

Abstract

Bryostatin 1, a complex macrocyclic lactone isolated from Bugula neritina, has been the subject of multiple clinical trials for cancer. Although it functions as an activator of protein kinase C (PKC) in vitro, bryostatin 1 paradoxically antagonizes most responses to the prototypical PKC activator, the phorbol esters. The bottom half of the bryostatin 1 structure has been shown to be sufficient to confer binding to PKC. In contrast, we have previously shown that the top half of the bryostatin 1 structure is necessary for its unique biological behavior to antagonize phorbol ester responses. Neristatin 1 comprises a top half similar to that of bryostatin 1 together with a distinct bottom half that confers PKC binding. We report here that neristatin 1 is bryostatin 1-like, not phorbol ester-like, in its biological activity on U937 promyelocytic leukemia cells. We conclude that the top half of the bryostatin 1 structure is largely sufficient for bryostatin 1-like activity, provided the molecule also possesses an appropriate PKC binding domain.
Natural products have afforded a treasure trove of potent, biologically active structures, which have served as therapeutic leads, as probes identifying novel biological targets, and as tools for dissecting biological mechanisms.1−9 The bryostatins, isolated from the sea moss Bugula neritina, were initially identified as potential cancer chemotherapeutic agents on the basis of their activity in the P388 leukemia cell system.10,11 Bryostatin 1 has since been the subject of numerous clinical trials for cancer and is beginning clinical trials for Alzheimer’s disease (www.clinicaltrials.gov). Unfortunately, the development of the bryostatins has faced significant obstacles. Their supply has been severely limited due to the scarcity of source material. Their chemical complexity—they are macrocyclic lactones with 11 chiral centers—has made structure–activity studies problematic. The lack of detailed mechanistic understanding has so far precluded the restriction of clinical trials to those cancer patients for whom the underlying molecular basis for their cancer would predict that bryostatin 1 would be a rational therapy.
Protein kinase C (PKC) and six other families of proteins fulfill a critical role in cellular signaling, transducing signals for the many receptor–ligand combinations that trigger activation of phospholipase C isoforms, which in turn leads to the release of the lipophilic second messenger diacylglycerol (DAG).12−15 PKC and these other proteins all contain one or more C1 domains, which act as the recognition module for DAG. DAG inserts into a hydrophilic cleft in the C1 domain, completing a hydrophobic surface at the top of the C1 domain and driving its membrane insertion.16 This membrane insertion is associated with translocation of the protein and, in the case of PKC, conformational change leading to enzymatic activation.
Reflecting the great biological importance of the PKC pathway for cells, organisms have identified multiple potent templates that can function as DAG mimetics, interacting in a similar fashion with C1 domains but doing so with orders of magnitude enhanced potency.17 The phorbol esters, tetracyclic diterpenes, have been most extensively studied, but other examples include the teleocidins, which are indole alkaloids, aplysiatoxin, which is a polyacetate, the iridals, which are triterpenes, and the bryostatins, macrocyclic lactones.17−19 All of these agents act in a similar fashion, binding to and activating PKC.
The initial basis for the intense interest directed at the phorbol esters was that they represented the prototype for tumor promoters, agents that themselves are not carcinogenic but that induce cancer following prior exposure to a single subthreshold dose of carcinogen.20 With the recognition that the phorbol esters act as diacylglycerol analogues, these compounds have proven to be critical tools for understanding the signaling role of PKCs and the other DAG targets. We now appreciate that PKC is an important contributor to cancer at many levels and a promising therapeutic target.14
Bryostatin 1 is unique among C1 domain ligands in that, whereas it binds to and activates PKC in a fashion similar to that of the phorbol esters, paradoxically it fails to induce most typical phorbol ester responses and, if coapplied with phorbol ester, it acts as a functional antagonist to block the phorbol ester action.21 We have recently shown that a series of 12 known PKC activators, which spanned some 8 powers of 10 in logP, showed biological responses that largely differed in degree but not in kind when compared to the unique action of bryostatin 1.22 Of particular importance from a therapeutic perspective, bryostatin 1 was not tumor promoting and inhibited tumor promotion by the phorbol esters.23
Despite the formidable challenges, great progress has been made in recent years with the total synthesis of bryostatin 124,25 as well as in the preparation of structurally simplified bryostatin 1 analogues.26−28 Using binding to PKC as a measure of functional activity, Wender and colleagues showed that dramatic simplification of the upper half of bryostatin 1 could be achieved with retention of binding activity.26,29 They therefore proposed the concept that the upper portion of the bryostatin 1 structure served as a “spacer domain”, which functioned primarily to hold the critical lower portion of the molecule in the correct conformation for binding. These same simplified structures retained biological activity in cell lines in which both phorbol ester and bryostatin 1 were growth inhibitory.
We have focused on a distinct functional issue. The unique behavior of bryostatin 1 among PKC ligands is not that it binds to PKC but rather that it functionally antagonizes many PKC-mediated responses in many systems. We have therefore probed the structural requirements responsible for this unique behavior. Using the U937 promyelocytic leukemia cell system, one of the well-studied systems in which the differential behavior of bryostatin 1 and phorbol ester has been characterized, we showed that the bryologue Merle 23, a bryostatin 1 derivative differing only in the absence of substituents on the top half of the molecule, in fact functioned like phorbol ester, not like bryostatin 1.30 These results indicated that for the unique biological activity of bryostatin 1 the top half was necessary. Left unresolved was whether the top half was sufficient, provided that the top half retained an appropriate conformation and was coupled to a suitable PKC binding domain.
Results and Discussion
Neristatin 1 has provided the opportunity to address this question. Previously, we reported the isolation of neristatin 1 from B. neritina.31 Structurally, it possesses a top half identical to that of bryostatin 1, except for a larger ester group at C7 (Figure 1) (notably, this same group is found in bryostatin 14). The bottom half, however, is substantially rearranged. In bryostatin 1, the C26 secondary hydroxyl group plays a critical role in the C1 domain interactions, fulfilling a role analogous to that of the primary hydroxyl group of DAG or of the primary C20 hydroxyl group of the phorbol ester, forming hydrogen bonds with Leu251 and Thr242 of the C1 domain (numbering as in the C1b domain of PKCδ).32 The adjacent C25 oxygen of bryostatin 1 forms the ester linkage of the macrocycle. In neristatin 1, among other differences, the ring has been expanded by migration of the ester to the C26 oxygen, generating a free secondary hydroxyl group at C25. We have previously characterized the behavior of a bryologue, Merle 43, which shows a similar rearrangement alongside other modifications designed to probe the influence of the B ring on biological activity (Figure 1).33 Despite the differences in the lower half of the molecule, neristatin 1 had still been found to bind to PKC with a Ki = 21.2 ± 1.3 nM.31 This is similar to the Ki for Merle 43 of 13.8 nM33 and compares to the Ki for bryostatin 1 of 0.48 nM.34
Figure 1.

Comparison of the structures of bryostatin 1, neristatin 1, and Merle 43. The structures of the typical phorbol ester PMA and of Merle 23, which differs from bryostatin 1 in the substitutions on the A and B rings, are also included.
After confirming >95% purity of the neristatin 1, we first analyzed its activity for inhibition of growth of Toledo cells (Figure 2). The Toledo cell system provides a convenient measure of activity; it is highly sensitive to phorbol esters, and both phorbol esters and bryostatin 1 cause a similar extent of growth inhibition. Consistent with its PKC binding activity, neristatin 1 inhibited Toledo cell growth with an IC50 of approximately 1.58 ± 0.23 nM. It was thus approximately 17-fold less potent than bryostatin 1 and about 2.5-fold more potent than Merle 43.
Figure 2.
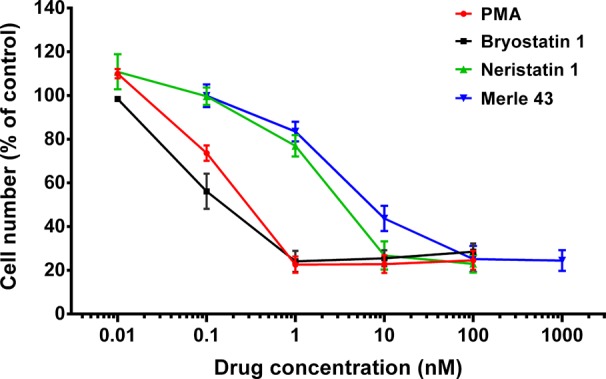
Inhibition of the growth of Toledo cells (72 h treatment). Values are mean ± SEM of triplicate independent experiments.
In the U937 cell system, PMA caused approximately 50% inhibition of cell growth; bryostatin 1 showed very little inhibition (Figure 3A). Neristatin 1 was similar to bryostatin 1, although the extent of growth inhibition was slightly greater. As described before, Merle 43 was like PMA.33 Bryostatin 1 is able to block the inhibition of the U937 cell growth induced by PMA. Likewise, neristatin 1 largely blocked the PMA response. Consistent with the activity of Merle 43 by itself, growth inhibition by the combination of Merle 43 and PMA was similar to that by either alone.
Figure 3.
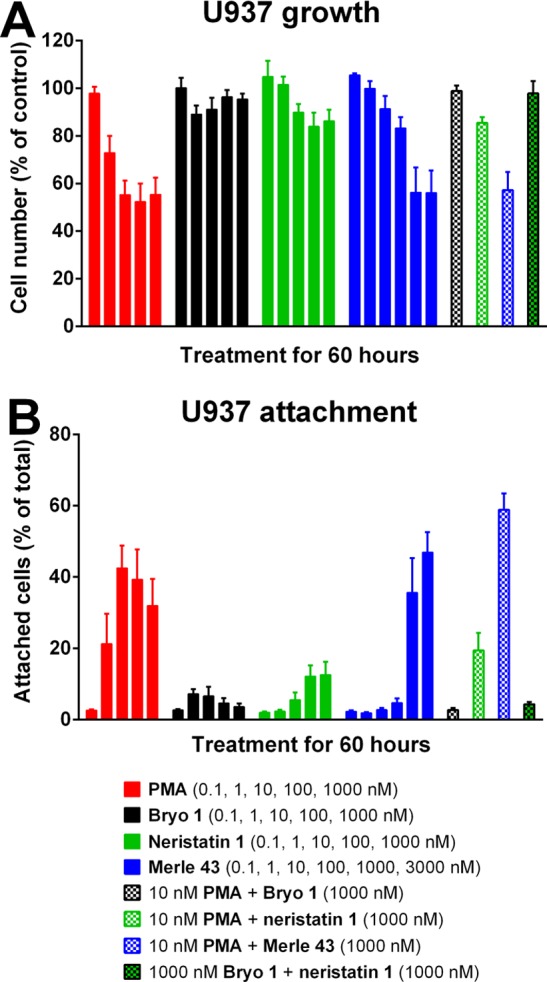
Comparison of activities on U937 cell growth and attachment. (A) Inhibition of proliferation; (B) induction of attachment; values are the mean ± SEM of triplicate experiments.
A similar pattern of behavior was seen for the induction of attachment of U937 cells (Figure 3B). PMA induced cell attachment; bryostatin 1 induced very little; neristatin 1 again resembled bryostatin 1 in inducing little attachment, although the level was slightly greater than that for bryostatin 1. Bryostatin 1 inhibited the attachment induced by PMA; neristatin 1 partially inhibited the attachment induced by PMA; Merle 43 was like PMA. As illustrated, the response to bryostatin 1 is slightly biphasic, with a lower response at higher concentrations. Because of the scarcity of material, we could use neristatin 1 only over a dose range similar to that for bryostatin 1, despite its somewhat lower potency.
PMA induces secretion of TNFα by the U937 cells; bryostatin 1 induces much less.35 Neristatin 1 resembles bryostatin 1, although again the level is slightly higher (Figure 4). The curves were slightly biphasic. Extending the concentration of neristatin 1 to 3000 nM brought the level of TNFα induction down to the level of that induced by bryostatin 1. Merle 43 induced TNFα induction to approximately 60% of that by PMA. In each case, the combination of PMA and bryostatin 1 or bryologue yielded a level of TNFα secretion comparable to that by bryostatin 1 or bryologue alone.
Figure 4.
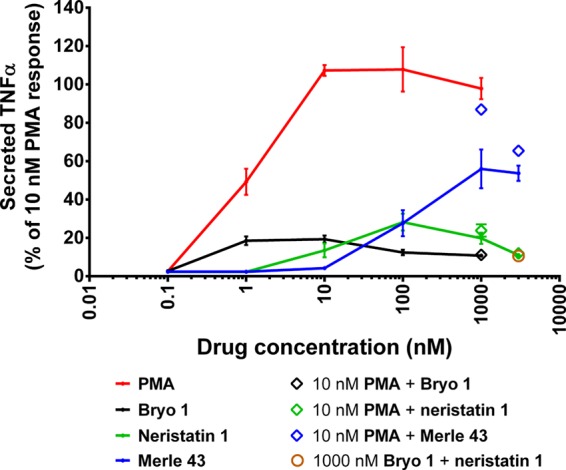
Comparison of activities on TNFα secretion by U937 cells. Values are the mean ± SEM of triplicate experiments.
Our findings with neristatin 1 complement our previous findings with Merle 23.30 Merle 23 had shown that the top half of the bryostatin 1 structure was necessary for the unique functional behavior in the U937 cells of bryostatin 1 compared to other PKC ligands. Neristatin 1 shows that the top half of the bryostatin 1 molecule is largely sufficient for the unique functional behavior of bryostatin 1 compared to other PKC ligands. It is important, however, not to overinterpret our findings. First, we have already clearly shown that bryostatin 1-like or phorbol ester-like behavior is not a quantal property.35 Rather, different bryologues displayed different extents of bryostatin 1-like behavior in the U937 (or LNCaP) cells. Further, the extent of bryostatin 1-like or phorbol ester-like behavior depended not only on the bryologue but also on the specific cell and the specific response.36 For cell proliferation and attachment of the U937 cells, neristatin 1 was largely similar to bryostatin 1, but proliferation was inhibited a little more and the level of attachment was a little higher. Second, the role of the lower half of the bryostatin 1 molecule in maintaining the correct conformation of the top half for activity should not be neglected. Along with the C3 hydroxyl, the C19 hydroxyl of bryostatin 1 contributes to the internal hydrogen-bonding network with the pyran oxygens that we and others have described.32
Within these limitations, a working model is that the bryostatin 1 structure can be visualized in terms of two separate domains conferring distinct functions. The bottom half confers binding potency, necessary for activity. For this function, the top half is a spacer domain to the first approximation. Once again, a cautionary note is that the top half is known to actually contribute somewhat to binding, as illustrated by the reduced binding activity of WN-1, which significantly differs from bryostatin 1 only in the deletion of the B ring.37 The function conferring the unique pattern of biological response for bryostatin 1, as distinct from its PKC binding activity, is provided by the top half of the molecule, with the bottom half providing a generic binding domain. This concept, to the degree that it is valid, provides a powerful strategy for functional simplification, because substantially less complicated structural solutions for C1 domain binding, such as the DAG-lactones, are known.
This model accords with our current, albeit incomplete, understanding of what may contribute to the antagonistic activity of bryostatin 1. We have shown that bryostatin 1 and PMA initially induce a similar pattern of gene expression but that the duration of expression is more transient for bryostatin 1, with the extent of transience depending on the specific gene.35 The extent to which various bryologues were bryostatin 1-like or PMA-like correlated with the degree to which they induced the more transient response like that of bryostatin 1. Candidate mechanisms for the abrogated response to bryostatin 1 are more rapid down-regulation of the classic PKC isoforms (α and β) and the loss of the novel PKC isoforms δ and ε from the nuclear fraction. At least for one of the unique responses to bryostatin 1, namely, its biphasic down-regulation of PKCδ, we showed that this unique response did not depend on some special characteristic of the C1 domains of PKCδ.38 Rather, if we made a series of chimeric constructs between the N-terminal regulatory (C1 domain containing) and C-terminal catalytic halves of PKCα and PKCδ, the unique biphasic down-regulation in response to bryostatin 1 was dependent on the C-terminal catalytic half of PKCδ, not on the regulatory half, which contains the bryostatin 1 binding sites. Unlike the other PKC ligands, bryostatin 1 forms a cap over the top of the C1 domain, anchored by insertion of the lower half of the bryostatin 1 into the binding cleft.32,39 This new surface, unique to the bryostatins, potentially could promote novel interactions with the C-terminal portion of the PKC, altering the conformation of the enzyme and influencing its down-regulation and nuclear localization. While still speculative, the predictions of this model are testable.
Experimental Section
The collection and isolation of neristatin 1 have been previously described.31 The purity of neristatin 1 was confirmed to be >95% by NMR at the time of the current experiments.
The growth of Toledo cells and the determination of growth inhibition, induction of attachment, and the secretion of TNFα by the U937 cells were all measured as previously described.34,37
This research was supported by NIH grant GM28961 to G.E.K., by grants from the Arizona Biomedical Research Commission and the Robert B. Dalton Endowment Fund (to G.R.P.), by Outstanding Investigator Grant CA44344-01-01 from the Division of Cancer Treatment and Diagnostics, NCI (to G.R.P.), and in part through the Intramural Research Program, Center for Cancer Research, NCI, NIH (Project Z1A BC 005270).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Cragg G. M.; Grothaus P. G.; Newman D. J. J. Nat. Prod. 2014, 77, 703–723. [DOI] [PubMed] [Google Scholar]
- Brown D. G.; Lister T.; May-Dracka T. L. Bioorg. Med. Chem. Lett. 2014, 24, 413–418. [DOI] [PubMed] [Google Scholar]
- Cragg G. M.; Grothaus P. G.; Newman D. J. Chem. Rev. 2009, 109, 3012–3043. [DOI] [PubMed] [Google Scholar]
- Cragg G. M.; Boyd M. R.; Cardellina J. H. 2nd.; Newman D. J.; Snader K. M.; McCloud T. G. Ciba Found. Symp. 1994, 185, 178–190. [DOI] [PubMed] [Google Scholar]
- Cragg G. M.; Newman D. J. Phytochem. Rev. 2009, 8, 313–331. [Google Scholar]
- Pettit G. R.; Melody N.; Hempenstall F.; Chapuis J.-C.; Groy T. L.; Williams L. J. Nat. Prod. 2014, 77, 863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascolutti M.; Quinn R. J. Drug Discovery Today 2014, 19, 215–221. [DOI] [PubMed] [Google Scholar]
- Kinghorn A. D.; Chin Y.-W.; Swanson S. M. Curr. Opin. Drug Discovery Dev. 2009, 12, 189–196. [PMC free article] [PubMed] [Google Scholar]
- Pettit G. R. J. Nat. Prod. 1996, 59, 812–821. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Herald C. L.; Doubek D. L.; Arnold E.; Clardy J. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar]
- Pettit G. R. Fortschr. Chem. Org. Naturst. 1991, 57, 153–195. [DOI] [PubMed] [Google Scholar]
- Dempsey E. C.; Newton A. C.; Mochly-Rosen D.; Fields A. P.; Reyland M. E.; Insel P. A.; Messing R. O. Am. J. Lung Cell Mol. Physiol. 2000, 279, 429–438. [DOI] [PubMed] [Google Scholar]
- Antal C. E.; Newton A. C. Biochem. Soc. Trans. 2014, 42, 1477–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griner E. M.; Kazanietz M. G. Nat. Rev. Cancer 2007, 7, 281–294. [DOI] [PubMed] [Google Scholar]
- Reyland M. E. Front. Biosci. 2009, 14, 2386–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Kazanietz M. G.; Blumberg P. M.; Hurley J. H. Cell 1995, 81, 917–924. [DOI] [PubMed] [Google Scholar]
- Blumberg P. M.; Kedei N.; Lewin N. E.; Yang D.; Czifra G.; Pu Y.; Peach M. L.; Marquez V. E. Curr. Drug Targets 2008, 9, 641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki H.; Sigumura T. Adv. Cancer Res. 1987, 49, 223–264. [DOI] [PubMed] [Google Scholar]
- Shao L.; Lewin N. E.; Lorenzo P. S.; Hu Z.; Enyedy I. J.; Garfield S. H.; Stone J. C.; Marner F. J.; Blumberg P. M.; Wang S. J. Med. Chem. 2001, 44, 3872–3880. [DOI] [PubMed] [Google Scholar]
- Hecker E. Cancer Res. 1968, 28, 2338–2349. [PubMed] [Google Scholar]
- Ruan B. F.; Zhu H. L. Curr. Med. Chem. 2012, 19, 2652–2664. [DOI] [PubMed] [Google Scholar]
- Kedei N.; Lubart E.; Lewin N. E.; Telek A.; Lim L.; Mannam P.; Garfield S. H.; Kraft M. B.; Keck G. E.; Jelinek R.; Blumberg P. M. ChemBioChem 2011, 12, 1242–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennings H.; Blumberg P. M.; Pettit G. R.; Herald C. L.; Shores R.; Yuspa S. H. Carcinogenesis 1987, 8, 1343–1346. [DOI] [PubMed] [Google Scholar]
- Keck G. E.; Poudel Y. B.; Cummins T. J.; Rudra A.; Covel J. A. J. Am. Chem. Soc. 2011, 133, 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manaviazar S.; Hale K. J. Angew. Chem., Int. Ed. 2011, 50, 8786–8789. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Verma V. A.; Paxton T. J.; Pillow T. H. Acc. Chem. Res. 2008, 41, 40–49. [DOI] [PubMed] [Google Scholar]
- Hale K. J.; Manaviazar S. Chem.—Asian J. 2010, 5, 704–754. [DOI] [PubMed] [Google Scholar]
- Hale K. J.; Hummersone M. G.; Manaviazar S.; Frigerio M. Nat. Prod. Rep. 2002, 19, 413–453. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Hinkle K. W.; Koehler M. F.; Lippa B. Med. Res. Rev. 1999, 19, 388–407. [DOI] [PubMed] [Google Scholar]
- Keck G. E.; Kraft M. B.; Truong A. P.; Li W.; Sanchez C. C.; Kedei N.; Lewin N. E.; Blumberg P. M. J. Am. Chem. Soc. 2008, 130, 6660–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit G. R.; Gao F.; Herald C. L.; Blumberg P. M.; Lewin N. E.; Nieman R. A. J. Am. Chem. Soc. 1991, 113, 6693–6695. [Google Scholar]
- Keck G. E.; Poudel Y. B.; Rudra A.; Stephens J. C.; Kedei N.; Lewin N. E.; Peach M. L.; Blumberg P. M. Angew. Chem., Int. Ed. 2010, 49, 4580–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft M. B.; Poudel Y. B.; Kedei N.; Lewin N. E.; Peach M. L.; Blumberg P. M.; Keck G. E. J. Am. Chem. Soc. 2014, 136, 13202–13208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedei N.; Lewin N. E.; Geczy T.; Selezneva J.; Braun D. C.; Chen J.; Herrmann M. A.; Heldman M. R.; Lim L.; Mannan P.; Garfield S. H.; Poudel Y. B.; Cummins T. J.; Rudra A.; Blumberg P. M.; Keck G. E. ACS Chem. Biol. 2013, 8, 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedei N.; Telek A.; Michalowski A. M.; Kraft M. B.; Li W.; Poudel Y. B.; Rudra A.; Petersen M. E.; Keck G. E.; Blumberg P. M. Biochem. Pharmacol. 2013, 85, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedei N.; Telek A.; Czap A.; Lubart E. S.; Czifra G.; Yang D.; Chen J.; Morrison T.; Goldsmith P. K.; Lim L.; Mannan P.; Garfield S. H.; Kraft M. B.; Li W.; Keck G. E.; Blumberg P. M. Biochem. Pharmacol. 2011, 81, 1296–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews I. P.; Ketcham J. M.; Blumberg P. M.; Kedei N.; Lewin N. E.; Peach M. L.; Krische M. J. J. Am. Chem. Soc. 2014, 136, 13209–13216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo P. S.; Bogi K.; Acs P.; Pettit G. R.; Blumberg P. M. J. Biol. Chem. 1997, 272, 33338–33343. [DOI] [PubMed] [Google Scholar]
- Kimura K.; Mizutani M. Y.; Tomioka N.; Endo Y.; Shudo A.; Itai A. Chem. Pharm. Bull. 1999, 47, 1134–1137. [Google Scholar]