1474–1485. [DOI] [PubMed] [Google Scholar]
- Various Z-alkenylmetal reagents have been used in cross coupling reactions leading to Z-olefins. For example, see:; a Wilke G.; Müller H. Ann. Chem. 1960, 629, 222–240. [Google Scholar]; b Alexakis A.; Normant J.; Villiéras J. Tetrahedron Lett. 1976, 38, 3461–3462. [Google Scholar]; c Takami K.; Yorimitsu H.; Oshima K. Org. Lett. 2002, 4, 2993–2995. [DOI] [PubMed] [Google Scholar]; d Akiyama K.; Gao F.; Hoveyda A. H. Angew. Chem., Int. Ed. 2010, 49, 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hatakeyama T.; Nakagawa N.; Nakamura M. Org. Lett. 2009, 11, 4496–4499. [DOI] [PubMed] [Google Scholar]; f Krasovskiy A. L.; Haley S.; Voigtritter K.; Lipshutz B. H. Org. Lett. 2014, 16, 4066–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchilla R.; Nájera C. Chem. Rev. 2014, 114, 1783–1826. [DOI] [PubMed] [Google Scholar]
- For the recent examples of Z-selective semihydrogenation of alkynes, see:; a Drost R. M.; Bouwens T.; van Leest N. P.; de Bruin B.; Elsevier C. J. ACS Catal. 2014, 4, 1349–1357. [Google Scholar]; b Slack E. D.; Gabriel C. M.; Lipshutz B. H. Angew. Chem., Int. Ed. 2014, 53, 14051–14054. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gieshoff T. N.; Welther A.; Kessler M. T.; Prechtl M. H. G.; von Wangelin A. J. Chem. Commun. 2014, 50, 2261–2263. [DOI] [PubMed] [Google Scholar]
- Handbook of metathesis; Grubbs R. H., Ed.; Wiley-CH: Weinheim, Germany, 2003. [Google Scholar]
- For recent examples on Z-selective olefin metathesis, see:; a Bronner S. M.; Herbert M. B.; Patel P. R.; Marx V. M.; Grubbs R. H. Chem. Sci. 2014, 5, 4091–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mangold S. L.; O’Leary D. J.; Grubbs R. H. J. Am. Chem. Soc. 2014, 136, 12469–12478. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang H.; Yu E. C.; Torker S.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2014, 136, 16493–16496. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Koh M. J.; Khan R. K. M.; Torker S.; Yu M.; Mikus M. S.; Hoveyda A. H. Nature 2015, 517, 181–186. [DOI] [PubMed] [Google Scholar]
- a Murakami K.; Yorimitsu H. Beilstein J. Org. Chem. 2013, 9, 278–302. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mueller A. J.; Jennings M. P. A. Org. Lett. 2007, 9, 5327–5329. [DOI] [PubMed] [Google Scholar]; c Fujii Y.; Terao J.; Kambe N. Chem. Commun. 2009, 1115–1117. [DOI] [PubMed] [Google Scholar]; d Kambe N.; Moriwaki Y.; Fujii Y.; Iwasaki T.; Terao J. Org. Lett. 2011, 13, 4656–4659. [DOI] [PubMed] [Google Scholar]
- For recent works on Fe-catalyzed reductive coupling with C=C double bonds, see:; a Greenhalgh M. D.; Thomas S. P. ChemCatChem. 2014, 6, 1520–1522. [Google Scholar]; b Jones A. S.; Paliga J. F.; Greenhalgh M. D.; Quibell J. M.; Steven A.; Thomas S. P. Org. Lett. 2014, 16, 5964–4967. [DOI] [PubMed] [Google Scholar]; c Lo J. C.; Gui J.; Yabe Y.; Pan C.-M.; Baran P. S. Nature 2014, 516, 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fe-catalyzed reductive coupling between ArBr and alkenyl bromide using Mg as reductant has been reported, see:Czaplik W. M.; Mayer M.; von Wangelin A. J. ChemCatChem 2011, 3, 135–138. [Google Scholar]
- a Fürstner A.; Martin R. Chem. Lett. 2005, 34, 624–629. [Google Scholar]; b Sherry B. D.; Fürstner A. Acc. Chem. Res. 2008, 41, 1500–1511. [DOI] [PubMed] [Google Scholar]
- a Fürstner A.; Martin R.; Krause H.; Seidel G.; Goddard R.; Lehmann C. W. J. Am. Chem. Soc. 2008, 130, 8773–8787. [DOI] [PubMed] [Google Scholar]; b Adams C. J.; Bedford R. B.; Carter E.; Gower N. J.; Haddow M. F.; Harvey J. N.; Huwe M.; Cartes M. Á.; Mansell S. M.; Mendoza C.; Murphy D. M.; Neeve E. C.; Nunn J. J. Am. Chem. Soc. 2012, 134, 10333–10336. [DOI] [PubMed] [Google Scholar]
- For alkyl halides, reduction and homocoupling occurred to some degree. For terminal alkynes, no reduction or dimerization was observed. The conversion of alkynes was between 80 and 100%. The isolation and purification of products were straightforward.
- The low to moderate yields for arylalkynes bearing carbonyl groups might be partially due to pinacol coupling reaction.
- The modified protocol is applicable to the coupling of secondary and tertiary alkyl halides as well, having similar efficiency as the unmodified protocol.
- Sun C.-L.; Krause H.; Fürstner A. Adv. Synth. Catal. 2014, 356, 1281–1291. [Google Scholar]
- Tron G. C.; Pirali T.; Sorba G.; Pagliai F.; Busacca S.; Genazzani A. A. J. Med. Chem. 2006, 49, 3033–3044. [DOI] [PubMed] [Google Scholar]
- Nagaoka T.; Banskota A. H.; Tezuka Y.; Saiki I.; Kadota S. Bioorg. Med. Chem. 2002, 10, 3351–3359. [DOI] [PubMed] [Google Scholar]
- The coupling of caffeic acid 6-iodohexyl ester would be a more direct method to prepare the target compound. However, the enoate and phonol groups might not be compatible. Moreover, an excess of alkyl halide is required in this step, which is unfavorable for using a highly functionalized alkyl halide such as caffeic acid 6-iodohexyl ester.
- Buckle D. R. (Beecham Group p.l.c., England) Novel Compounds. U.S. Patent US4713486, December 15, 1987.
- Jiao X.-Y.; Bentrude W. G. J. Am. Chem. Soc. 1999, 121, 6088–6089. [Google Scholar]
- The pKa of acidic C(sp)-H by phenylacetylene and water in DMSO are 28.8 and 31.4, respectively (http://www.chem.wisc.edu/areas/reich/pkatable/) (accessed on March 26, 2015).
- Huo S. Org. Lett. 2003, 5, 423–425. [DOI] [PubMed] [Google Scholar]
- The reduction of FeBr2 to Fe(0) particle could not be ruled out; see ref (8c).
- Wille U. Chem. Rev. 2013, 113, 813–853. [DOI] [PubMed] [Google Scholar]
- Water (5 equiv) was added in the reaction mixture before the reaction started. However, this addition quenched the catalysis. Excess HCl (aq) was added after the reaction to promote the protonation of alkenylzinc, but the yield of Z-olefin was only ∼80% (GC yield), which is ∼10% less than the yield using the standard procedure.
- Zhang N.; Samanta S. R.; Rosen B. M.; Percec V. Chem. Rev. 2014, 114, 5848–5958. [DOI] [PubMed] [Google Scholar]
- The Fe/Co co-catalyzed carbomagnesiation of alkynes has been reported, see:Shirakawa E.; Yamagami T.; Kimura T.; Yamaguchi S.; Hayashi T. J. Am. Chem. Soc. 2005, 127, 17164–17165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.