

Abstract
Muscular diseases lead to muscle fiber degeneration, impairment of mobility, and in some cases premature death. Many of these muscular diseases are largely idiopathic. The goal of this study was to identify biomarkers based on their functional role and possible mechanisms of pathogenesis, specific to individual muscular disease. We analyzed the muscle transcriptome from five major muscular diseases: acute quadriplegic myopathy (AQM), amyotrophic lateral sclerosis (ALS), mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), dermatomyositis (DM) and polymyositis (PM) using pairwise statistical comparison to identify uniquely regulated genes in each muscular disease. The genome-wide information encoded in the transcriptome provided biomarkers and functional insights into dysregulation in each muscular disease. The analysis showed that the dysregulation of genes in forward membrane pathway, responsible for transmitting action potential from neural excitation, is unique to AQM, while the dysregulation of myofibril genes, determinant of the mechanical properties of muscle, is unique to ALS, dysregulation of ER protein processing, responsible for correct protein folding, is unique to DM, and upregulation of immune response genes is unique to PM. We have identified biomarkers specific to each muscular disease which can be used for diagnostic purposes.
INTRODUCTION
Muscular diseases affect millions of individuals each year, causing muscle atrophy, weakness and/or pain, and often lead to muscle fiber degeneration, impairment of mobility and even premature death (1). Past studies attempting to identify biomarkers and elucidate mechanisms of pathogenesis have improved our understanding of these diseases, although several, including acute quadriplegic myopathy (AQM), amyotrophic lateral sclerosis (ALS), dermatomyositis (DM) and polymyositis (PM), still remain largely idiopathic. For instance, AQM (also known as critical illness myopathy) has been associated with preferential loss of myosin-associated thick filament proteins and non-excitability of the affected muscle (2,3). Although many factors such as mechanical ventilation and neuromuscular blockers might be triggers for AQM, the underlying pathophysiological mechanisms remain largely unknown (2). Similarly, ALS is a motor neuron disease with progressive neurodegeneration leading to muscle atrophy (4). Several genes such as SOD1 and ALS2, whose mutations cause familial-ALS, have also been identified. Nevertheless, mechanisms for sporadic-ALS (∼80–90% of ALS cases) are still unclear (4,5). Invasion of immune cells such as lymphocytes in DM and PM indicates that they may both be immune-related diseases, and yet disease mechanisms are not completely understood (6,7). Additionally, besides the mutation of nuclear DNA, the mutations of mitochondrial DNA are also responsible for myopathies such as mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) is caused by mitochondrial DNA 3243A>G mutation in the MT-TL1 gene (8).
Clinical therapies for muscular diseases vary case by case and involve testing drugs which often target the symptoms of the diseases. Only few treatments have been successful. For example, so far, only Riluzole has been shown to be effective in slowing the progression of ALS, but without any subjective improvement (5). In AQM, patients often recover completely. However, treatments are limited to controlling the triggering factors (e.g. neuromuscular blockers and steroids) and nutritional compensation for the negative protein balance (2). In DM and PM, corticosteroids are very effective and used as standard treatment. However, long-term treatment with corticosteroids leads to various side effects and disabilities (9). Past studies were mostly focused on individual disease in comparison only to a healthy muscle, and hence the discovered traits/targets may be neither unique nor optimal in discriminating related but distinct muscular diseases which share clinically recognizable common features. For example, because widespread myosin loss is seen in AQM, myosin-to-actin ratio is used for diagnosing AQM (2). However, focal myosin loss has been reported in other disorders, such as DM (2), making the diagnosis ambiguous. Stress and inflammatory pathways involving mitogen-activated protein kinase and TGFβ are activated in AQM, ALS and DM (2,10–12); and mitochondrial biogenesis and muscle hypertrophy pathways are downregulated in most muscular diseases (13). The two myositis, PM and DM, are similar in attributes (i.e. muscle inflammation and varying degrees of muscle weakness) (1). Thus, a current challenge for clinical diagnosis and treatment of muscular diseases depends on identification of unique disease-specific genes/biomarkers.
Unique disease-specific genes/biomarkers are defined as the genes which are extremely up or downregulated in that disease with respect to all other conditions. This list of uniquely regulated genes can contain genes affected by both primary dysregulation and secondary/compensatory mechanisms. The identification of unique disease-specific genes has 2-fold advantages. First, it removes the commonly regulated genes (have common regulation in two or more diseases) and provides the filtered list of biomarker genes that improve differential diagnosis. Second, as this list contains primary dysregulated genes, further analysis of these uniquely regulated genes can provide insights into unique pathway modules and functional differentiation of the as yet poorly understood pathogenesis, and identify possible drug targets for future treatments.
Recent advances in molecular phenotyping technologies combined with novel bioinformatics strategies provide a unique opportunity to better understand these muscular diseases at a systems level. Gene Expression Omnibus (GEO, a transcriptome repository) contains extensive transcriptomic data of muscle from patients with muscular diseases and serves as an excellent molecular phenotype for functional analysis (14). These transcriptomic data of muscle represent functional states of muscle and the comparison of the data in diseased and normal patients will allow identification of factors involved in the diseases. However, as these microarrays were collected from patients of heterogeneous backgrounds, ethnicities, age groups, etc., they are subject to high levels of noise/variance. Thus, analysis of this type of high variance data requires a robust statistical method. While simple t-test is generally used to find significance in high variance data, it requires large sample size. In many cases, however, large numbers of patient data are not available and the t-test may lead to unreliable estimates of variances, and increase the number of false positives. Therefore, a modified t-test based on variance modeling approaches must be utilized to handle small sample size studies (15,16). Such methods borrow information from genes across the array and overcome above limitations. In variance modeling approaches, the variance estimates are derived from global or local model of the intrinsic variance structure of the microarray data. The model describes the relationship between expression intensity and variance (σ2) and can be written as:
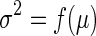 |
(1) |
where μ denotes ‘true’ expression level. In general, μ is not known prior to observing array measurements, thus the observed sample mean is often used as a surrogate.
In this study, microarray data from muscle tissue in five muscular diseases, namely, AQM, ALS, MELAS, DM and PM, were analyzed using a variance modeling approach. The five muscular diseases were chosen based on three criteria. First, we ensured that our study covers a range of different muscular diseases, namely, inflammatory, non-inflammatory, acquired and inherited muscular diseases. The second criterion was to account for the high adaptive nature of muscle and the crucial role of age in muscle degradation and disease progression. To avoid age-related discrepancies, we focused on subjects whose ages were between 20 and 60 years. Thus adolescent muscular diseases and sarcopenia were excluded from our study. The third criterion was the availability of raw data; single platform, GPL96 (Affymetrix HG-U133A), was used to avoid platform variation. Data were most available on this platform for our five muscular diseases, making them the optimal subjects for the study. There was no gender preference in the raw data or in the study as gender plays no significant role in the progression or acquisition of the diseases. The genome-wide information encoded in the large expression data was reduced to provide unique biomarkers and possible mechanistic insights into dysregulation in major muscle functions in each muscular disease (Fig. 1). Our results show: calcium handling and forward membrane path dysregulations in AQM patients, unique downregulation of major thin and thick filament in ALS, dysfunctional protein synthesis in DM patients and upregulated immune response in PM. These uniquely regulated genes associated with the dysfunction can be used to diagnose patients with the muscular diseases and assess progress after post-treatments.
Figure 1.

Functional dysregulation of muscle in AQM, ALS, DM and PM. Each dashed-rectangle represents gene expression profiling of individual disease indicated by the disease name. Red, light red, green and light green nodes represent uniquely upregulated, upregulated, uniquely downregulated and downregulated genes, respectively. The dysregulation of genes in the forward membrane path, responsible for transmitting action potential from neural excitation, is specific to AQM, while the dysregulation of myofibril genes, responsible for mechanical support of the muscle, is specific to ALS, dysregulation of ER protein processing, responsible for correct protein folding, is specific to DM, and upregulation of immune response genes is specific to PM.
RESULTS
AQM
The muscle of AQM patients demonstrates altered muscle membrane excitability, which disappears along with the recovery process in weeks to months (2,3). In excitation–contraction coupling, acetylcholine (ACh) released from the axon diffuses across the synapse and binds to the nicotinic cholinergic receptors (AChR) on the myocyte. Then AChR opens the Na/K channel and causes the depolarization of the sarcolemma. The action potential, through voltage-gated Na+ and K+ channels, propagates through the entire sarcolemma and the t-tubule. This activates dihydropyridine receptors (DHPR) which in turn open RyR1 [the calcium channel on sarcoplasmic reticulum (SR)] through their mechanical linkage, causing the release of Ca2+ stored in SR into the cytosol. Our result shows that the non-excitability of the muscle associated with AQM patients can occur at two levels: at the forward membrane path and at the SR [Fig. 1A (AQM), Supplementary Material, Table S1]. In the forward membrane path, the observed unique downregulation of DHPR (CACNA1S) limits the transmission of the muscle excitation signal from the sarcolemma into the cytosol [Fig. 1A (AQM)]. The downregulation of ion channels, i.e. potassium (K+), sodium (Na+) etc., which are responsible for transmitting action potential, further reduces the activation of DHPR. At the SR, unique downregulation of SR-related proteins such as ANK1 (ankyrin 1, the membrane protein in SR essential for maintaining its size), CAPN3 (calpain 3, a non-lysosomal cysteine protease whose absence causes decreases in SR Ca2+ storage), and CASQ1 (calsequestrin 1, a high-affinity calcium-binding protein) reduces Ca2+ storage in SR in AQM patients. When SR's calcium storage capacity decreases—due to reduced SR size and/or reduced Ca2+ buffering—the amount of Ca2+ available to be released through RyR1 from SR into the sarcoplasm [Fig. 1A (AQM)] will decrease as well. However, it is difficult to determine whether this is a result of adaptation to the loss of signals from DHPR according to the ‘use it or lose it’ rule, or another contributor to the non-excitability of muscle in AQM.
ALS
Motor neurons progressive degradation followed by muscle atrophy and weakness due to long-term denervation are a few typical symptoms of ALS. Since transcriptomic data were obtained from muscle biopsy, we could not find dysregulation in motorneurons. However, we found unique biomarkers showing dysregulation of myofibril genes in ALS, i.e. unique downregulation of major proteins in thin filaments and thick filaments. This unique downregulation weakens the mechanical support affecting the entire myofibril in ALS [Fig. 1A (ALS), Supplementary Material, Table S2]. Specifically, the unique downregulation of nebulin and ACTA1 (alpha F-actin) indicate the decreased length of thin filaments in ALS patients (17). Concomitantly, upregulated actin-capping proteins (CAPZA1, CAPZB and TMOD1), responsible for increasing the ‘capping’ of actin filament's polymerization and elongation, further suggest shorter actin filaments in ALS. This conclusion is supported by the downregulation of tropomyosin (TPM-1/2/3) and unique downregulation of troponins (TNN-C1/C2/I2/T1), which may be an autoregulation in response to shorter nebulin and actin. Thick filaments, composed mainly of titin, myosins and some M-band proteins, also showed an overall downregulation as shown in Figure 1A (ALS). Our results showed that unique downregulation of titin, a major component of thick filament, and most myosin light chains in muscle (MYL1, MYL2 and MYLPF) and downregulation of myosin heavy chains (MYH2 and MYH7) can lead to structural failure and weakened muscle movement in ALS. The downregulated myomesin, responsible for anchoring myosin to titin, decreases structural alignment of the thick filament. The combination of these factors can lead to loss of actin–myosin interaction, and ultimately decreases the myofibril's capacity to generate force and reduce passive stiffness in the muscle. Additionally, the unique upregulation of electron transport chain (ETC) is consistent with hypermetabolism, a well-documented characteristic of ALS (18).
DM
Unlike ALS and AQM, the symptomatic similarities in DM and PM present challenges in clinical diagnosis. However, using our approach, we were able to identify key molecular differences. Our results showed that DM patients can suffer from significant dysfunctional protein synthesis occurring on four different levels. First is at a spliceosomal level, shown by unique downregulation of genes involved in spliceosome components and Prp19 complex of the spliceosome pathway, indicating that mRNA production is in deficit in DM and thus, less available for protein translation. Second is at a ribosomal level, shown by the uniquely downregulated ribosome including both large and small ribosome subunits, indicating decreased protein translation in DM patients (Supplementary Material, Fig. S1). Third occurs at an endoplasmic reticulum (ER) level, shown by uniquely downregulated RRBP1, ribosome receptor on ER [Fig. 1A (DM)]. Less ribosome receptor implies less binding of ribosome to ER, impeding protein transport into the ER for folding. Furthermore, unique downregulation of GlcII complex (GANAB and PRKCSH), CRT complex (CALR) and ERGIC53 complex (LMAN1) in the protein folding pathway impairs ER's ability to fold its proteins properly (Supplementary Material, Fig. S2). Hence, reduced protein synthesis can occur in DM patients due to unique downregulation of ribosome, decreased transport of synthesized protein to ER owing to low binding of ribosome to ER, and dysfunctional protein folding of proteins that reach ER. Lastly, the degradation of the mis-folded proteins occur at ubiquitination level, and uniquely downregulated ubiquitin mediated proteolysis pathway indicates that incorrectly folded proteins are less efficiently degraded in DM patients.
Our study also suggests that mitochondrial size and its functional integrity are also affected in DM patients. Two variables are known to influence the ATP production rate of mitochondria—the size of mitochondria and the amount of enzymes in them; the downregulation of protein synthesis would suggest that DM patients suffer from both. The reduced mitochondria size is suggested by the unique downregulation of TOMM20, TOMM22, TIMM17A (an essential component of TIM23 complex) and TIMM44 (an essential component of PAM complex). While TOMM20 and TOMM 22 form a receptor at the mitochondrion outer membrane surface for importing mitochondrial proteins into the mitochondrial intermembrane space, TIM23 complex and PAM complex translocate transit peptide-containing proteins from the intermembrane space across the inner membrane into the mitochondrial matrix (19). Decreased mitochondrial biogenesis and protein transport both limit ATP production in DM patients. The unique downregulation of glycolytic pathway and TCA (tricarboxylic acid) cycle also suggest that overall energy metabolism is significantly downregulated in DM.
PM
The etiology of PM remains largely unknown with only its inflammatory response as its main indicator. Previous studies have also suggested that PM may be an autoimmune disease (7,20). Our results support inflammation as PM's key feature due to increased immune response [Fig. 1B (PM)]. These include unique upregulation of antigen processing and presentation pathway, phagosome pathway, natural killer (NK) cell mediated cytotoxicity pathway, chemokine signaling pathway and cytokine–cytokine receptor interaction pathway (Supplementary Material, Figs. S3–S7). Major histocompatibility complex-I and II (MHC-I and MHC-II) were also uniquely upregulated in PM, where the uniquely upregulated MHC-I recognizes and activates the NK cell mediated cytotoxicity pathway. Unique upregulation of MHC-I genes, such as HLA-A/B/C/F/G, observed in our results is consistent with the fact that MHC-I staining is used for screening and diagnosis of PM (21). Many PM patients also have a history of being diagnosed with Epstein–Barr virus (EBV) (22). As EBV uses MHC-II (expressed by genes HLA-DR/DP/DQ/DM) to enter into host, our analysis reveals that the uniquely upregulated MHC-II seen in our results is arguably the main cause of increased EBV infection of PM patients.
Studies have shown that chemokines, a special migratory subset of cytokines which aid immune cells to reach the sites of inflammation, play a role in PM (23). Unique upregulation of several chemokines, such as CCL4, CCL13, CXCL9 and CXCL13, were observed in PM patients. Furthermore, we observed increased transcripts in cell adhesion, focal adhesion and extra cellular matrix (ECM) receptor pathways, reflecting increased leukocyte migration/trafficking and consequently increased immune response in PM. While the primary causes of DM and PM are not elucidated, our study offers unique biomarkers to differentiate the two myositides.
MELAS
We were able to find uniquely regulated genes for MELAS, however, the enrichment analysis did not provide any mechanistic insights into MELAS in our study. It can be attributed to the fact that MELAS is caused by mitochondrial DNA rather than nuclear DNA (8).
DISCUSSION
Muscle atrophy and weakness are common symptoms of most muscle diseases. Our analysis suggests that different genes/parts of muscle are uniquely dysregulated in each distinct muscular disease. For example, ALS shows atrophy due to unique downregulation of thin filaments and thick filaments whereas AQM shows atrophy due to unique downregulation of thick filaments and the Z-disk. To test the uniqueness of the disease-specific genes, we included Duchenne muscular dystrophy (DMD) microarray data and performed test analysis (six-way Venn). The analysis showed that dystrophin gene was uniquely downregulated while ECM genes were uniquely upregulated in DMD. These results were consistent with previous studies (24,25). Further, our results on other myopathies were unaffected by the inclusion of DMD data (Supplementary Material, Table S3). In summary, inclusion of DMD data into our analysis validated our approach.
Our statistical approach to analyzing patient data provides unique biomarkers and preliminary insights into possible mechanisms that are associated with each muscular disease. Table 1 provides the list of gene biomarkers that can be used effectively to diagnose patients with AQM, ALS, DM and PM. We have identified downregulation of forward membrane path and calcium homeostasis resulting in loss of muscle excitability in AQM patients. In ALS, we showed that myofibril was significantly degraded, mostly due to shortened thin filaments and deteriorated thick filaments. In DM, protein dysregulation was found on four different levels: downregulation of spliceosome, ribosomes, protein processing in ER pathway and degradation of mis-folded proteins. Accumulation of mis-folded proteins can lead to ER stress, among other responses. The ER stress may be the cause of inflammation witnessed in DM. We were also able to identify inflammatory and immune responses as unique biomarkers for PM. Our results showed that we can differentiate DM and PM based on transcriptional changes.
Table 1.
Genes involved in each disease and their functions ( = uniquely downregulated, ↓=downregulated,
= uniquely downregulated, ↓=downregulated,  =uniquely upregulated and ↑=upregulated)
=uniquely upregulated and ↑=upregulated)
| Disease | Genes | Functions/pathways |
|---|---|---|
| AQM |
 CASQ1, CASQ1,  ANK1, ANK1, 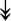 CAPN3 CAPN3 |
SR-related proteins: indicative of SR size and Ca2+ storage. |
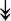 CACNA1S, ↓CACNB1, ↓CACNG1 CACNA1S, ↓CACNB1, ↓CACNG1 |
Serve as gates for Ca2+ from the t-tubule to SR needed to activate RyR1. Activated RyR1 allows release of Ca2+ into the sarcoplasm. | |
| ALS |
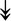 ACTA1, ACTA1, 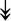 TNNI2, TNNI2, 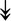 TNNC1, TNNC1, 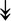 TNNC2, TNNC2, 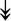 TNNT1, TNNT1, 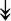 NEB, ↓TPM-1/2/3 NEB, ↓TPM-1/2/3 |
Actin-related proteins: tropomyosins, which wrap around actin, prevent actin–myosin interaction. Troponins serve as binding sites on tropomyosin for Ca2+ and help strip tropomyosin from actin to allow myosin to interact with actin. |
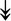 MYH11, MYH11, 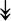 MYL1, MYL1, 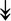 MYL2, MYL2, 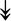 MYLPF, MYLPF, 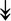 TTN, ↓MYH2, ↓MYH7, ↓MYOM1 TTN, ↓MYH2, ↓MYH7, ↓MYOM1 |
Myosin-related proteins: myosin is comprised of heavy and light chains. Titin binds to the myosin, providing muscle integrity during its contraction and relaxation. Myomesins are responsible for anchoring myosin to titin. | |
| DM |
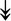 PQBP1, PQBP1, 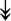 USP39, USP39, 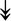 SNW1, SNW1, 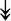 LSM4, LSM4, 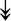 PRPF31, PRPF31, 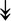 PRPF19, PRPF19, 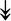 NAA38, NAA38,  XAB2, XAB2,  SNRPD2, SNRPD2,  U2AF1, U2AF1,  PRPF4, PRPF4, 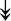 RBM8A RBM8A |
Responsible for splicing of pre-mRNA. |
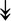 RSL24D1, RSL24D1, 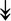 RPL11, RPL11, 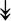 RPL37, RPL37, 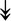 RPL37A, RPL37A, 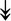 RPL36A, RPL36A, 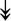 RPS8, RPS8, 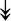 RPS20, RPS20, 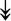 RPL23 RPL23 |
Large and small ribosome subunits. | |
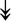 GANAB, GANAB, 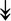 LMAN1, LMAN1, 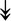 PRKCSH, PRKCSH,  RRBP1, RRBP1,  CALR, CALR,  EIF2AK2, ↑EIF2AK1 EIF2AK2, ↑EIF2AK1 |
Proteins related to protein processing in ER. | |
 UBE4B, UBE4B,  PRPF19, PRPF19, 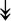 ANAPC5, ANAPC5, 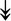 TCEB2, TCEB2, 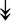 UBE2E1, UBE2E1, 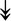 UBE2M UBE2M |
Responsible for proper degradation of incorrectly folded proteins. | |
| PM |
 HLA-A/B/C/F/G, ↑HLA-DR/DP/DQ/DM HLA-A/B/C/F/G, ↑HLA-DR/DP/DQ/DM |
MHC-I and MHC-II related proteins: MHC-I and MHC-II proteins are immune response proteins responsible for displaying antigen fragments. |
 CXCL13, CXCL13,  CXCR6, CXCR6,  GNB5, GNB5,  DOCK2, DOCK2,  CXCL9, CXCL9,  RAC2, RAC2,  CCL4, CCL4,  CCL13, CCL13,  CCL18, CCL18,  XCL1 XCL1 |
Chemokines, responsible for signaling immune cells. | |
 ITGA6, ITGA6,  ITGA4, ITGA4,  SIGLEC1, SIGLEC1,  COL4A6, COL4A6,  ITGB7 ITGB7 |
Proteins involved in cell adhesion molecules, focal adhesion and ECM receptor interaction, all of which aid leukocyte trafficking and migration. |
MATERIALS AND METHODS
Microarray data
Data for five muscular disease (three AQM patients, nine ALS patients, four MELAS patients, five DM patients and six PM patients) and control (120 healthy subjects) were downloaded from GEO on Platform GPL96 (Affymetrix HG-U133A). Platform GPL96 contained 22 215 probes representing 13 262 unique genes. Data was downloaded in the format of either CEL files or probe intensity values, and Affymetrix Expression Console (MAS5) was used to process the CEL files. Quantile normalization was used to normalize all data. To test the uniqueness of the disease-specific genes, we also downloaded DMD microarray data (23 patients) and included this in the test analysis (see Table 2 for more information).
Table 2.
Description of the data downloaded from GEO
| Condition | Description | Study name |
|---|---|---|
| Normal | 120 Microarray, 62 male, 55 female, 3 unknown, age 20–70 and 35 unknown, muscle 114 quadriceps and 6 unknown | GSE674, GSE6011 |
| Diseased | ||
| Amyotrophic lateral sclerosis (ALS) | 9 Microarrays, 9 male, mean age 50, unknown muscle | GSE3307 |
| Acute quadriplegic myopathy (AQM) | 3 Microarrays, 1 male and 2 female, mean age 72.3, unknown muscle | GSE1017 |
| Dermatomyositis (DM) | 5 Microarrays, gender unknown, mean age 57.7 (one microarray with unknown age), unknown muscle | GSE5370 |
| Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) | 4 Microarrays, 3 male and 1 female, mean age 37.3, unknown muscle | GSE1462 |
| Polymyositis (PM) | 6 Microarrays, unknown gender, unknown age, unknown muscle | GSE3112 |
| Duchenne muscular dystrophy (DMD) | 23 Microarray, male, age 1.5–61 months, muscle quadriceps | GSE6011 |
Skeletal muscle function protein (SMFP) and KEGG classification data
The study utilized two different forms of classifications in order to enrich the identified biomarkers in each muscular disease, allowing us to understand their respective roles. The first, and primary, classification used, which we refer to as SMFP Classification (26,27), was based on SMFPs, while the second classification was based on Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The functional protein/gene parts list of skeletal muscle, generated in a past study, consisted of four key functional families: EA family, MECH family, METB family and SIGP family, each containing numerous subfamilies (Supplementary Material, Table S4). As for the KEGG based classification, the needed KEGG pathway data were downloaded from the KEGG database.
Identification of unique genes
Pairwise comparison was then performed for all combination of five muscular diseases and the controls to find differentially regulated genes between any two compared conditions. In order to find the ‘unique genes,’ or biomarkers, for a condition, the intersection of five pairwise comparisons, obtained between the condition and all other conditions, was taken. This cross-intersection allowed us to remove the commonly regulated genes. For example, to find ‘unique genes’ of AQM, we first applied the t-test between AQM and control (normal muscle), giving a list of genes differentially regulated in AQM versus control. Then the same t-test was applied between AQM and ALS; AQM and DM; AQM and PM; and AQM and MELAS, respectively, each of which returned a list of genes differentially regulated in AQM versus the compared condition. P-value cutoff ≤0.05 was used for all the cases. The intersection of these lists represented the genes differentially regulated in AQM compared with all other conditions (Fig. 2). We denoted this intersection as ‘unique genes’ for AQM. Similarly, this method was repeated for ALS, MELAS, DM and PM, revealing the unique genes for each disease (Supplementary Material, Fig. S8). The unique genes are henceforth referred to as ‘uniquely down- or up-regulated’ genes, while differentially regulated genes with respect to healthy only are referred to as either ‘downregulated’ or ‘upregulated’ genes.
Figure 2.

Identification of unique genes for AQM using Venn diagram. Each set (circle) represents a list of differentially regulated genes found between AQM and another conditions. The intersection of all five sets represents the list of uniquely regulated genes for AQM.
Functional enrichment of unique genes
We further performed functional enrichment of these lists of unique genes to find their physiological significance using KEGG pathways and SMFP Classification (26,27). This was done by comparing the ‘unique’ list of genes to a ‘background’. List of all the genes probed in the microarray was considered as a background. Hypergeometric distribution was used to compute enrichment probabilities:
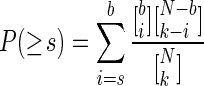 |
(2) |
where, N is the total number of ‘background’ genes, b is the number of ‘background’ genes annotated with the pathway/function in N, k is the total number of ‘selected’ genes and s is the number of selected genes annotated with the term in k. P-value of <0.05 was used for cutoff. Depending on the general direction of the genes, the pathway was considered either up- or down-regulated.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work is supported by grants from National Heart, Lung and Blood Institute (grant numbers R33 HL087375-02, R01 HL113601, 5R01HL106579 and 1R01HL108735-10A1 to S.S.); National Science Foundation (collaborative grant DBI-0835541 and STC 0939370 to S.S.).
REFERENCES
- 1.Engel A., Franzini-Armstrong C. Myology: Basic and Clinical. New York: McGraw-Hill, Medical Pub. Division; 2004. [Google Scholar]
- 2.Larsson L. Acute quadriplegic myopathy: an acquired ‘myosinopathy. Adv. Exp. Med. Biol. 2008;642:92–98. doi: 10.1007/978-0-387-84847-1_8. [DOI] [PubMed] [Google Scholar]
- 3.Rich M.M., Teener J.W., Raps E.C., Schotland D.L., Bird S.J. Muscle is electrically inexcitable in acute quadriplegic myopathy. Neurology. 1996;46:731–736. doi: 10.1212/wnl.46.3.731. [DOI] [PubMed] [Google Scholar]
- 4.Shi P., Gal J., Kwinter D.M., Liu X., Zhu H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim. Biophys. Acta. 2010;1802:45–51. doi: 10.1016/j.bbadis.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller R.G., Mitchell J.D., Lyon M., Moore D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND) Cochrane Database Syst. Rev. 2007:CD001447. doi: 10.1002/14651858.CD001447.pub2. [DOI] [PubMed] [Google Scholar]
- 6.Dalakas M.C. Polymyositis, dermatomyositis, and inclusion-body myositis. New Engl. J. Med. 1991;325:1487–1498. doi: 10.1056/NEJM199111213252107. [DOI] [PubMed] [Google Scholar]
- 7.Ghirardello A., Zampieri S., Tarricone E., Iaccarino L., Gorza L., Doria A. Cutting edge issues in polymyositis. Clin. Rev. Allergy Immunol. 2011;41:179–189. doi: 10.1007/s12016-010-8238-7. [DOI] [PubMed] [Google Scholar]
- 8.Karicheva O.Z., Kolesnikova O.A., Schirtz T., Vysokikh M.Y., Mager-Heckel A.M., Lombes A., Boucheham A., Krasheninnikov I.A., Martin R.P., Entelis N., et al. Correction of the consequences of mitochondrial 3243A>G mutation in the MT-TL1 gene causing the MELAS syndrome by tRNA import into mitochondria. Nucl. Acids Res. 2011;39:8173–8186. doi: 10.1093/nar/gkr546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choy E.H., Isenberg D.A. Treatment of dermatomyositis and polymyositis. Rheumatology. 2002;41:7–13. doi: 10.1093/rheumatology/41.1.7. [DOI] [PubMed] [Google Scholar]
- 10.Kim E.K., Choi E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Ilzecka J., Stelmasiak Z., Dobosz B. Transforming growth factor-Beta 1 (tgf-Beta 1) in patients with amyotrophic lateral sclerosis. Cytokine. 2002;20:239–243. doi: 10.1006/cyto.2002.2005. [DOI] [PubMed] [Google Scholar]
- 12.Confalonieri P., Bernasconi P., Cornelio F., Mantegazza R. Transforming growth factor-beta 1 in polymyositis and dermatomyositis correlates with fibrosis but not with mononuclear cell infiltrate. J. Neuropathol. Exp. Neurol. 1997;56:479–484. doi: 10.1097/00005072-199705000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Chabi B., Adhihetty P.J., Ljubicic V., Hood D.A. How is mitochondrial biogenesis affected in mitochondrial disease? Med. Sci. Sports Exerc. 2005;37:2102–2110. doi: 10.1249/01.mss.0000177426.68149.83. [DOI] [PubMed] [Google Scholar]
- 14.Edgar R., Domrachev M., Lash A.E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucl. Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramaniam S., Hsiao G. Gene-expression measurement: variance-modeling considerations for robust data analysis. Nat. Immunol. 2012;13:199–203. doi: 10.1038/ni.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baldi P., Long A.D. A Bayesian framework for the analysis of microarray expression data: regularized t -test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- 17.Pappas C.T., Krieg P.A., Gregorio C.C. Nebulin regulates actin filament lengths by a stabilization mechanism. J. Cell. Biol. 2010;189:859–870. doi: 10.1083/jcb.201001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouteloup C., Desport J.C., Clavelou P., Guy N., Derumeaux-Burel H., Ferrier A., Couratier P. Hypermetabolism in ALS patients: an early and persistent phenomenon. J. Neurol. 2009;256:1236–1242. doi: 10.1007/s00415-009-5100-z. [DOI] [PubMed] [Google Scholar]
- 19.Rehling P., Brandner K., Pfanner N. Mitochondrial import and the twin-pore translocase. Nat. Rev. Mol. Cell Biol. 2004;5:519–530. doi: 10.1038/nrm1426. [DOI] [PubMed] [Google Scholar]
- 20.Nagaraju K. Update on immunopathogenesis in inflammatory myopathies. Curr. Opin. Rheumatol. 2001;13:461–468. doi: 10.1097/00002281-200111000-00002. [DOI] [PubMed] [Google Scholar]
- 21.van der Pas J., Hengstman G.J., ter Laak H.J., Borm G.F., van Engelen B.G. Diagnostic value of MHC class I staining in idiopathic inflammatory myopathies. J. Neurol. Neurosurg. Psychiatry. 2004;75:136–139. [PMC free article] [PubMed] [Google Scholar]
- 22.Chen D.Y., Chen Y.M., Lan J.L., Chen H.H., Hsieh C.W., Wey S.J., Lu J.J. Polymyositis/dermatomyositis and nasopharyngeal carcinoma: the Epstein–Barr virus connection? J. Clin. Virol. 2010;49:290–295. doi: 10.1016/j.jcv.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 23.Szodoray P., Alex P., Knowlton N., Centola M., Dozmorov I., Csipo I., Nagy A.T., Constantin T., Ponyi A., Nakken B., et al. Idiopathic inflammatory myopathies, signified by distinctive peripheral cytokines, chemokines and the TNF family members B-cell activating factor and a proliferation inducing ligand. Rheumatology. 2010;49:1867–1877. doi: 10.1093/rheumatology/keq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haslett J.N., Sanoudou D., Kho A.T., Han M., Bennett R.R., Kohane I.S., Beggs A.H., Kunkel L.M. Gene expression profiling of Duchenne muscular dystrophy skeletal muscle. Neurogenetics. 2003;4:163–171. doi: 10.1007/s10048-003-0148-x. [DOI] [PubMed] [Google Scholar]
- 25.Pescatori M., Broccolini A., Minetti C., Bertini E., Bruno C., D'Amico A., Bernardini C., Mirabella M., Silvestri G., Giglio V., et al. Gene expression profiling in the early phases of DMD: a constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007;21:1210–1226. doi: 10.1096/fj.06-7285com. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y., Winters J., Subramaniam S. Functional classification of skeletal muscle networks. I. Normal physiology. J. Appl. Physiol. 2012;113:1884–1901. doi: 10.1152/japplphysiol.01514.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y., Winters J., Subramaniam S. Functional classification of skeletal muscle networks. II. Applications to pathophysiology. J. Appl. Physiol. 2012;113:1902–1920. doi: 10.1152/japplphysiol.01515.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.