

Abstract
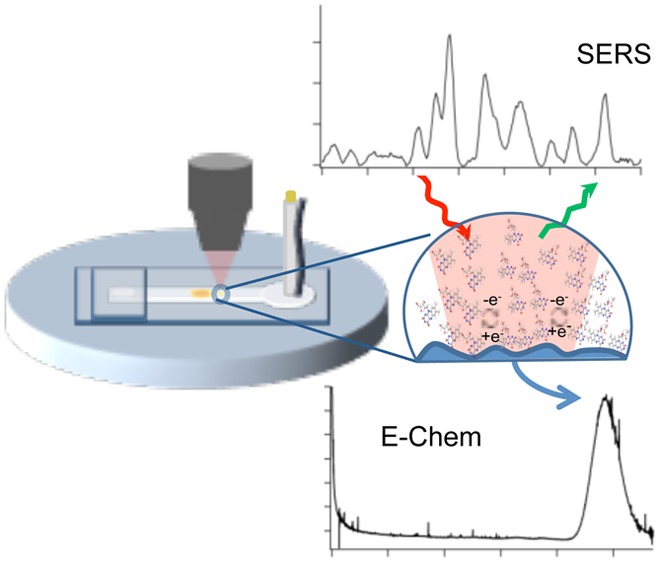
The combination of hydrodynamic focusing with embedded capillaries in a microfluidic device is shown to enable both surface enhanced Raman scattering (SERS) and electrochemical characterization of analytes at nanomolar concentrations in flow. The approach utilizes a versatile polystyrene device that contains an encapsulated microelectrode and fluidic tubing, which is shown to enable straightforward hydrodynamic focusing onto the electrode surface to improve detection. A polydimethyslsiloxane (PDMS) microchannel positioned over both the embedded tubing and SERS active electrode (aligned ∼200 μm from each other) generates a sheath flow that confines the analyte molecules eluting from the embedded tubing over the SERS electrode, increasing the interaction between the Riboflavin (vitamin B2) and the SERS active electrode. The microfluidic device was characterized using finite element simulations, amperometry, and Raman experiments. This device shows a SERS and amperometric detection limit near 1 and 100 nM, respectively. This combination of SERS and amperometry in a single device provides an improved method to identify and quantify electroactive analytes over either technique independently.
Microfluidic-based devices are increasingly being used for analysis in biological, chemical, and biomedical applications.1,2 Reducing dimensions down to the micrometer scale results in numerous advantages including small sample volumes, high throughput detection, and the ability to combine multiple processes. However, there is a strong need for the integration of sensitive detection methods into these microfluidic devices. Detection methods in microfluidics include optical, electrochemical, and mass spectrometric detection.3,4 The most widely used techniques are electrochemical and optical.
Electrochemical detection is a popular technique for a wide range of analytes due to the small sample volumes required, the low cost, and the high sensitivity it provides. A range of methods can be used including amperometry,5−7 potentiometry,8 and conductometry.9 These electrochemical methods provide great quantitative information about the electroactive species being examined. The drawback is the lack of chemical specificity. The exact chemical identity of the analyte is difficult to discern.
Optical detection offers complementary information to that of electrochemical techniques. The common methods include fluorescence,10 absorbance,11 luminescence,12 surface plasmon resonance (SPR),13 and surface-enhanced Raman spectroscopy (SERS).14−18 All of these techniques offer unique advantages to one another. SERS has become an attractive method due to the ability to identify analytes from unique Raman signatures. The signal enhancements associated with SERS provide low detection limits, while its insensitivity to water facilitates detection in many environments,19 making it a useful tool for examining species in situ.
Sensitive SERS detection in a microfluidic device, with flow, remains difficult due to the distance dependence of the localized surface plasmon resonance (LSPR) that originates from metallic nanostructures.20,21 The analyte of interest must be located near the enhancing surface to gain any SERS signal, and that signal decays rapidly. This problem is inconsequential when dropping a solution onto a metallic nanostructure and allowing it to evaporate. In this case, the molecules are adsorbed to the surface, but in solution, the analyte has the ability to diffuse away from the surface.22 Adding flow to the system limits the time the analyte is near the metallic nanostructure, further limiting detection and typically requiring micromolar or larger concentrations.23
The challenge with SERS detection in flow is improving the interaction between the metallic nanostructure and the analyte. One approach is to mix the sample with nanocolloids,15−18,24−27 but this has drawbacks associated with spectral reproducibility and sophisticated mixing schemes.28 Planar substrates can avoid these problems,29 but the limit of detection is controlled by transport of the analyte to the surface. Recently, a simple and effective approach for SERS detection in solution was demonstrated for a capillary-based system using hydrodynamic focusing to promote analyte–substrate interactions.30 The analyte slowly exiting a capillary is confined within a region above the SERS surface by a faster moving sheath fluid in a macroscopic flow channel. This region is defined by the flow rate ratio between sheath flow and sample streams. The technique is commonly used in flow cytometry,31 capillary electrophoresis with laser-induced fluorescence,32,33 and other detection methods. This strategy has been successfully used for online SERS detection with capillary zone electrophoresis (CZE) to separate the structural isomers of rhodamine34 and the detection of the 20 proteinogenic l-amino acids35 and peptides.36
The increased control of fluid dynamics and sampling volumes intrinsic to microfluidic devices suggests a route to further improvements. A unique method to fabricate microchip devices with integrated electrodes was shown using both epoxy and polystyrene to encapsulate the electrode. These devices integrate multiple components into a simple microfluidic platform including microchip electrophoresis with electrochemical detection and the investigation of cellular processes using both optical and electrochemical methods.37−40 The detection limits are improved with the deposition of metals onto the embedded electrodes to create pillars, increasing the surface area of the electrodes.37 This approach, incorporating both encapsulated microelectrodes and fluidic tubing,41 was shown to have superior analytical performance when compared to other fluidic connections with considerable dead volume.
Incorporating hydrodynamic focusing into a microfluidic platform with embedded fluidic tubing and a SERS active electrode suggests a new route to ultrasensitive detection. The combination of SERS and electrochemical experiments allows a unique advantage of combining multiple detection methods to obtain complementary information about analytes. In this study, the combination of finite element analysis, amperometry, and Raman experiments is used to demonstrate improved electrochemical and SERS detection of the redox-active metabolite riboflavin. The results show the SERS activity of the deposited electrodes and the ability to detect riboflavin at low concentrations during amperometric experiments by confining it onto the SERS-active electrode using hydrodynamic focusing.
Experimental Section
Materials and Reagents
Device and Electrode Fabrication
The fabrication of the polystyrene-encapsulated electrodes was performed as previously reported37 with the addition of embedded fluidic tubing.41 Figure 1 illustrates the resulting device. Using colloidal silver, a 25 μm gold wire electrode was connected to an extending wire and shrink tubing was fastened around the two for security. Alignment holes for the tubing and electrodes were punched into an aluminum weighing dish with a syringe needle. To ensure the gold electrode was against the outside wall of the fused silica capillary tubing, the gold wire was wrapped around the capillary and both were threaded through the hole. By adjoining the electrode and tubing, this allowed for the electrode to be approximately 100–200 μm away from the i.d. of the capillary. To prevent polystyrene powder from clogging the capillary while heating, a PDMS plug was used to seal the capillary opening on the backside of the weighing dish. Polystyrene powder was then poured into the dish, which was heated to 250 °C until the powder was completely melted. The device was left to cool until reaching room temperature before removing from the hot plate and weighing dish. The encapsulated gold (Au) electrode was made SERS-active by electrodepositing silver (Ag) onto the surface. A PDMS reservoir was positioned around the electrode, and the Ag plating solution was placed inside. A potential of −1.2 V vs Ag/AgCl was applied for 200 s to form the roughened surface. In situ Raman measurements were performed using a 250 μm wide by 100 μm high channel molded in a thin film of PDMS. A 20:1 mixture of the elastomer base was poured onto a silicon master and heated at 75 °C for 1 h. The PDMS chip was cut to form a reservoir for solution collection and positioned over the SERS-active electrode in line with the capillary and SERS-active electrode (Figure 1c).
Figure 1.
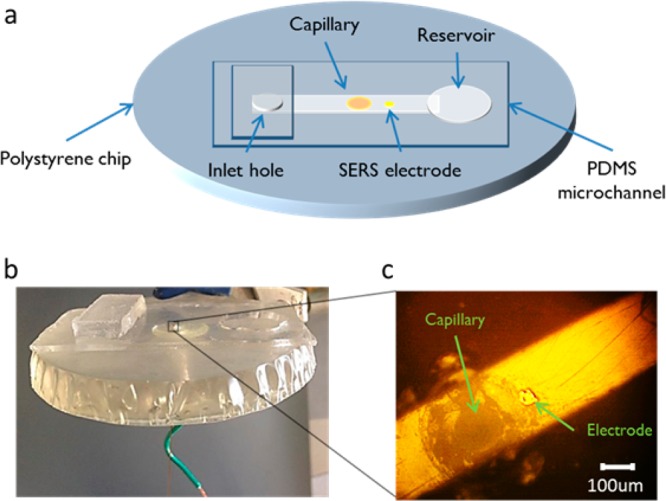
(a) Diagram of polystyrene chip with embedded 150 μm capillary and 25 μm SERS-active electrode. The addition of a 250 μm PDMS channel over the top allows for solution flow and in situ experiments. (b) Shows the actual chip while (c) is a brightfield image showing the capillary and electrode in the microchannel.
Raman Measurements
Raman spectroscopy was performed using a previously described home-built system42 along with a commercial Raman microscope (InVia, Renishaw, Inc.). Laser excitation for both was provided by a 632.8 nm HeNe laser. Flow experiments were completed using the home-built system. The sample was illuminated through a 40× water immersion objective (Olympus, NA = 0.8), and the power measured at the sample was 1 mW. Raman scattering was collected through the same objective and transmitted back to the spectrograph and EMCCD (Newton 970, Andor). Spectra were recorded in kinetic series with varying acquisition times. Maps of the SERS electrodes were done with the commercial Raman microscope. A 20× objective (Leica, NA = 0.4) was used to illuminate the sample, and the laser power measured at the sample was ∼0.8 mW. A spectrum was recorded at each point of the map with a 1 s acquisition time.
Electrochemical Measurements
Electrochemical measurements were made using a CH Instruments Model 660D Potentiostat. The embedded electrode functions as the working electrode with a platinum wire auxiliary electrode and an Ag/AgCl reference electrode. Both electrodes are placed at the end of the microchannel, in a reservoir. All potentials in this manuscript are referenced versus Ag/AgCl and 0.1 M NaOH is the supporting electrolyte. Cyclic voltammetry and riboflavin (diffusion coefficient of 4.0 × 10–6 cm2/s43) were used to determine the surface area of both the bare Au microelectrode and the surface after the deposition of Ag. The scan rate was 0.1 V/s and the potential was swept from −0.3 to −1.0 V. The concentration of riboflavin (in a 0.1 M NaOH supporting electrolyte) was 5 mM.
Electrode Roughening
The Raman signal of the deposited Ag SERS electrode was improved by running an oxidation–reduction cycle (ORC), with flow, to electrochemically roughen the surface and remove any contaminants. The roughening was done in 0.1 M NaOH at a flow rate of 10 μL/min. To perform the ORC, the potential was swept from −0.5 to 0.6 V at 5 mV/s for 3 scans. The PDMS microchannel was removed and the electrode was rinsed with nanopure water.
Flow Assembly
Figure S-1 in the Supporting Information shows a schematic diagram of the experimental setup for the flow experiments. The PDMS microchannel is positioned over both the encapsulated capillary and electrode so that the analyte of interest can be delivered into the detection regime on the electrode. Solution is driven via a syringe pump (Model NE-1000, New Era Pump Systems Inc., Farmingdale, NY) through both the inlet-hole in the PDMS channel and the capillary. Hydrodynamic focusing of the capillary solution is achieved by pumping the sheath flow continuously through the inlet. The sample is injected through a Valco 4-port injector with an internal 100 nL sample loop (Vici) and enters the channel through the capillary. The liquid collects at the end of the flow channel in a reservoir cut out of the PDMS where the counter and reference electrodes are positioned to allow for electrochemical detection. When placed under the home-built microscope system, simultaneous spectro-electrochemical experiments can be completed.
COMSOL Simulations
Commercial finite element analysis software, Comsol Multiphysics 4.4a (COMSOL Inc., Burlington, MA), was used to model the fluid dynamics inside the microchannel. A 3-D model of the flow channel was designed in the CAD setting of the program. The geometry consisted of a cylindrical tube intersecting with a rectangular channel with dimensions matching the capillary and microchannel. Laminar flow was modeled using Navier–Stokes equations at steady state. The mass transport of the solute molecules inside the capillary and microchannel was modeled using Fick’s law for steady state transport. A concentration boundary condition of 1 mM was applied to the capillary (sample) inlet and 0 mM to the inlet of the microchannel.
Results
SERS Electrode Characterization
The SERS-active electrode was formed by electrodepositing Ag onto an Au microelectrode embedded into a polystyrene chip. The surface area of the deposited SERS electrode was determined using cyclic voltammetry and the Randles-Sevcik equation as follows:44
Voltammograms of 5 mM riboflavin were completed, and the peak current was used to calculate the area before and after the electrodeposition. The resulting area after a 200 s electrodeposition of silver is 1.59 × 10–5 cm2, which is 3 times larger than with the flat microelectrode (5.00 × 10–6 cm2). The height of the electrode is ∼2 μm.
To assess the SERS activity of the deposited electrode, a self-assembled monolayer of thiophenol was formed on the surface and a Raman map of the electrode was obtained as shown in Figure 2a–d. Figure 2a shows the brightfield image with the corresponding mapped section highlighted. Several characteristic bands associated with thiophenol are seen at 1000, 1022, and 1075 cm–1 in Figure 2d. Figure 2c shows a map of the band area at 1075 cm–1. The Raman map demonstrates that the highest signals occur around the edges of the electrode surface. Furthermore, the strong scattering around the edges of the electrode can be seen from the darkfield image in Figure 2b.
Figure 2.
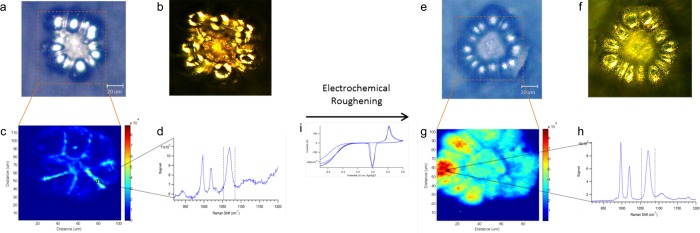
A SERS-active electrode is formed by electrodepositing silver onto a gold microelectrode. The left portion shows the (a) brightfield, (b) darkfield, (c) heatmap of the 1075 cm–1 band area in thiophenol, and (d) representative spectrum of thiophenol. The Raman signal was improved by running an oxidation–reduction cycle with flow to electrochemically roughen the surface. The right figure demonstrates the results with the (e) brightfield, (f) darkfield, (g) heatmap, and (h) spectrum. The heatmap illustrates not only an increase in the overall signal but also a rise with relative signal uniformity across the surface. The cyclic voltammogram from the ORC is shown in (i).
Electrochemically pretreating the surface using an oxidation–reduction cycle (ORC) with flow was found to dramatically increase the SERS signal. Figure 2e–h shows the SERS response from a self-assembled monolayer of thiophenol after cycling. The brightfield image with the mapped section highlighted is shown in Figure 2e and the darkfield image in Figure 2f. Comparison of the bright and dark field images before and after electrochemical cycling show subtle differences; however, dramatic differences are observed in the Raman maps. Figure 2g is a map of the 1075 cm–1 band area in thiophenol. The heatmap shows how the overall SERS intensity across the substrate has increased, demonstrating an improved SERS response is obtained with electrochemical cycling after metal deposition. ORC in chloride has been traditionally used to roughen silver electrodes;45 the pretreatment here may act to either roughen or displace adsorbed CN– from the electroplating process.
Depositing and electrochemically cycling silver produces a highly enhancing SERS-active electrode.45−47 The identity of the underlying wire does not seem to matter, as we have successfully formed SERS microelectrodes with gold, copper, silver, and other embedded wires. Multiple electrode materials can be embedded into the polystyrene,38 and either silver or gold can be deposited to observe SERS. The substrate can also be regenerated easily by polishing down the surface and electrodepositing new nanostructures.
Modeling of Hydrodynamic Focusing in the Microfluidic Device
To determine the confinement effects from sheath flow in this experimental setup, we modeled our configuration using COMSOL simulations. These simulations model the interaction between a sheath flow and a sample eluting from the capillary embedded in the polystyrene base. The ratio of the sheath liquid to the sample liquid was varied to see how the sample flow is confined. The geometry consisted of a cylindrical tube intersecting with a rectangular channel. The dimensions of both matched that of the capillary (150 μm i.d.) and microchannel (250 μm wide by 100 μm high).
Figure 3 presents the results of the COMSOL simulations from a view normal to the surface in the xz-plane (left) and a top-view in the xy-plane (right). The first image in each row diagrams the position of the electrode relevant to the sample eluting from the capillary under the influence of the sheath flow. The SERS microelectrode position is limited by the outer diameter of the embedded capillary. The experiment was modeled with a constant sample flow rate of 1 μL/min, while increasing the sheath flow to sample flow rate ratio from 1:1 to 2:1, 5:1, 10:1, 20:1, and 30:1, respectively. Figure S-2, Supporting Information, shows wide-field fluorescence images from the top-view of the capillary with Rhodamine-6G eluting at different flow ratios, which help substantiate the xy-plane of the simulations.
Figure 3.

xz (left) and xy (right) view of the fluid dynamics inside the flow channel modeled using COMSOL. A schematic representation of the simulations with the location of the capillary and microelectrode is on top of each view, followed by varying sheath to capillary flow ratios. The concentration intensity scales from zero (blue) to 1 mM (red) in these simulations.
The confinement of the analyte relative to the position of the SERS electrode changes as the sheath flow rate increases. Using a 1:1 sheath to capillary flow ratio, the simulation predicts little confinement of the analyte at the SERS electrode. The most concentrated portion of the stream does not interact near the area where the electrode is positioned. The optimum sample confinement occurs at a sheath flow to sample flow ratio of 5:1. At this flow rate ratio, there is sufficient confinement to maximize the sample concentration to 200 μm after the capillary, which is well within where the SERS-active electrode is positioned. When the flow ratio is 10:1 or greater, the COMSOL simulations seem to show a rapid mixing of the two flows, leading to much quicker dilution of the eluting analyte.
Depending on the ratio between the sheath and capillary flow rates, the concentration of the analyte decays rapidly from the capillary inlet. Figure S-3, Supporting Information, shows how this signal decays at the different flow ratios, on the basis of the distance from the capillary. The highlighted region is the approximate distance that the electrode can be placed. In that region, at a 5:1 flow ratio, the concentration is ∼0.987 mM, still near the 1 mM initial concentration modeled in the simulation. This indicates that the SERS electrode is close enough to benefit from the focusing effect. The average concentration at each flow ratio within the electrode region is shown in Figure 4a, plotted versus the sheath to capillary flow rate, showing the sample concentration at the SERS electrode is optimized at the 5:1 flow ratio.
Figure 4.
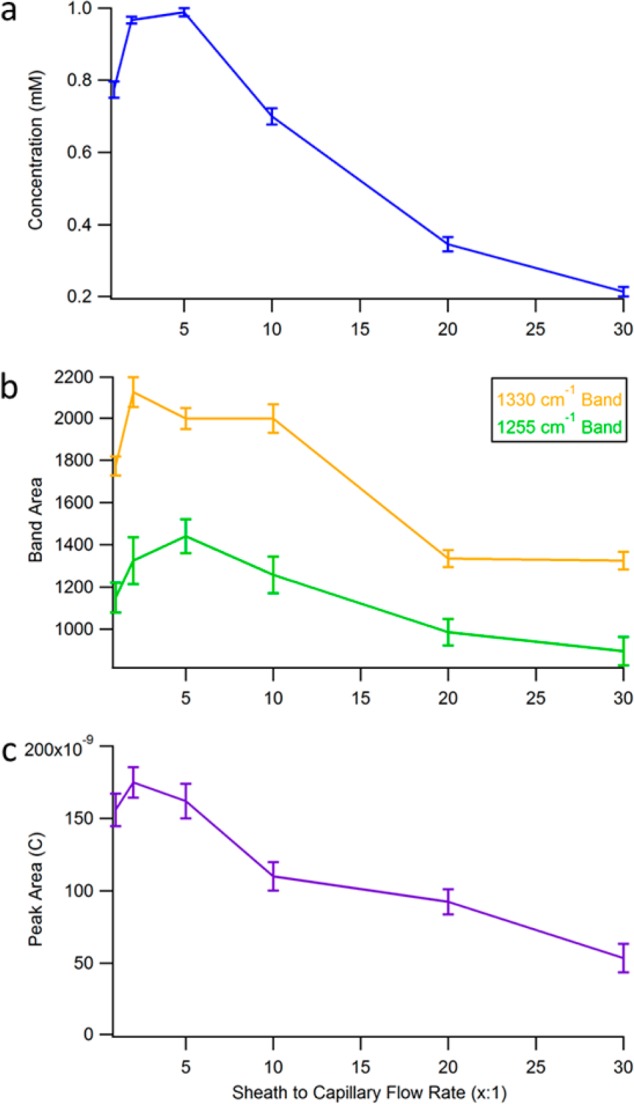
A comparison of the (a) COMSOL simulation, (b) amperometry, and (c) SERS detection results with varying sheath to capillary flow rates demonstrating the focusing effect. (a) Shows a plot of the average concentration within the electrode regime at each flow ratio from the simulations. (b) Shown here is a plot of the band area of two characteristic frequencies of 1.0 μm riboflavin, 1255 and 1330 cm–1, versus the flow ratio. (c) A plot of the area of the amperometric peak obtained from 100 nL injections of 100 μM riboflavin at varying flow ratio is shown. All three plots demonstrate that the optimal sample confinement comes from a flow ratio of between 2:1 and 5:1.
The model indicates that using a sample capillary embedded within the bottom of a larger microfluidic channel can produce flow focusing in three dimensions. The position of the electrode with respect to the outer diameter of the sample capillary limits the obtainable focusing; however, the simulation predicts 98.7% of the eluted sample concentration is maintained at the optimum flow rate ratio.
On-Chip Sheath-Flow SERS and Amperometric Detection
Raman measurements were obtained at varying sheath flow rates to determine the effect on the observed SERS signal. 1.0 μM riboflavin was eluted from the capillary with varying sheath to capillary flow rates and the signal measured. Figure 4b shows the absolute band areas at 1255 and 1330 cm–1 from the SERS spectrum of riboflavin, plotted as a function of sheath flow rate. The bands used are associated with the ring III stretching modes coupled with a N–H bending mode and ring II modes of riboflavin, respectively.48,49
The trend demonstrates that the highest SERS intensity is observed at a sheath flow to sample flow rate ratio near 5:1. This agrees with the COMSOL simulation results in Figure 3a. As the sheath flow is increased further, there is a noticeable drop in the SERS intensity that is nonlinear. The drop matches the COMSOL simulations.
The focusing effect on electrochemical detection was also studied. The reduction of riboflavin in the sample was monitored by amperometry. Amperometric detection was performed with 100 nL injections of 100 μm riboflavin injected into the microchannel at varying sheath to capillary flow rates while the electrode was held at a constant potential of −1.2 V. The faradaic current is related to the charge transferred across the electrified interface as a result of the reaction and can be expressed by the equation as follows:44
where Q is coulombs (or Amperes/second), n is the number of electrons transferred, F is Faraday’s constant, and N is the number of moles. Increased current corresponds to improved transport and more analyte molecules reacting at the electrode surface.50−53
The charge passed at the electrode was determined by integrating the peak area in each amperogram and plotted versus the flow ratio, as shown in Figure 4c. The plot shows an increase from the 1:1 to the 2:1 ratio. After, there is a steady, nonlinear decrease in the peak area. The results observed indicate that the highest currents are seen between the 2:1 and 5:1 sheath to capillary flow rates. The trend is consistent with the COMSOL simulations. However, when compared to the SERS intensity plot, while the trend is similar, the signal decreases more rapidly in the electrochemical experiment. This may represent differences in electrochemical vs spectroscopic detection associated with analyte absorption.
SERS and Electrochemical Sensitivity and Limit of Detection
The results from the COMSOL simulations, SERS, and amperometry indicate that a 5:1 sheath to capillary flow ratio is optimal for obtaining the highest signals. Using this ideal flow, the SERS and electrochemical limit of detections were examined.
Amperometric experiments were done at −1.2 V, appropriate for riboflavin reduction. 100 nL injections of riboflavin at varying concentrations were completed at a 5:1 sheath to capillary flow ratio. Figure 5 shows a plot of the peak height versus concentration ranging from 1 mM to 100 nM. Using this platform, the limit of detection (3 × slope/noise) is 89 nM. As expected, the plot shows a linear increase in current with increasing concentration. The slope of the line is 13 nA/mM. This suggests a concentration per electrode area of 1.1 nA/μm radius/mM. For riboflavin (D0 = 4.0 × 10–6 cm2/s43), the steady state current at a disk can be expressed by the equation as follows:44
The maximum current is 1.5 nA/μm radius/mM. This value suggests we are not at steady state and that higher analyte flux to the electrode could further improve detection.
Figure 5.
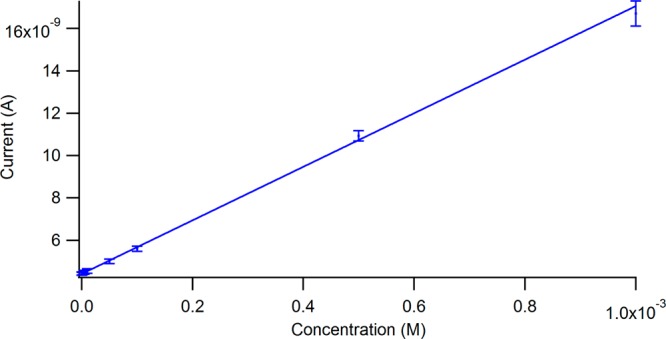
100 nL injections of varying concentrations of riboflavin were examined with a 5:1 sheath to capillary flow rate while applying −1.2 V. Shown is a plot of the peak height versus the concentration. The limit of detection is 100 nM with the focusing effect.
The electrodeposition of silver not only provides a SERS substrate but also increases the surface area by a factor of 3, after a 200 s deposition. This will lead to an increase in current and a lower limit of detection when compared to the flat electrode. The question becomes is the increased electrochemical signal seen a result of a larger surface area alone or also due to the hydrodynamic focusing effect employed. Without embedded tubing or the use of focusing, an examination of catechol resulted in a limit of detection of 600 nM with the flat microelectrode.37 The observation of 100 nM riboflavin here is 6 times lower than that. Experiments suggest surface area accounts for a 3× increase in signal. The additional increase of 2× is consistent with our COMSOL prediction, indicating that focusing the analyte onto the electrode also improves the amperometric limit of detection.
Under the same optimized conditions, SERS experiments were done to access the limit of detection. This study was done concurrently with the amperometry. Figure 6a shows the SERS spectrum at varying riboflavin concentrations with 250 ms acquisition times while applying −1.2 V. The heatmaps in Figure 6b–d show the observed SERS intensity at each Raman shift as a function of time for each 100 nL injection of riboflavin at each corresponding concentration. Figure S-6, Supporting Information, shows a zoom and closer inspection shows the rapid absorption and desorption observed in the SERS signals with time. Consecutive injections showed the same magnitude of SERS intensity, demonstrating the stability of the SERS electrode.
Figure 6.

(a) Spectrum of varying concentrations of riboflavin with a 5:1 sheath to capillary flow rate. −1.2 V is applied. The heatmap showing the SERS intensity as a function of time for the (b) 100 nM, (c) 10 nM, and (d) 1 nM injection of riboflavin.
Using an electrode array embedded in PS devices, concentrations down to 4 nM have been successfully detected electrochemically,54 indicating that a lower limit of detection is possible, albeit it with more electrochemically active species than riboflavin (such as catechol and nitric oxide). The SERS spectra at each concentration show variation in peak intensity commonly associated with SERS; however, the observed peak frequencies are assigned to the flavin moiety (Table S-1, Supporting Information).
Discussion
A common approach to hydrodynamic focusing in microfluidic devices utilizes focusing streams that are on the same plane as other channels in the device.55,56 A unique feature of this PS-based encapsulation approach is that both the electrodes and tubing can be aligned within close proximity and in a manner where they are perpendicular to the resulting chip surface. This results in a 3-dimensional focusing effect, where the incoming flow stream (from an off-chip injector) is focused onto the pillar detection electrode. While there have been other fabrication intensive approaches for 3-dimensional focusing,57 this approach is relatively straightforward and easy to fabricate. For the devices described in this work, the resulting encapsulated base, once developed, can be used for long periods of time, with a PDMS channel being used to seal over the tubing/electrode surface and a fresh electrode surface being made as desired with a polishing step.
The microfluidic device used here for flow analysis has several advantages over the initial sheath-flow SERS detector developed30 and other microchip-based analytical methods that use off-chip injection methods to introduce the analyte. Microfluidic channels provide improved control of flow conditions over the larger flow channel used previously. Additionally, the SERS microelectrode embedded into the polystyrene allows for simultaneous electrochemical and Raman measurements. By positioning embedded tubing within 200 μm of the microelectrode and using a fast moving sheath flow, hydrodynamic focusing can be used to confine the analyte eluting from the capillary over a short distance. The embedded tubing also allows for low dead volume analysis and has been shown to improve the reproducibility and analytical performance when compared to other off-chip methods.41
The inherent drawback to this is that if the SERS electrode is not close enough to the embedded capillary, then no advantages are seen. The flux to the electrode is limited by this geometric constraint. It is important to align the PDMS microchannel over the capillary and electrode. The stream of analyte is focused over a region governed by the ratio between the sheath and capillary flow rates (Figure 3). If the electrode is not centered in the flow channel, then the analyte stream will not be focused onto the electrode where the sensing is done. We have been able to reproducibly get the electrode within 200 μm of the embedded capillary, illustrating the feasibility of this, as well as lining up the capillary and electrode within the channel. Differences in flow sensitivity from experiment to experiment are due to challenges in alignment of the flow channel with the electrode. It is worth noting that by embedding/fixing the sample inlet capillary and detection electrode in polystyrene, realigning the flow channel is straightforward.
A unique advantage to this microfluidic device is the ability to couple SERS with electrochemical quantification. The embedded microelectrode allows for the ability to examine redox active species and gain quantitative information. Electrodeposition of Ag is done to make a roughened surface capable of giving rise to SERS to obtain chemical specific information about the electron transfer events. In SERS, a hotspot can dominate and good quantitative information is difficult to gain at higher concentrations, but since the charge is directly related to the number of molecules using electrochemical techniques, we can obtain that quantitative information.
A challenge for this device is surface heterogeneity. The substrate after electrodeposition is not very uniform, leading to lower enhancements across the surface when compared to other SERS substrates. This can be improved by electrochemically roughening the surface; however, there are still some spots on the surface that do not give rise to much enhancement. Finding the “hotspot” enables detection of concentrations down to 1 nM with SERS; however, this is accompanied by some fluctuations in peak intensity. Other approaches to preparing SERS electrodes may help address this challenge going forward.58
As seen in Figure 6, the signal-to-noise ratio observed in the SERS measurements is very high, even at nanomolar concentrations. This could be a result of several factors. The confinement of the analyte near the SERS electrode by the sheath flow should increase the interaction between the two, leading to higher signals. Another contributor is the time the analyte is interacting with the SERS substrate. The slower moving the sheath fluid, the more time the analyte will be near the substrate. The confinement is related to the overall channel dimensions, such that slower flow rates are sufficient in the microchannel geometry.
Along with the high signal-to-noise ratio, Figure 6 also shows the enhancement of different vibrational bands of riboflavin relative to one another at each concentration. The SERS spectrum of riboflavin is concentration and potential dependent, as well as mode specific.59−63 While the slight shift in some peak positions (Table S-1, Supporting Information) could be explained by concentration-dependent changes,59 the significant change in certain bands being enhanced over others does not appear to be attributed to that. This could result from how the molecule is orientated on the surface during the flow injection experiments. The orientation of the molecule can be affected by the potential applied to the SERS electrode,60−63 and different orientations would result in certain bands being enhanced over others. The surface heterogeneity likely results in a variety of plasmonic environments, which have been shown to alter the observed SERS spectrum.64
Conclusions
A unique microfluidic approach using hydrodynamic focusing and combining SERS with amperometry for high throughput detection has been demonstrated. The microfluidic device maintains the advantages of sheath-flow SERS detection in a compact design. These advantages include fast detection, high throughput, better signal-to-noise, lower limits of detection, and nonfouling of the analyte to the SERS electrode. This means that appreciable signal can be observed with small sample volumes. The addition of a PDMS microchannel over a polystyrene embedded capillary and microelectrode provides a smaller total volume, a reduced dead volume, and a controlled fluidic environment to confine the analyte eluting from the capillary onto the microelectrode for simultaneous SERS and amperometric detection. SERS detection of riboflavin was demonstrated at a concentration of 1 nM, while the electrochemical detection limit is 89 nM. This device suggests a straightforward route to improving trace detection both spectroscopically and electrochemically.
Acknowledgments
The authors acknowledge support from the National Institute of General Medical Sciences Awards R21GM107893 (Z.D.S.) and R15GM084470-03 (R.S.M.) and the University of Notre Dame Advanced Diagnostics and Therapeutics Initiative.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kovarik M. L.; Gach P. C.; Ornoff D. M.; Wang Y.; Balowski J.; Farrag L.; Allbritton N. L. Anal. Chem. 2011, 84, 516–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salieb-Beugelaar G. B.; Simone G.; Arora A.; Philippi A.; Manz A. Anal. Chem. 2010, 82, 4848–4864. [DOI] [PubMed] [Google Scholar]
- Baker C. A.; Duong C. T.; Grimley A.; Roper M. G. Bioanalysis 2009, 1, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasas S. A.; Fogarty B. A.; Huynh B. H.; Lacher N. A.; Carlson B.; Martin R. S.; Vandeveer W. R. I.; Lunte S. M. In Separation Methods in Microanalytical Systems, Kutter J. P., Fintschenko Y., Eds.; Taylor & Francis: Boca Raton, FL, 2006; pp 433–498. [Google Scholar]
- Cai X.; Klauke N.; Glidle A.; Cobbold P.; Smith G. L.; Cooper J. M. Anal. Chem. 2002, 74, 908–914. [DOI] [PubMed] [Google Scholar]
- Woolley A. T.; Lao K. Q.; Glazer A. N.; Mathies R. A. Anal. Chem. 1998, 70, 684–688. [DOI] [PubMed] [Google Scholar]
- Martin R. S.; Gawron A. J.; Lunte S. M.; Henry C. S. Anal. Chem. 2000, 72, 3196–3202. [DOI] [PubMed] [Google Scholar]
- Tantra R.; Manz A. Anal. Chem. 2000, 72, 2875–2878. [DOI] [PubMed] [Google Scholar]
- Galloway M.; Stryjewski W.; Henry A.; Ford S. M.; Llopis S.; McCarley R. L.; Soper S. A. Anal. Chem. 2002, 74, 2407–2415. [DOI] [PubMed] [Google Scholar]
- Ross D.; Gaitan M.; Locascio L. E. Anal. Chem. 2001, 73, 4117–4123. [DOI] [PubMed] [Google Scholar]
- Petersen N. J.; Mogensen K. B.; Kutter J. P. Electrophoresis 2002, 23, 3528–3536. [DOI] [PubMed] [Google Scholar]
- Filanoski B.; Rastogi S. K.; Cameron E.; Mishra N. N.; Maki W.; Maki G. Luminescence 2008, 23, 22–27. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Yu F.; Zare R. N. Lab Chip 2008, 8, 694–700. [DOI] [PubMed] [Google Scholar]
- Connatser R. M.; Cochran M.; Harrison R. J.; Sepaniak M. J. Electrophoresis 2008, 29, 1441–1450. [DOI] [PubMed] [Google Scholar]
- Tong L.; Righini M.; Gonzalez M. U.; Quidant R.; Kall M. Lab Chip 2009, 9, 193–195. [DOI] [PubMed] [Google Scholar]
- Huh Y. S.; Chung A. J.; Cordovez B.; Erickson D. Lab Chip 2009, 9, 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park T.; Lee S.; Seong G. H.; Choo J.; Lee E. K.; Kim Y. S.; Ji W. H.; Hwang S. Y.; Gweon D.-G.; Lee S. Lab Chip 2005, 5, 437–442. [DOI] [PubMed] [Google Scholar]
- Strehle K. R.; Cialla D.; Rösch P.; Henkel T.; Köhler M.; Popp J. Anal. Chem. 2007, 79, 1542–1547. [DOI] [PubMed] [Google Scholar]
- Antonio K. A.; Schultz Z. D. Anal. Chem. 2014, 86, 30–46. [DOI] [PubMed] [Google Scholar]
- Dieringer J. A.; McFarland A. D.; Shah N. C.; Stuart D. A.; Whitney A. V.; Yonzon C. R.; Young M. A.; Zhang X.; Van Duyne R. P. Faraday Discuss. 2006, 132, 9–26. [DOI] [PubMed] [Google Scholar]
- Lal S.; Grady N. K.; Goodrich G. P.; Halas N. J. Nano Lett. 2006, 6, 2338–2343. [DOI] [PubMed] [Google Scholar]
- Asiala S. M.; Schultz Z. D. Chem. Commun. 2013, 49, 4340–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White I. M.; Yazdi S. H.; Yu W. W. Microfluid. Nanofluid. 2012, 13, 205–216. [Google Scholar]
- Wilson R.; Bowden S. A.; Parnell J.; Cooper J. M. Anal. Chem. 2010, 82, 2119–2123. [DOI] [PubMed] [Google Scholar]
- Wang M.; Benford M.; Jing N.; Coté G.; Kameoka J. Microfluid. Nanofluid. 2009, 6, 411–417. [Google Scholar]
- Wang M.; Jing N.; Chou I. H.; Cote G. L.; Kameoka J. Lab Chip 2007, 7, 630–632. [DOI] [PubMed] [Google Scholar]
- Nirode W. F.; Devault G. L.; Sepaniak M. J.; Cole R. O. Anal. Chem. 2000, 72, 1866–1871. [DOI] [PubMed] [Google Scholar]
- Abalde-Cela S.; Abell C.; Alvarez-Puebla R. A.; Liz-Marzan L. M. J. Phys. Chem. Lett. 2014, 5, 73–79. [DOI] [PubMed] [Google Scholar]
- DeVault G. L.; Sepaniak M. J. Electrophoresis 2001, 22, 2303–2311. [DOI] [PubMed] [Google Scholar]
- Negri P.; Jacobs K. T.; Dada O. O.; Schultz Z. D. Anal. Chem. 2013, 85, 10159–10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro H. M.Practical Flow Cytometry; Wiley: New York, 1994. [Google Scholar]
- Cheng Y.; Dovichi N. J. Science 1988, 242, 562–564. [DOI] [PubMed] [Google Scholar]
- Dada O. O.; Huge B. J.; Dovichi N. J. Analyst 2012, 137, 3099–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri P.; Flaherty R. J.; Dada O. O.; Schultz Z. D. Chem. Commun. 2014, 50, 2707–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri P.; Schultz Z. D. Analyst 2014, 139, 5989–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri P.; Sarver S. A.; Schiavone N. M.; Dovichi N. J.; Schultz Z. D. Analyst 2015, 140, 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimovic A.; Johnson A. S.; Kiss I. Z.; Martin R. S. Electrophoresis 2011, 32, 822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A. S.; Anderson K. B.; Halpin S. T.; Kirkpatrick D. C.; Spence D. M.; Martin R. S. Analyst 2013, 138, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. B.; Halpin S. T.; Johnson A. S.; Martin R. S.; Spence D. M. Analyst 2013, 138, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A. S.; Selimovic A.; Martin R. S. Electrophoresis 2011, 32, 3121–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becirovic V.; Doonan S. R.; Martin R. S. Anal. Methods 2013, 5, 4220–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asiala S. M.; Schultz Z. D. Analyst 2011, 136, 4472–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. D.; Renslow R.; Babauta J.; Ahmed B.; Beyenal H. Sens. Actuators, B: Chem. 2012, 161, 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical methods: Fundamentals and applications; Wiley: New York, 1980. [Google Scholar]
- Tuschel D. D.; Pemberton J. E.; Cook J. E. Langmuir 1986, 2, 380–388. [Google Scholar]
- Pettinger B.; Philpott M. R.; Gordon J. G. J. Chem. Phys. 1981, 74, 934–940. [Google Scholar]
- Jeanmaire D. L.; Van Duyne R. P. J. Electroanal. Chem. 1977, 84, 1–20. [Google Scholar]
- Bowman W. D.; Spiro T. G. Biochemistry 1981, 20, 3313–3318. [DOI] [PubMed] [Google Scholar]
- Lee N. S.; Sheng R. S.; Morris M. D.; Schopfer L. M. J. Am. Chem. Soc. 1986, 108, 6179–6183. [Google Scholar]
- Yoon S. K.; Choban E. R.; Kane C.; Tzedakis T.; Kenis P. J. A. J. Am. Chem. Soc. 2005, 127, 10466–10467. [DOI] [PubMed] [Google Scholar]
- Hasenbank M. S.; Fu E.; Nelson J. B.; Schwartz D. T.; Yager P. Lab Chip 2007, 7, 441–447. [DOI] [PubMed] [Google Scholar]
- Xuan J.; Leung M. K. H.; Leung D. Y. C.; Ni M.; Wang H. Int. J. Hydrogen Energy 2011, 36, 11075–11084. [Google Scholar]
- Ebrahimi Khabbazi A.; Richards A. J.; Hoorfar M. J. Power Sources 2010, 195, 8141–8151. [Google Scholar]
- Selimovic A.; Martin R. S. Electrophoresis 2013, 34, 2092–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehlenbrock M. J.; Price A. K.; Martin R. S. Analyst 2006, 131, 930–937. [DOI] [PubMed] [Google Scholar]
- Dziubinski M. In Advances in Microfluidics; Kelly R., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Daniele M. A.; Boyd D. A.; Mott D. R.; Ligler F. S. Biosens. Bioelectron. 2015, 67, 25–34. [DOI] [PubMed] [Google Scholar]
- Alam R.; Lightcap I. V.; Karwacki C. J.; Kamat P. V. ACS Nano 2014, 8, 7272–7278. [DOI] [PubMed] [Google Scholar]
- Liu F.; Gu H.; Lin Y.; Qi Y.; Dong X.; Gao J.; Cai T. Spectrochim. Acta, Part A 2012, 85, 111–119. [DOI] [PubMed] [Google Scholar]
- Xu J.; Birke R. L.; Lombardi J. R. J. Am. Chem. Soc. 1987, 109, 5645–5649. [Google Scholar]
- Wang X.-m.; Yan M.-d.; Zhu J.-j.; Chen H.-Y. J. Electroanal. Chem. 1998, 451, 187–192. [Google Scholar]
- Dendisová-Vyškovská M.; Kokaislová A.; Ončák M.; Matějka P. J. Mol. Struct. 2013, 1038, 19–28. [Google Scholar]
- Abdelsalam M.; Bartlett P. N.; Russell A. E.; Baumberg J. J.; Calvo E. J.; Tognalli N. G.; Fainstein A. Langmuir 2008, 24, 7018–7023. [DOI] [PubMed] [Google Scholar]
- Ye J.; Wen F. F.; Sobhani H.; Lassiter J. B.; Van Dorpe P.; Nordlander P.; Halas N. J. Nano Lett. 2012, 12, 1660–1667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.