

Abstract
Aims
The mitochondrial permeability transition pore (mPTP) plays a central role for tissue damage and cell death during ischaemia–reperfusion (I/R). We investigated the contribution of mitochondrial inorganic polyphosphate (polyP), a potent activator of Ca2+-induced mPTP opening, towards mPTP activation and cardiac cell death in I/R.
Methods and results
A significant increase in mitochondrial free calcium concentration ([Ca2+]m), reactive oxygen species (ROS) generation, mitochondrial membrane potential depolarization (ΔΨm), and mPTP activity, but no cell death, was observed after 20 min of ischaemia. The [Ca2+]m increase during ischaemia was partially prevented by the mitochondrial Ca2+ uniporter (MCU) inhibitor Ru360 and completely abolished by the combination of Ru360 and the ryanodine receptor type 1 blocker dantrolene, suggesting two complimentary Ca2+ uptake mechanisms. In the absence of Ru360 and dantrolene, mPTP closing by polyP depletion or CSA decreased mitochondrial Ca2+ uptake, suggesting that during ischaemia Ca2+ can enter mitochondria through mPTP. During reperfusion, a burst of endogenous polyP production coincided with a decrease in [Ca2+]m, a decline in superoxide generation, and an acceleration of hydrogen peroxide (H2O2) production. An increase in H2O2 correlated with restoration of mitochondrial pHm and an increase in cell death. mPTP opening and cell death on reperfusion were prevented by antioxidants Trolox and MnTBAP [Mn (III) tetrakis (4-benzoic acid) porphyrin chloride]. Enzymatic polyP depletion did not affect mPTP opening during reperfusion, but increased ROS generation and cell death, suggesting that polyP plays a protective role in cellular stress response.
Conclusions
Transient Ca2+/polyP-mediated mPTP opening during ischaemia may serve to protect cells against cytosolic Ca2+ overload, whereas ROS/pH-mediated sustained mPTP opening on reperfusion induces cell death.
Keywords: Inorganic polyphosphate, Mitochondrial permeability transition pore, Ischaemia–reperfusion injury, Oxidative stress, Mitochondrial ryanodine receptor
1. Introduction
It is generally accepted that mitochondrial permeability transition (PT) is associated with the opening of the mitochondrial permeability transition pore (mPTP),1 a large non-specific channel in the inner mitochondrial membrane (IMM). PT represents a dramatic increase in ion conductance of the IMM. Under physiological conditions, IMM permeability is relatively low. This is required for maintenance of the tight coupling of the oxidative phosphorylation machinery. Opening of the mPTP leads to cell and tissue damage during ischaemia–reperfusion (I/R) injury (IRI). Ca2+ and reactive oxygen species (ROS) are considered to be two key activators of PT during IRI.2 However, recent data indicate that, depending on the complex balance between cellular stress inducers and antioxidant defence systems, mPTP can undergo transient (low-conductance) or long-lasting (high-conductance) openings.3,4 Transient mPTP opening has been suggested to be involved in physiological processes such as intracellular Ca2+ homeostasis, NAD+ trafficking, and transient formation of ROS,5–7 and it could also play a role in cardioprotection by ischaemic preconditioning.8–10 While a transient pore opening is typically a reversible event which is not associated with cell death, long-lasting pore opening is followed by profound alterations of cellular bioenergetics that are considered irreversible; it results in increased mitochondrial permeability to ions and solutes with mol. wt of up to 1.5 kDa, matrix swelling, loss of critical electrochemical gradients, and depolarization of mitochondrial membrane potential (ΔΨm). In this condition, the F1F0-ATP synthase actively hydrolyzes rather than synthesizes ATP, leading inevitably to cell death.11–13 Our recent studies indicate that inorganic polyphosphate (polyP),14–16 a polymer of orthophosphates linked by phosphoanhydride bonds, plays a major role in activation of Ca2+-induced mPTP opening in cardiomyocytes. Furthermore, we found that polyP depletion prevented a transient drop in ΔΨm associated with Ca2+-induced mPTP opening.15,16 This discovery together with the fact that polyP plays multiple roles in cellular and mitochondrial function,16 including its possible participation in mitochondrial energy metabolism14–18 and in regulation of the respiratory chain activity,18 led us to hypothesize that polyP is a mediator of a transient mPTP opening in low-conductance mode. Here, we aimed to dissect the role played by polyP in mitochondrial function, mPTP activation, and cell death at various stages of simulated I/R. We found that polyP depletion prevented mPTP opening during ischaemia. However, polyP depletion caused an increase in ROS production, did not inhibit mPTP opening, and potentiated ROS-mediated cell death during reperfusion, suggesting that polyP could play a protective role against oxidative stress during reperfusion. Results are discussed in the context of the existence of two potential modes of mPTP activation during I/R: (i) a transient Ca2+/polyP-induced mode observed during ischaemia which may serve to protect cells against cytosolic Ca2+ overload and (ii) sustained ROS/pH-induced mode observed during reperfusion associated with necrotic cell death.
2. Methods
2.1. Cell isolation and culture
Left ventricular myocytes were isolated from adult New Zealand White rabbits (3–4 month old, 2.5 kg, Myrtle's Rabbitry, Thompsons Station, TN, USA). Rabbits were anaesthetized with sodium pentobarbital (50 mg/kg), and hearts were quickly excised, mounted on a Langendorff apparatus, and retrogradely perfused via the aorta as previously described.15 For adenoviral gene transfer, myocytes were cultured for 24–48 h on laminin-coated glass coverslips in PC-1 medium. All protocols were in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23; revised 2011) and approved by the institutional Animal Care and Use Committee.
2.2. PolyP depletion in mitochondria
To decrease mitochondrial polyP, mitochondria-targeted green fluorescent protein (GFP)-tagged exopolyphosphatase (PPX)15 that specifically hydrolyzes polyP into inorganic phosphate was adenovirally expressed in cardiomyocytes. PPX overexpression resulted in an ∼80% decrease in mitochondrial polyP levels after 24 h in culture.15 To account for the potential non-specific effects of protein overexpression in the mitochondrial matrix, control myocytes were infected with mitochondrially targeted GFP (control) adenovirus and cultured under the same conditions as PPX overexpressing cells.
2.3. Simulated I/R
Ischaemia was simulated by acidosis (pH 6.4), inhibition of glycolysis (glucose replaced with 20 mM deoxyglucose), and inhibition of mitochondrial respiration (with complex IV inhibitor sodium cyanide, NaCN) by cell exposure to glucose-free modified Tyrode solution containing (in mM) 20 2-deoxyglucose, 2 NaCN, 135 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, and 10 Hepes, pH 6.4 for 20 min.19 Reperfusion was simulated by 15 min superfusion with standard Tyrode solution consisting of (in mM) 135 NaCl, 4 KCl, 10 glucose, 10 Hepes, 1 MgCl2, and 2 CaCl2; pH 7.4.
2.4. Measurement of mitochondrial function
Laser scanning confocal microscopy (A1R, Nikon) was used to follow the changes in mitochondrial free calcium concentration ([Ca2+]m),20–22 ΔΨm,20,21 ROS generation,23–25 mPTP activity,24–26 mitochondrial glutathione redox status,21,27 and mitochondrial pH (pHm)27 using specific fluorescent indicators. All fluorescence signals were recorded from individual cells and background corrected. All fluorescent indicators were obtained from Molecular Probes/Life Technologies (Grand Island, NY, USA) unless noted otherwise. Detailed experimental protocols of these experiments are described in Supplementary material online.
2.5. PolyP detection in cardiomyocytes
PolyP levels were measured in control (GFP) and polyP-depleted (PPX) cells loaded with 5 µg/mL of 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) for 30 min at 37°C.14,15,28 DAPI was excited with 403 nm laser light, and emitted fluorescence was measured at 552–617 nm. Data are presented as background subtracted fluorescence collected from the whole cell and normalized to the basal level (F/F0).
2.6. Cell death
Release of lactate dehydrogenase (LDH) into the extracellular medium was measured after 20 min of simulated ischaemia and 15 min of reperfusion as an index of cell death.19 LDH release (expressed as percentage of enzyme release with respect to total cellular content and normalized to the levels of LDH release in the non-ischaemic conditions to account for cell death induced by mechanical stress due to solution replacement and suction) was measured spectrophotometrically (BioTek Synergy Mx multiplate reader) at 490 nm. The LDH assay based on the conversion of yellow tetrazolium salt by LDH into a red, formazan-class dye was used according to manufacturer specifications (Clontech).
2.7. Statistical analysis
Results are reported as mean ± S.E.M. for the indicated number (n) of cells or separate experiments for the LDH release assay. Individual comparisons are made using paired or unpaired t-tests; group comparisons are made using analysis of variances with post hoc comparisons using Tukey's test. Differences are considered statistically significant at P < 0.05.
3. Results
3.1. ROS formation, [Ca2+]m, and pHm during I/R
We monitored simultaneously how I/R affects [Ca2+]m and ROS generation. For [Ca2+]m measurements, the mitochondrially targeted Ca2+-sensitive protein, Mitycam,22 was adenovirally expressed in ventricular myocytes. The mitochondrial localization of Mitycam was confirmed by colocalization with mitochondria-entrapped tetramethylrhodamine methyl ester (TMRM; Figure 1A). Control experiments using permeabilized cells confirmed that this probe was sensitive to increases in mitochondrial matrix Ca2+ and removal of the driving force for Ca2+ uptake using the protonophore carbonyl cyanide p-(tri-fluromethoxy)phenylhydrazone (FCCP; Figure 1B). Generation of superoxide () was measured with the fluorescent probe MitoSOX Red, a cationic probe that distributes to the mitochondrial matrix, and is rapidly oxidized primarily by , and to much lesser extent by H2O2 or other ROS.21,23 As shown in Figure 1C, [Ca2+]m increased almost immediately with initiation of ischaemia which was followed by an increase in production. Upon reperfusion, [Ca2+]m slowly declined (from R = 0.51 ± 0.05–0.34 ± 0.06, n = 11, Figure 1C and D) and no significant additional increase in generation was observed (the rate of generation actually decreased from 220 ± 21% during ischaemia to 107 ± 6% during reperfusion; n = 11, P < 0.05; Figure 1C and D). Using 2′,7′-dichlorofluorescein, a fluorescent probe which preferentially detects H2O2, we found that H2O2 generation (165 ± 15% increase over the basal rate) followed the increase in generation already during ischaemia; however, significantly higher rates (286 ± 46%, n = 10, P < 0.05 compared with ischaemia) of H2O2 generation were observed during reperfusion (Figure 1C and D). We also monitored the mitochondrial glutathione redox state using the mitochondrially targeted fluorescent protein roGFP1 (mito-roGFP1).27 Mito-roGFP1 specifically senses changes in mitochondrial glutathione redox state. Under basal conditions, the mito-roGFP1 sensor was 51 ± 3% oxidized (n = 10; Figure 1E and F). During ischaemia, mito-roGFP1 oxidized further (10 ± 2% increase from basal level, n = 10) reflecting the oxidative environment of the mitochondrial matrix associated with ischaemia. Additional oxidation was detected during reperfusion (19 ± 4% increase from the ischaemia level, n = 10), which paralleled the enhanced H2O2 generation. Using the mitochondrially targeted pHm sensor mito-SypHer,27 we could demonstrate that the mitochondrial environment was significantly acidified (the mito-SypHer ratio signal decreased from R = 8.90 ± 0.64 under basal conditions to 1.66 ± 0.09 during ischaemia; n = 7; Figure 1E and F). During reperfusion, pHm slowly recovered towards basal level and eventually became more alkaline (R = 9.58 ± 1.04, n = 7) by the end of 15 min reperfusion. These data indicate that, similar to earlier reports,19 this model mimics conditions of I/R observed in vivo.
Figure 1.

(A) Mitochondrial localization of Ca2+-sensitive protein Mitycam (left) was confirmed by colocalization with mitochondrial membrane potential-sensitive dye TMRM (middle) as reflected in the overlay image (right). (B) [Ca2+]m changes measured with Mitycam in permeabilized ventricular myocytes in response to elevation of extramitochondrial [Ca2+] ([Ca2+]em) from 0.1 to 0.8 and 2 µM. Addition of the mitochondrial respiratory chain uncoupler FCCP resulted in release of Ca2+ from mitochondria. (C) Recordings of [Ca2+]m changes monitored with Mitycam, superoxide, and H2O2 generation monitored by MitoSox Red and 2′,7′-dichlorofluorescein, respectively, during I/R in intact control myocytes. (D) Summary of [Ca2+]m (n = 11 cells, four animals), superoxide (n = 11 cells, four animals), and H2O2 (n = 10 cells, three animals) changes during I/R. (E) Recordings of mitochondrial pH (pHm) and redox changes during I/R monitored by genetically encoded mitochondria-targeted probes mito-SypHer and mito-roGFP1, respectively, in intact control myocytes. (F) Summary of mitochondrial pHm (n = 7 cells, three animals) and redox (n = 10 cells, three animals) changes during I/R.
3.2. [Ca2+]m kinetics during I/R in control and polyP-depleted cells
Mitochondrial Ca2+ overload is an important factor contributing to mPTP activation; however, the mechanisms that lead to mitochondrial Ca2+ increase during I/R are still controversial. Therefore, we studied [Ca2+]m changes in control and polyP-depleted cells during I/R. [Ca2+]m was monitored using X-Rhod-1 as a Ca2+ reporter with Co2+ quenching of the cytosolic Ca2+ signal.21 We found that [Ca2+]m increased almost immediately with the onset of ischaemia, continued to increase during the entire period of ischaemia, and slowly declined during reperfusion in both control and polyP-depleted cells (Figure 2A and B). The [Ca2+]m increase during ischaemia was significantly higher in control cells compared with polyP-depleted cells (Figure 2C). Blocking MCU with 1 µM Ru360 reduced the [Ca2+]m increase during I/R by ∼50% in both control and polyP-depleted cells, but was not able to prevent the [Ca2+]m increase completely, suggesting incomplete inhibition of the MCU or additional mechanisms for Ca2+ entry during ischaemia. Cell treatment with Ru360 also changed the kinetics of the [Ca2+]m increase during ischaemia in both control and polyP-depleted cells. A sharp initial [Ca2+]m increase was observed, followed by an additional slow increase in [Ca2+]m. It has been reported previously that similar kinetics of [Ca2+]m in Ru360-treated cells were mediated by additional Ca2+ entry through ryanodine receptor type 1 channels located in the IMM (mRyR1, ryanodine receptor type 1 channels located in the IMM).29,30 To test the possibility of mRyR1 contribution to the elevated [Ca2+]m during ischaemia, Ru360-treated cells were preincubated with 10 µM of the RyR1 blocker dantrolene. Simultaneous cell treatment with Ru360 and dantrolene nearly completely eliminated the increase in [Ca2+]m during I/R in both control and polyP-depleted cells (Figure 2C), whereas dantrolene alone did not affect [Ca2+]m significantly during I/R (data not shown). Next, we tested for the involvement of ROS in the increase of [Ca2+]m. Cell treatment with 50 µM MnTBAP [Mn (III) tetrakis (4-benzoic acid) porphyrin chloride], a membrane permeable superoxide dismutase mimetic, did not affect the increase of [Ca2+]m during I/R in both control and polyP-depleted cells (Figure 2C), suggesting that Ca2+ entry during ischaemia was independent from the observed ROS formation. Furthermore, we found that cell treatment with 1 µM of the mPTP desensitizer CSA significantly attenuated the [Ca2+]m increase in control cells, but did not affect [Ca2+]m in polyP-depleted cells (Figure 2C). Moreover, the increase in cytosolic [Ca2+]i that is typically observed during ischaemia (see Supplementary material online, Figure S1) was further enhanced when mPTP opening was prevented either by CSA treatment or polyP depletion, suggesting that during ischaemia Ca2+ can enter mitochondria through mPTP. To conclude, these data indicate that three different mechanisms contribute to the elevated [Ca2+]m levels during ischaemia: (i) Ca2+ uptake through MCU, (ii) Ca2+ entry through mRyR1 when MCU activity is inhibited, and (iii) additional Ca2+ entry through the mPTP presumably opening in a low-conductance mode.
Figure 2.

(A and B) Recordings of [Ca2+]m during I/R in control (A, n = 36 cells; 10 animals) and polyP-depleted cells (B, PPX, n = 34 cells; 9 animals) in the presence of Ru360 alone (inhibits MCU; n = 11 cells, three animals in control + Ru360 and n = 7 cells, three animals in the PPX + Ru360 group), Ru360 together with 10 µM dantrolene (inhibits mRyR1, n = 5 cells, three animals in control and n = 7 cells, three animals in the PPX group), CSA (desensitizes mPTP; n = 11 cells, three animals in control + Ru360 and n = 7 cells, three animals in the PPX + Ru360 group), and MnTBAP (superoxide dismutase mimetic, scavenges superoxide; n = 6 cells, three animals in control + MnTBAP and n = 9 cells, three animals in the PPX + MnTBAP group). (C) Average amplitudes for the [Ca2+]m increase at the end of ischaemia and reperfusion in control (black bars) and polyP-depleted (grey bars) cells.
3.3. Blocking mPTP opening during ischaemia by either polyP depletion or CSA increased superoxide ( ) levels in mitochondria
Oxidative stress is a well-known important component of I/R injury. Therefore, we evaluated the source of generation in the mitochondrial matrix during I/R and the effect of polyP depletion on generation. Using MitoSOX Red as a sensor for , we detected an increase in generation during ischaemia with only a small additional increase in fluorescence observed during reperfusion (Figure 3). In both control and polyP-depleted cells, the fluorescence increase was significantly attenuated by MnTBAP (Figure 3A and B), confirming the fidelity of detection with MitoSOX Red. Blocking mitochondrial Ca2+ uptake through MCU with 1 µM Ru360 completely prevented ROS generation during I/R in control (Figure 3A) and polyP-depleted (Figure 3B) cells. This indicates that Ca2+ entering via MCU stimulates mitochondrial generation during ischaemia. The mitochondrial respiratory chain appeared to be the main source of generation since exposure to a ‘mock’ ischaemia solution lacking sodium cyanide prevented the increase in MitoSOX Red fluorescence (Figure 3C). Moreover, we found that during ischaemia either polyP depletion or CSA treatment led to an increased mitochondrial accumulation (Figure 3A and B). Taking into account that this effect was further enhanced in polyP-depleted cells in the presence of CSA (Figure 3B and C), it is likely that polyP regulation of production is not directly linked to its ability to inhibit Ca2+-induced mPTP. Taken together, these data indicate that mitochondrial Ca2+ uptake through MCU was stimulating production in the mitochondrial matrix; however, polyP was protecting mitochondria from the excessive generation.
Figure 3.

(A and B) Superoxide generation measured with MitoSox Red during I/R in control (A, n = 30 cells, 10 animals) and polyP-depleted (B, PPX, n = 27 cells, 8 animals) cells in the absence and presence of 1 µM Ru360 (n = 10 cells from four animals in control + Ru360; n = 7 cells, three animals in the PPX + Ru360 group), 1 µM CSA (n = 10 cells, three animals in control + CSA; n = 9 cells, three animals in the PPX + CSA group), and 50 µM MnTBAP (n = 7 cells, three animals in control + MnTBAP; n = 8 cells, three animals in the PPX + MnTBAP group). ‘Mock’ ischaemia solution did not contain NaCN (n = 7 cells, three animals in control; n = 8 cells, three animals in the PPX group). (C) Average amplitude of MitoSox Red fluorescence changes at the end of ischaemia and at the end of reperfusion in control and polyP-depleted cells.
3.4. Role of polyP in mPTP activity and cell death under I/R conditions
While it is established that both ischaemia and reperfusion can facilitate activation of mPTP, the relative contribution of these two conditions to mPTP activation and cell death is not well understood. Here, we investigated the kinetics of mPTP activity at the various stages of I/R in control cells and cells with depleted levels of the mitochondrial polyP (a known potent endogenous activator of Ca2+-induced mPTP14,15) in the absence and presence of the mPTP desensitizer CSA. mPTP activity was quantified as calcein red-orange release from the mitochondrial matrix compartment. As shown in Figure 4A and B, ischaemia induced a profound increase in the rate of calcein release (305 ± 17%, n = 23) in control cells, indicative of enhanced mPTP opening. This calcein release was almost completely prevented by polyP depletion (103 ± 4%, P < 0.001 vs. control, n = 22); however, only partially decreased by CSA treatment (181 ± 13%, P < 0.001, n = 11). Switching from ischaemia to reperfusion solution was accompanied by continued calcein release (255 ± 22%); however, in contrast to ischaemic conditions, polyP depletion had no effect on mPTP activity during reperfusion (208 ± 28%), whereas CSA was very effective in blocking calcein release (123 ± 28%) at this stage. This suggests different mechanisms of action for polyP and CSA with respect to mPTP opening during I/R. Surprisingly, despite mPTP inhibition during ischaemia, polyP-depleted cells did not show protection against necrotic cell death at the end of reperfusion (measured as percentage change in LDH release; Figure 4C). In fact, cell death was even more pronounced compared with control conditions. On the other hand, cell death was inhibited by 1 µM CSA in control cells; however, in polyP-depleted cells, CSA treatment brought cell death to the level observed in control untreated cells but did not decrease it further. In both control and polyP-depleted myocytes, cell death was significantly inhibited by prevention of Ca2+ entry or by scavenging of ROS (Figure 4C). No cell death was observed after 20 min of ischaemia in either group (not shown), suggesting that mPTP opening during ischaemia was limited, possibly occurring in a low-conductance mode or in a transient mode that was insufficient to disturb mitochondrial homeostasis to the extent to result in cell death.
Figure 4.
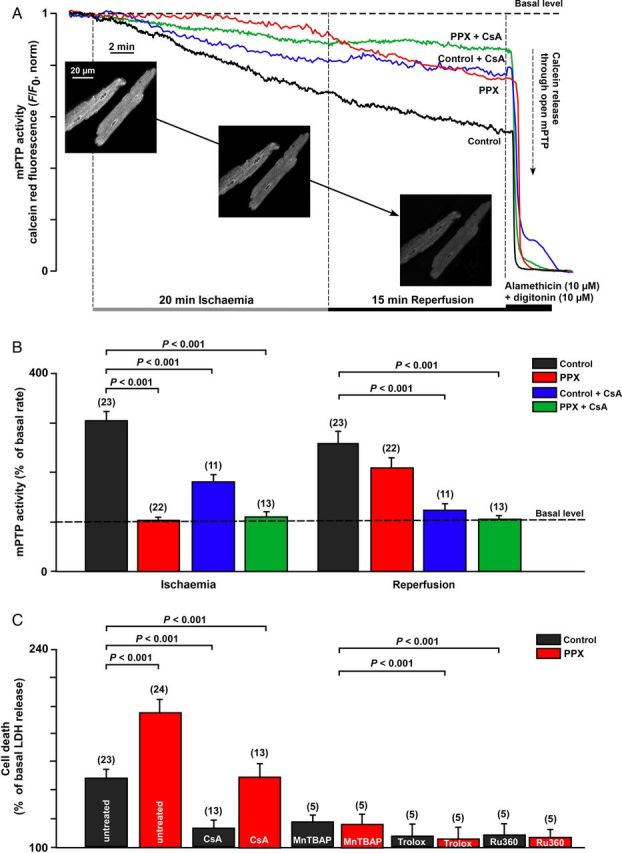
Effects of polyP depletion and CSA on mPTP activity and necrotic cell death in I/R. (A) Normalized traces of calcein red fluorescence changes during 20 min ischaemia followed by 15 min of reperfusion in control (n = 23 cells, six animals) and polyP-depleted (PPX, n = 19 cells, eight animals) cells in the absence or presence of 1 µM of CSA (n = 11 cells, three animals for control + CSA; n = 13 cells, four animals for PPX + CSA group). Inserts show images of calcein-loaded cardiomycytes before and after exposure to I/R. (B) Summary of calcein red release from mitochondria (as percentage of basal rate) at the end of ischaemia and reperfusion. (C) Cell death (percentage of LDH release with respect to basal LDH release rates) measured at the end of I/R in control and polyP-depleted (PPX) cells in the absence or presence of 1 µM CSA, 50 µM MnTBAP, 1 µM Trolox (scavenges ROS), and 1 µM Ru360. n is the number of hearts used in each experiment.
3.5. PolyP depletion and CSA protect against ΔΨm depolarization during ischaemia
One of the interesting and unexpected findings described in the previous section is that mPTP inhibition did not strictly correlate with protection from cell death. Thus, we hypothesized that, in ischaemia and reperfusion, mPTP was operating in different modes where transient mPTP opening potentially in a low-conductance state during ischaemia may serve to maintain Ca2+ homeostasis and protect cells. Since mPTP opening in a low-conductance mode might escape the detection with the calcein assay, we further test this hypothesis by monitoring ΔΨm using TMRM fluorescence. In this case, mPTP opening even in a low-conductance mode is expected to induce ΔΨm depolarization albeit to a lesser degree. As shown in Figure 5A, on average, ΔΨm decreased (depolarized) significantly to 0.79 ± 0.07 (normalized values; n = 23) during ischaemia. mPTP closing by polyP depletion was associated with an attenuated ΔΨm depolarization to 0.95 ± 0.06 (P < 0.05, n = 19). mPTP desensitization with CSA diminished ΔΨm depolarization in control (0.93 ± 0.05; P < 0.05, n = 10) and polyP-depleted (0.93 ± 0.05; P < 0.05, n = 9) cells. During reperfusion, an initial re-polarization was observed in all four groups (Figure 5B), with a higher degree of hyperpolarization detected in polyP-depleted cells (1.25 ± 0.08, P < 0.05, n = 19). However, at the end of reperfusion, ΔΨm in all groups was depolarized, albeit to substantially different degrees. The most pronounced depolarization of ΔΨm was observed in control (0.44 ± 0.07) and CSA-treated polyP-depleted (0.46 ± 0.07) cells. Cell treatment with CSA alone (without polyP depletion) significantly attenuated ΔΨm depolarization (0.901 ± 0.03; P < 0.05, n = 10) on reperfusion. However, polyP depletion was not as effective as CSA in preserving ΔΨm (0.67 ± 0.08; P < 0.05; n = 19) at the end of reperfusion. In summary, the ΔΨm data are consistent with the mPTP activity results obtained with the calcein red-orange assay, with the exception of the observation made in polyP-depleted cells in the presence of CSA. Under the latter condition, minimal loss of calcein red-orange was observed, while the TMRM data suggested substantial ΔΨm depolarization. This could be explained by (i) mPTP opening with a pore size of <790 Da which would not allow calcein red-orange release from mitochondria (see also Discussion on heterogeneous mPTP pore sizes) or (ii) mPTP-independent ΔΨm depolarization associated with disturbed mitochondrial metabolism in polyP-depleted cells. We found that endogenous polyP levels monitored by DAPI fluorescence14,15,28 increased dramatically during reperfusion in control myocytes (Figure 6A and B), demonstrating that polyP generation is associated with response to stress conditions such as I/R. Measurements in PPX-overexpressing cells did not reveal significant changes in DAPI fluorescence, confirming that these changes were indeed mediated by polyP formation. Moreover, we found that polyP generation in cardiomyocytes depended on the activity of the mitochondrial respiratory chain: (i) stimulation of the respiratory chain activity with complex I substrates 5 mM pyruvate and 2 mM glutamate led to that of polyP production, while uncoupling of the mitochondrial respiratory chain with protonophore FCCP (1 µM) decreased polyP levels (Figure 6C and E). At the same time, we found that inhibition of F1F0-ATP synthase (mitochondrial complex V) by addition of 5 µg/mL of oligomycin induced a significant increase in polyP levels (Figure 6D and E), while at the same time produced a depolarization of ΔΨm after the initial small hyperpolarization (Figure 6F and G). In cells with normal oxidative phosphorylation, ΔΨm is maintained by the proton pumping activity of the mitochondrial respiratory chain. However, if respiration is impaired as it is observed during the ischaemic insult (Figure 5), the thermodynamic equilibrium favours activity of the mitochondrial complex V in reverse mode, making this enzyme acting as a proton motive F1F0-ATPase that consumes ATP and translocates protons from the mitochondrial matrix to the cytosol in order to maintain ΔΨm as long as the glycolytic supply of ATP is maintained.31 Since in our experimental ischaemic conditions, where glycolysis was suppressed by glucose removal and 2-deoxyglucose supplementation, the initial small increase in polyP levels during ischaemia could be explained by the decreased polyP consumption by the F1F0-ATPase. Switching from ischaemia to the reperfusion solution would restore mitochondrial respiratory chain activity and glycolysis, and therefore increase polyP generation by the respiratory chain explaining the observed ‘burst’ in polyP generation (Figure 6A). Altogether, these data suggest that polyP production is tightly linked to the activity of the mitochondrial F1F0-ATP synthase and ATP generation/consumption. Moreover, different degrees of CSA-sensitive ΔΨm depolarization during I/R support the hypothesis of two conductance modes, which is further supported by the data shown in Supplementary material online, Figure S2. Switching pH from the acidic (pH 6.4) back to physiological levels (pH = 7.4) increased the degree of calcein red-orange release induced by oxidative stress almost three-fold, suggesting a dramatic increase in the mPTP conductivity.
Figure 5.

(A) Changes in ΔΨm during I/R assessed by TMRM fluorescence in control and polyP-depleted (PPX) cells in the absence or presence of 1 µM CSA. (B) Average values of TMRM fluorescence at the end of ischaemia and at different reperfusion times in control (n = 23 cells, four animals), PPX (n = 19 cells, four animals), control + CSA (n = 10 cells, three animals), and PPX + CSA (n = 9 cells, three animals) groups.
Figure 6.

Measurements of polyP levels during I/R. (A) Recordings of DAPI fluorescence in control (with overexpressed GFP) and polyP-depleted (PPX) cardiomyocytes exposed to 20 min of simulated chemical ischaemia followed by a 15 min reperfusion period in normal Tyrode solution. (B) Average values of maximal DAPI fluorescence at the end of 20 min ischaemia and 15 min reperfusion in control (n = 6 cells, three animals) and polyP-depleted (PPX, n = 6 cells, three animals) myocytes. (C) PolyP level changes in control cells upon stimulation of the respiratory chain with 5 mM pyruvate and 2 mM glutamate and subsequent application of 1 µM FCCP. (D) PolyP level changes in control cells upon F1F0-ATP synthase inhibition with 5 µg/mL of oligomycin and subsequent application of 1 µM FCCP. (E) Average values of maximal DAPI fluorescence upon application of oligomycin, pyruvate/glutamate, and FCCP. (F) Changes in ΔΨm during addition of 5 µg/mL of oligomycin and subsequent application of 1 µM FCCP in control cells. (G) Average values of TMRM fluorescence at the beginning and end of oligomycin application and at the end of FCCP application.
4. Discussion
The main focus of the present work was to investigate the role of polyP and its interplay with Ca2+, ROS, and pH in the induction of mPTP under stress conditions simulating I/R injury. The model which we used takes advantage of the fact that conditions of I/R can be mimicked by depriving cells of glucose and inhibiting the respiratory chain, referred to as “chemical ischaemia” conditions (for the detailed original description of the model, see Ruiz-Meana et al.19). Even though our experiments were performed at normoxic oxygen levels, we determined that conditions of “chemical ischaemia” induced a substantial increase in Ca2+-dependent ROS generation and a depletion of the glutathione antioxidant system, which was further exacerbated during reperfusion. As a limitation of our study, we did not monitor the status of the thioredoxin system, another major antioxidant system in the mitochondrial matrix32,33 which plays a role in H2O2 scavenging. However, we found that both cell death and mPTP activity could be prevented by blocking ROS and [Ca2+]m increases during ischaemia. This indicates that the chemical I/R model is appropriate for the investigation of these two major inducers of cell damage during I/R.
Our data confirmed a previously described finding that polyP stimulates Ca2+-induced opening of mPTP. On the other hand, we found that polyP does not prevent mPTP opening when ROS contribution begins to play a major role. The different effects of polyP on mPTP activity during I/R could be explained by the complex role that polyP plays in regulation of the mitochondrial function. As it was demonstrated previously polyP participates in formation of channel structures with properties mimicking mPTP,34 raising the possibility that the effects of polyP in Ca2+-induced mPTP opening could indeed be a direct participation of the polymer in formation of the mPTP channel. This model involves formation of a non-protein complex of two polymers—polyP and poly-β-hydroxybutyrate (PHB)—which localize to the membrane as a stable complex of these molecules and Ca2+ (polyP/Ca2+/PHB complex). In this scenario, depletion of mitochondrial polyP will naturally lead to the inhibition of complex formation and induction of the mPTP. Recent data suggest that dimers of the F1F0-ATP synthase can form channels with characteristics similar to the mPTP;35 however, the molecular details of channel formation by the F1F0-ATP synthase remain unclear. Particular attention has been brought to the subunit c of the F1F0-ATP synthase as a potential component of the mPTP.36,37 This is supported by a recent report which shows, using a patch-clamp approach, that subunit c is likely involved as a key part of the channel.38 Notably, there is evidence that the polyP/Ca2+/PHB channel forming complex is closely associated with subunit c.34 It is possible that polyP interaction with subunit c is required for Ca2+-induced formation of the supra-molecular complex and formation of mPTP. Moreover, our data demonstrate (Figure 6) that inhibition of the F1F0-ATP synthase by oligomycin increased polyP levels in cardiomyocytes, suggesting an interaction between polyP and F1F0-ATP synthase.
The effects of polyP on ROS-induced mPTP on the other hand appear to be very different. Physiological roles of polyP in mitochondrial function are not well established (see Dedkova and Blatter39 for a comprehensive review). However, it is highly possible that polymers that contain phosphate bonds which are similar to ones found in ATP and have a high density of negative charges might have a crucial role in mammalian physiology as well as pathology. For example, polyP plays an important role in Ca2+ signal transduction via P2Y1 purinergic receptors in the mammalian brain,40 and increased polyP accumulation has been associated with mutations that are related to Parkinson's disease.41 Therefore, one of the possibilities is that polyP can play an integral role in mitochondrial physiology and thus its depletion makes mitochondria more susceptible to stress conditions. Another very attractive possibility might involve a previously unrecognized role of polyP as a protective component actively involved in stress–response mechanisms. The phenomenon of a protective role of polyP is well known in bacteria, but has never been investigated in mammalian mitochondria. Recent studies performed in bacteria indicate that, during oxidative stress, the organisms can respond by a dramatic accumulation of polyP which is used for cell protection.42 In this scenario, the proposed mechanisms of polyP action is through chaperone interactions with membrane proteins which prevent their misfolding.42 The concept of misfolded proteins as key triggers of ROS-induced mPTP opening is well known.43,44 Lemasters's group44 introduced the concept that mPTP forms by aggregation of misfolded integral membrane proteins damaged by oxidative stress. This misfolding exposes hydrophilic residues to the bilayer phase. These hydrophilic surfaces cluster and enclose aqueous channels that conduct low mol. wt solutes. Initially, chaperone-like proteins block conductance through these misfolded protein clusters.44 However, when protein clusters exceed the ability of chaperons to block conductance45 during increased oxidative stress observed in reperfusion, unregulated pore opening44 in high-conductance mode occurs and Ca2+ is released out of mitochondria.46 Taking into account that we were able to detect a similar increase in polyP content during reperfusion as was seen in bacteria, it is tantalizing to suggest that polyP might play a universal protective role and that this mechanism was conserved from bacteria to mammalian mitochondria through evolution (see also Discussion in Gray et al.42).
Our study results are compatible with the existence of two distinct modes of mPTP activation during I/R: (i) a Ca2+/polyP-induced mode observed during ischaemia and (ii) an ROS/pH-induced mode observed during reperfusion. [Ca2+]m levels increased almost immediately during initiation of ischaemia which was followed by a gradual increase in mitochondrial superoxide accumulation and a massive increase in H2O2 generation during reperfusion (Figures 1 and 2). Despite the fact that mPTP opening during ischaemia is not favoured because of acidosis (Figure 1F), our study (Figures 4 and 5) and others47,48 clearly demonstrated that mPTP opening occurred already during ischaemia. Mitochondria with reduced ability to sustain a polarized ΔΨm, due to reduced electron transport power and/or increased IMM leak, are more susceptible to mPTP opening because depolarization directly increases mPTP open probability.49,50 By monitoring the rate of calcein release from mitochondria and changes in ΔΨm, we determined two phases of mPTP opening during I/R which were differently affected by CSA and polyP depletion. CSA only partially affected the rate of calcein release during ischaemia, but provided an effective protection against mPTP opening during reperfusion (Figure 4A and B). Depletion of polyP, on the other hand, was very effective against mPTP opening only during ischaemia, but failed to provide protection during reperfusion. Despite the fact that both CSA treatment and polyP depletion partially decreased [Ca2+]m and inhibited calcein release (i.e. mPTP opening) during ischaemia, we also found increased mitochondrial superoxide accumulation under these conditions (Figures 2 and 3). CSA treatment of polyP-depleted cells provided maximal protective effect against mPTP opening during ischaemia (Figure 4B); however, it also led to maximal accumulation within mitochondria (Figure 3B and C) and collapse of ΔΨm (Figure 5). The fact that mPTP inhibition during ischaemia by different interventions led to increased accumulation and cell death on reperfusion suggests that transient mPTP opening during ischaemia may be beneficial and serves as a protective mechanism against Ca2+ and ROS accumulation and subsequent cell damage. Two different (low and high) conductance modes of mPTP have been shown in experiments on isolated mitochondria.7,51–53 Several sub-conductance levels of mPTP were detected in patch-clamp experiments,54,55 where channel flickering was detected at a half-conductance state of ∼500 pS.54 This flickering 500 pS mPTP state observed in patch-clamp experiments was attributed to the low-conductance mode observed in isolated mitochondria and permeabilized cells. In our study, Ca2+/polyP-triggered mPTP opening during ischaemia resulted in small CSA-sensitive membrane depolarization (Figure 5), which failed to induce necrotic cell death, thus pointing towards mPTP opening in the low-conductance mode. Moreover, according to the ‘misfolded proteins’ theory, the conductance by individual PT pores may vary with their varying protein composition.44 This expectation is consistent with a kinetic analysis of mitochondrial swelling after the mPTP opening, which suggested that the diameters of individual pores are heterogeneous (within the 400–1450 Da range).56,57 If the polyP possess a chaperone-like activity as it was found in bacteria42 (see above), it could have different effects on mPTP activity depending on the length of polyP chain.
We presented evidence that the increase in [Ca2+]m was a trigger for superoxide generation during ischaemia. We found that several mechanisms contributed to mitochondrial Ca2+ uptake during ischaemia: Ca2+ uptake by MCU (blocked by Ru360), Ca2+ entry through mRyR1 (blocked by dantrolene), and Ca2+ entry through mPTP opening presumably in low-conductance mode (blocked by CSA in Figure 2). Mitochondrial Ca2+ uptake through mPTP under conditions of partially depolarized ΔΨm has been reported before.50 No additional increase in [Ca2+]m was observed during reperfusion. In fact, during reperfusion [Ca2+]m, slowly declined towards basal level (Figures 1B and 2A) while cytosolic [Ca2+]i increased in a CSA-sensitive manner (see Supplementary material online, Figure S1). These data are in agreement with previous reports46,58 that cytosolic Ca2+ overload during reperfusion is the consequence of bioenergetic failure after mPTP opening rather than its cause.
To conclude, our data demonstrate that mPTP opened during both ischaemia and reperfusion, although likely in different modes. We propose that a low-conductance or possibly transient mode of mPTP opening is mediated by a polyP–Ca2+ interaction during ischaemia and likely plays a protective role (Figures 4 and 5). The physiological importance of transient (low-conductance) mPTP opening, which does not lead to cell death,54 has been suggested to mediate ischaemic preconditioning-induced protection8,26 via (i) regulation of the mitochondrial matrix Ca2+ concentration;7 (ii) induction of mild mitochondrial uncoupling; 59 and (iii) regulation of mitochondrial ROS release.60 We now suggest that inorganic polyP also contributes to the mPTP opening in a low-conductance mode. During reperfusion, however, the high-conductance mPTP mode was triggered by the combination of pHm restoration and excessive H2O2 production (Figure 1 and see Supplementary material online, Figure S2). It has been reported previously that pH restoration upon reperfusion presents an independent contributing factor to cell hypercontracture and death.58 Reperfusion performed at low pH 6.2 led to prevention of hypercontracture and necrotic death,58,61 while it did not affect ROS generation itself.58 Moreover, silencing mitochondrial Na+/H+ exchanger 1 (NHE1) in rat cardiomyocytes significantly attenuated mPTP opening.62 Therefore, it appears that pHm can determine the mode of mPTP opening (low vs. high conductance), while polyP determines the Ca2+ sensitivity of mPTP. To date, translating findings from bench to bedside has been largely disappointing,63 as clinical studies involving Ca2+ channel blockers administered on the onset of myocardial reperfusion did not show any beneficial effects.64 Our data demonstrate that mitochondrial Ca2+ actually declines during reperfusion, and only inhibition of mitochondrial Ca2+ uptake before the ischaemic insult prevents ROS generation and subsequent cell death. Furthermore, our findings show that depletion of polyP was associated with enhanced cell death on reperfusion, indicating that stimulation of polyP production rather than inhibition of Ca2+ uptake on reperfusion could be beneficial for cardioprotection.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the National Institutes of Health Grants HL62231, HL80101, and HL101235, the Leducq Foundation (to L.A.B.), the American Heart Association (AHA) National Scientist Development Grant AHA 0735071N, Rush University Medical Center New Investigator Grant-in-Aid 31196 and RUMC Pilot Grant 31219 (to E.N.D.), and Heart and Stroke Foundation of Canada (to E.P.).
Acknowledgements
We thank Godfrey Smith from the University of Glasgow, Glasgow, Scotland, UK for providing the adenoviral Mitycam construct. The adenoviral construct of mito-roGFP1 was a kind gift from Jean-Christophe Jonas, Université Catholique de Louvain, Brussels, Belgium. The adenoviral mito-SypHer construct was kindly provided by Andreas Wiederkehr, University of Geneva, Geneva, Switzerland.
Conflict of interest: none declared.
References
- 1.Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol 2015;78:129–141. [DOI] [PubMed] [Google Scholar]
- 2.Halestrap AP. Mitochondria and reperfusion injury of the heart—a holey death but not beyond salvation. J Bioenerg Biomembr 2009;41:113–121. [DOI] [PubMed] [Google Scholar]
- 3.Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem 2001;276:12030–12034. [DOI] [PubMed] [Google Scholar]
- 4.Perrelli MG, Pagliaro P, Penna C. Ischemia/reperfusion injury and cardioprotective mechanisms: role of mitochondria and reactive oxygen species. World J Cardiol 2011;3:186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Lisa F, Ziegler M. Pathophysiological relevance of mitochondria in NAD(+) metabolism. FEBS Lett 2001;492:4–8. [DOI] [PubMed] [Google Scholar]
- 6.Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr 1996;28:131–138. [DOI] [PubMed] [Google Scholar]
- 7.Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997;89:1145–1153. [DOI] [PubMed] [Google Scholar]
- 8.Hausenloy DJ, Ong SB, Yellon DM. The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol 2009;104:189–202. [DOI] [PubMed] [Google Scholar]
- 9.Lim SY, Davidson SM, Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovasc Res 2007;75:530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol 2004;287:H841–H849. [DOI] [PubMed] [Google Scholar]
- 11.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta 2009;1787:1402–1415. [DOI] [PubMed] [Google Scholar]
- 12.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc Res 2004;61:372–385. [DOI] [PubMed] [Google Scholar]
- 13.Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 2015;78C:100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seidlmayer LK, Blatter LA, Pavlov E, Dedkova EN. Inorganic polyphosphate—an unusual suspect of the mitochondrial permeability transition mystery. Channels (Austin) 2012;6:463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seidlmayer LK, Gomez-Garcia MR, Blatter LA, Pavlov E, Dedkova EN. Inorganic polyphosphate is a potent activator of the mitochondrial permeability transition pore in cardiac myocytes. J Gen Physiol 2012;139:321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dedkova EN, Blatter LA. Role of beta-hydroxybutyrate, its polymer poly-beta-hydroxybutyrate and inorganic polyphosphate in mammalian health and disease. Front Physiol 2014;5:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abramov AY, Fraley C, Diao CT, Winkfein R, Colicos MA, Duchen MR, French RJ, Pavlov E. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc Natl Acad Sci USA 2007;104:18091–18096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pavlov E, Aschar-Sobbi R, Campanella M, Turner RJ, Gomez-Garcia MR, Abramov AY. Inorganic polyphosphate and energy metabolism in mammalian cells. J Biol Chem 2010;285:9420–9428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruiz-Meana M, Abellan A, Miro-Casas E, Agullo E, Garcia-Dorado D. Role of sarcoplasmic reticulum in mitochondrial permeability transition and cardiomyocyte death during reperfusion. Am J Physiol Heart Circ Physiol 2009;297:H1281–H1289. [DOI] [PubMed] [Google Scholar]
- 20.Davidson SM, Yellon D, Duchen MR. Assessing mitochondrial potential, calcium, and redox state in isolated mammalian cells using confocal microscopy. Methods Mol Biol 2007;372:421–430. [DOI] [PubMed] [Google Scholar]
- 21.Dedkova EN, Blatter LA. Measuring mitochondrial function in intact cardiac myocytes. J Mol Cell Cardiol 2012;52:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kettlewell S, Cabrero P, Nicklin SA, Dow JA, Davies S, Smith GL. Changes of intra-mitochondrial Ca2+ in adult ventricular cardiomyocytes examined using a novel fluorescent Ca2+ indicator targeted to mitochondria. J Mol Cell Cardiol 2009;46:891–901. [DOI] [PubMed] [Google Scholar]
- 23.Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc 2008;3:941–947. [DOI] [PubMed] [Google Scholar]
- 24.Dedkova EN, Blatter LA. Characteristics and function of cardiac mitochondrial nitric oxide synthase. J Physiol 2009;587:851–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dedkova EN, Seidlmayer LK, Blatter LA. Mitochondria-mediated cardioprotection by trimetazidine in rabbit heart failure. J Mol Cell Cardiol 2013;59:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hausenloy D, Wynne A, Duchen M, Yellon D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation 2004;109:1714–1717. [DOI] [PubMed] [Google Scholar]
- 27.Roma LP, Duprez J, Takahashi HK, Gilon P, Wiederkehr A, Jonas JC. Dynamic measurements of mitochondrial hydrogen peroxide concentration and glutathione redox state in rat pancreatic beta-cells using ratiometric fluorescent proteins: confounding effects of pH with HyPer but not roGFP1. Biochem J 2012;441:971–978. [DOI] [PubMed] [Google Scholar]
- 28.Aschar-Sobbi R, Abramov AY, Diao C, Kargacin ME, Kargacin GJ, French RJ, Pavlov E. High sensitivity, quantitative measurements of polyphosphate using a new DAPI-based approach. J Fluoresc 2008;18:859–866. [DOI] [PubMed] [Google Scholar]
- 29.Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem 2001;276:21482–21488. [DOI] [PubMed] [Google Scholar]
- 30.Tewari SG, Camara AK, Stowe DF, Dash RK. Computational analysis of Ca2+ dynamics in isolated cardiac mitochondria predicts two distinct modes of Ca2+ uptake. J Physiol 2014;592:1917–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, Tinker A, Duchen MR. Regulation of mitochondrial structure and function by the F1F0-ATPase inhibitor protein, IF1. Cell Metab 2008;8:13–25. [DOI] [PubMed] [Google Scholar]
- 32.Aon MA, Stanley BA, Sivakumaran V, Kembro JM, O'Rourke B, Paolocci N, Cortassa S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J Gen Physiol 2012;139:479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanley BA, Sivakumaran V, Shi S, McDonald I, Lloyd D, Watson WH, Aon MA, Paolocci N. Thioredoxin reductase-2 is essential for keeping low levels of H(2)O(2) emission from isolated heart mitochondria. J Biol Chem 2011;286:33669–33677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pavlov E, Zakharian E, Bladen C, Diao CT, Grimbly C, Reusch RN, French RJ. A large, voltage-dependent channel, isolated from mitochondria by water-free chloroform extraction. Biophys J 2005;88:2614–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA 2013;110:5887–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013;12:674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Azarashvili T, Odinokova I, Bakunts A, Ternovsky V, Krestinina O, Tyynela J, Saris NE. Potential role of subunit c of FF-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 2014;55:69–77. [DOI] [PubMed] [Google Scholar]
- 38.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA, Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the F1F0 ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci USA 2014;111:10580–10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dedkova EN, Blatter LA. Role of β-hydroxybutyrate, its polymer poly-β-hydroxybutyrate and inorganic polyphosphate in mammalian health and disease. Front Physiol 2014;5:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmstrom KM, Marina N, Baev AY, Wood NW, Gourine AV, Abramov AY. Signalling properties of inorganic polyphosphate in the mammalian brain. Nat Commun 2013;4:1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angelova PR, Agrawalla BK, Elustondo PA, Gordon J, Shiba T, Abramov AY, Chang YT, Pavlov EV. In situ investigation of mammalian inorganic polyphosphate localization using novel selective fluorescent probes JC-D7 and JC-D8. ACS Chem Biol 2014;9:2101–2110. [DOI] [PubMed] [Google Scholar]
- 42.Gray MJ, Wholey WY, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, Krieger AG, Smith EM, Bender RA, Bardwell JC, Jakob U. Polyphosphate is a primordial chaperone. Mol Cell 2014;53:689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun 2003;304:463–470. [DOI] [PubMed] [Google Scholar]
- 44.He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett 2002;512:1–7. [DOI] [PubMed] [Google Scholar]
- 45.He L, Lemasters JJ. Heat shock suppresses the permeability transition in rat liver mitochondria. J Biol Chem 2003;278:16755–16760. [DOI] [PubMed] [Google Scholar]
- 46.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 2009;1787:1395–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loor G, Kondapalli J, Iwase H, Chandel NS, Waypa GB, Guzy RD, Vanden Hoek TL, Schumacker PT. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim Biophys Acta 2011;1813:1382–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robin E, Guzy RD, Loor G, Iwase H, Waypa GB, Marks JD, Hoek TL, Schumacker PT. Oxidant stress during simulated ischemia primes cardiomyocytes for cell death during reperfusion. J Biol Chem 2007;282:19133–19143. [DOI] [PubMed] [Google Scholar]
- 49.Honda HM, Korge P, Weiss JN. Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci 2005;1047:248–258. [DOI] [PubMed] [Google Scholar]
- 50.Saotome M, Katoh H, Satoh H, Nagasaka S, Yoshihara S, Terada H, Hayashi H. Mitochondrial membrane potential modulates regulation of mitochondrial Ca2+ in rat ventricular myocytes. Am J Physiol Heart Circ Physiol 2005;288:H1820–H1828. [DOI] [PubMed] [Google Scholar]
- 51.Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 1979;195:460–467. [DOI] [PubMed] [Google Scholar]
- 52.Huser J, Blatter LA. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J 1999;343(Pt 2):311–317. [PMC free article] [PubMed] [Google Scholar]
- 53.Elustondo PA, Negoda A, Kane CL, Kane DA, Pavlov EV. Spermine selectively inhibits high-conductance, but not low-conductance calcium-induced permeability transition pore. Biochim Biophys Acta 2015;1847:231–240. [DOI] [PubMed] [Google Scholar]
- 54.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta 1995;1241:139–176. [DOI] [PubMed] [Google Scholar]
- 55.Zorov DB, Kinnally KW, Perini S, Tedeschi H. Multiple conductance levels in rat heart inner mitochondrial membranes studied by patch clamping. Biochim Biophys Acta 1992;1105:263–270. [DOI] [PubMed] [Google Scholar]
- 56.Massari S. Kinetic analysis of the mitochondrial permeability transition. J Biol Chem 1996;271:31942–31948. [PubMed] [Google Scholar]
- 57.Cassarino DS, Parks JK, Parker WD, Jr, Bennett JP., Jr The Parkinsonian neurotoxin MPP+ opens the mitochondrial permeability transition pore and releases cytochrome c in isolated mitochondria via an oxidative mechanism. Biochim Biophys Acta 1999;1453:49–62. [DOI] [PubMed] [Google Scholar]
- 58.Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol 2006;290:H2024–H2034. [DOI] [PubMed] [Google Scholar]
- 59.Zago EB, Castilho RF, Vercesi AE. The redox state of endogenous pyridine nucleotides can determine both the degree of mitochondrial oxidative stress and the solute selectivity of the permeability transition pore. FEBS Lett 2000;478:29–33. [DOI] [PubMed] [Google Scholar]
- 60.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000;192:1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bond JM, Herman B, Lemasters JJ. Protection by acidotic pH against anoxia/reoxygenation injury to rat neonatal cardiac myocytes. Biochem Biophys Res Commun 1991;179:798–803. [DOI] [PubMed] [Google Scholar]
- 62.Villa-Abrille MC, Cingolani E, Cingolani HE, Alvarez BV. Silencing of cardiac mitochondrial NHE1 prevents mitochondrial permeability transition pore opening. Am J Physiol Heart Circ Physiol 2011;300:H1237–H1251. [DOI] [PubMed] [Google Scholar]
- 63.Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth 2012;16:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bar FW, Tzivoni D, Dirksen MT, Fernandez-Ortiz A, Heyndrickx GR, Brachmann J, Reiber JH, Avasthy N, Tatsuno J, Davies M, Hibberd MG, Krucoff MW, Group CS. Results of the first clinical study of adjunctive CAldaret (MCC-135) in patients undergoing primary percutaneous coronary intervention for ST-elevation myocardial infarction: the randomized multicentre CASTEMI study. Eur Heart J 2006;27:2516–2523. [DOI] [PubMed] [Google Scholar]