

Abstract
Cervical cancer (CC) mortality is a major public health concern since it is the second cause of cancer-related deaths among women. Patients diagnosed with locally advanced CC (LACC) have an important rate of recurrence and treatment failure. Conventional treatment for LACC is based on chemotherapy and radiotherapy; however, up to 40% of patients will not respond to conventional treatment; hence, we searched for a prognostic gene signature able to discriminate patients who do not respond to the conventional treatment employed to treat LACC. Tumor biopsies were profiled with genome-wide high-density expression microarrays. Class prediction was performed in tumor tissues and the resultant gene signature was validated by quantitative reverse transcription–polymerase chain reaction. A 27-predictive gene profile was identified through its association with pathologic response. The 27-gene profile was validated in an independent set of patients and was able to distinguish between patients diagnosed as no response versus complete response. Gene expression analysis revealed two distinct groups of tumors diagnosed as LACC. Our findings could provide a strategy to select patients who would benefit from neoadjuvant radiochemotherapy-based treatment.
Introduction
Cervical cancer (CC) is the second leading cause of cancer-related deaths among women worldwide with an estimated 275,000 deaths in 2008; about 88% of them occur in developing countries. More than 80% of patients affected by CC have large tumors of advanced stage mainly those classified as locally advanced cervical cancer (LACC), for whom the mortality/incidence ratio is about 50% [1,2]. As with other cancers, treatment depends mainly on progression stage and some clinical characteristics such as tumor size [3,4]. LACC is defined by tumors confined to the pelvic wall; therefore, those patients have no distant metastasis. The standard treatment for patients diagnosed with LACC with International Federation of Gynecology and Obstetrics (FIGO) stages from IB2 to IVA [5] consists of radiotherapy in combination with cisplatin-based chemotherapy (40 mg/m2) followed by brachytherapy [5,6]; regrettably, the number of deceased patients due to disease progression after 5 years is as high at 50% [1].
Concomitant treatment based on chemotherapy and radiotherapy (CRT) has provided clinical benefits for pelvic control of CC; however, it has important toxicity in several patients, and some studies have shown that it could not significantly extend the overall survival in at least 40% of patients [7,8]; in addition, up to 35% of patients experience disease progression after CRT [9]. This scenario highlights the need for early detection of innate resistance to conventional or standard therapy, which would allow physicians to provide tailored treatment alternatives as early as possible. The advent of high-throughput technologies enables us to define patients’ tumors as a function of their gene expression profile and use this information to improve identification of patients that would benefit with conventional treatment and those in need of adjuvant therapy. Such an approach has been developed for breast cancer [10], leukemia [11], colon cancer [12], and B cell lymphoma [13]. Nevertheless, this approach is only currently applied in the clinic to breast cancer in the form of MammaPrint (www.agendia.com) and to prostate and colon cancers through Oncotype DX [14] (www.oncotypedx.com).
Patients who do not respond to conventional treatment could require other chemotherapy-based treatment schemes; therefore, their timely detection is crucial. To contribute to this aim, we searched for a gene expression signature able to predict the clinical outcome for LACC patients who receive conventional treatment as soon as at the time of diagnosis.
Thus far, there are no reports showing the use of microarrays to identify gene signatures associated with clinical response to CRT in LACC; here, by means of transcriptome profiling and machine learning algorithm, we identified a group of genes that can be used as molecular markers to predict the clinical outcome in those patients. Our rationale is that primary tumors that have not received any conventional treatment (virgin to treatment) carry expression patterns capable of predicting the potential tumor progression; hence, accurate identification of genes involved in the innate resistance could be employed as a prognosis signature associated with CRT treatment–derived clinical response. In this study, we analyzed the genome-wide expression profiles in a discovery group consisting of 89 LACC patients receiving conventional or standard treatment (CRT) by means of genome-wide high-density arrays, covering 45,000 expressed sequences. A nearest-mean classifier was trained for probe selection in a leave-one-out cross-validation process. We obtained a 27-gene signature capable of predicting with high significance the clinical response as complete response (CR) versus no response (NR). Next, gene expression values were confirmed by quantitative reverse transcription–polymerase chain reaction (qRT-PCR) on an independent validation group of 30 patients, confirming the gene expression signature.
Material and Methods
Tumor Samples
The population under this study included 119 patients prospectively enrolled into the National Cancer Institute of Mexico (INCAN) tumor-banking protocol at the time of diagnosis (April 2010 through August 2012). All patients included accept and signed informed consent; institutional ethics and scientific board committees approved the protocol. Immediately after punch biopsy, tumor samples were split into three pieces, one for pathologic confirmation of at least 80% of tumor cells that is mandatory for this type of molecular profiles and the remaining two for RNA and DNA isolation. RNA and DNA biopsies were frozen in liquid nitrogen until nucleic acid extraction. Eligibility criteria were 1) patients with a confirmed pathologic diagnosis of CC staged IB2 up to IIIB (LACC); 2) biopsies with pathology report with more than 80% of tumors cells; hence, the genomic analysis is mainly addressed for tumor cells; 3) age greater to 20 and less than 60 years; 4) high-quality DNA and RNA; 5) no presence of comorbidities; 6) without previous oncological treatment; and 7) patients able to receive standard or conventional therapy based on concurrent CRT. Chemotherapy was based on weekly cis-diamminedichloroplatinum(II) at 40 mg/m2 during five to six cycles. Radiotherapy consisted of external radiation and intracavitary brachytherapy, for a total dose of 64 to 66 Gy over 67 days [6]. Hence, all patients received the same conventional treatment. Clinical characteristics of patients are summarized in Table 1.
Table 1.
Clinical-Pathologic Status of CCLA Patients (n = 119)
| Characteristics | Patients |
|
|---|---|---|
| N | Percentage | |
| Age | ||
| Median | 48 | |
| Range | 29-69 | |
| Histologic type | ||
| Squamous cell carcinoma | 109 | 92.59% |
| Adenocarcinoma | 10 | 8.41% |
| Tumor size | ||
| ≤ 4 cm | 41 | 34.45% |
| ≥ 4 cm | 71 | 59.66% |
| Without data | 7 | 5.88% |
| Clinical stage (FIGO) | ||
| IB2 | 14 | 11.76% |
| IIA | 1 | 0.84% |
| IIB | 76 | 63.86% |
| IIIA | 1 | 0.84% |
| IIIB | 27 | 22.68% |
| HPV genotyping (frequency) | ||
| Type 16 | 57 | 37.74% |
| Type 18 | 28 | 18.54% |
| Type 45 | 16 | 10.59% |
| Type 33 | 8 | 6.72% |
| Others | 33 | 21.85% |
| Not determined | 9 | 5.96% |
| Patients with HPV co-infection | 33 | 27.73% |
| Patients without HPV co-infection | 77 | 64.70% |
| Not determined | 9 | 7.56 |
| Treatment outcome⁎ | ||
| CR | 79 | 66.38% |
| NR | 36 | 30.25% |
| Without date for desertion | 4 | 3.36% |
All patients received radiotherapy and cisplatin as coadjuvant (50-Gy external radiation, 35-Gy intracavitary brachytherapy, and six cycles of 40 mg/m2cis-diamminedichloroplatinum(II)).
Clinical Definitions
Staging was assessed according to the FIGO classification [15]. Clinical responses were evaluated by RECIST 1.1 criteria and computed axial tomography scan and were assigned as CR, defined as the disappearance of all signs of cancer in response to treatment, and NR, defined as patients with partial, progressive, or stable disease [16].
HPV Genotyping
DNA was obtained from cervical tumor biopsies by means of MagNAPure Compact Instrument following the manufacturer's recommendations (Roche Diagnostics GmbH, Roche Applied Science, Mannheim, Germany). HPV genotyping was assessed by two approaches, linear array HPV genotyping (Roche Diagnostics GmbH, Roche Molecular Biochemicals, Mannheim Germany) and nested multiplex PCR (MY/GP primers) with subsequent PCR-fragment direct sequencing [17].
RNA Purification and Microarray Hybridization
Eighty-nine samples obtained at the time of diagnoses were used to discover a gene expression signature associated with clinical response. We compared gene expression signatures from patients with CR against patients diagnosed as NR. The quality of RNA was assessed by means of 18S:28S ratio. Hybridization targets were prepared from 250 ng of total RNA and amplified with whole transcriptome amplification kit 2 (Sigma-Aldrich, St Louis, MO). Four micrograms of amplified and Cy3-labeled cDNA was used to hybridize onto high-density arrays containing 45,000 features according to the recommended protocol of Nimblegen Roche (Mannheim, Germany). After standard washes, arrays were scanned on the Nimblegen MS200 microarray scanner. Images were stored for further analyses.
Microarray Preprocessing and Statistical Analysis
Scanned images were gridded by using the NimbleScan v2.6 Software (Nimblegen Roche). Then, robust multi-array analysis background normalization and quantile normalization were performed for intra-array and inter-array normalization, respectively. Genes with signal intensities above a 95% random threshold were chosen [18]. Differential expression between clinical outcomes was assessed by moderated t tests and significance statistics for each gene were obtained by the empirical Bayes method implemented in limma package from Bioconductor [19]. Global differential expression was also examined by random sampling of class labels. We selected gene subsets on the basis of classifier optimal performance ranking, as in previous approaches [20,21]. A nearest mean classifier was trained for feature selection in a leave-one-out cross-validation process and feature selection was further tested by another leave-one-out cross-validation procedure to select the profile with the strongest association with clinical response. Graphics were generated using Genesis 2.1 software [22]. The total microarray raw and normalized data of this study are public available at the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE56303.
Validation of Gene Expression Profile by qRT-PCR
We employed the remaining 30 samples to validate the gene expression profile identified in the discovery tumor set. Twenty-seven differentially expressed genes were subjected to qRT-PCR. Each primer set was designed by an experimentally verified computer algorithm and then tested in a quality control assay to guarantee that they yield a single band of the predicted size by agarose gel electrophoresis. The sequence of primers and PCR conditions are shown in Supplemental Table S1. RT reactions were performed according to the MMLV protocol from Promega (Madison, WI) following the vendor's recommendations. Real-time PCR was performed using FastStart SYBR Green Master in Light Cycler 480 Instrument II (Roche, Mannheim, Germany) according to the manufacturer's protocol. Duplicate RT samples were used in each assay, data were normalized with β-actin housekeeping gene, and in a parallel way, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used. The comparative Ct method (ΔΔCt) was used to quantify gene expression, and relative quantification was calculated as 2− ΔΔCt for both housekeeping genes.
We employed the remaining 30 samples to validate the gene expression profile identified in the discovery tumor set. Twenty-seven differentially expressed genes were subjected to qRT-PCR. Each primer set was designed by an experimentally verified computer algorithm and then tested in a quality control assay to guarantee that they yield a single band of the predicted size by agarose gel electrophoresis. The sequence of primers and PCR conditions are shown in Supplemental Table S1. RT reactions were performed according to the MMLV protocol from Promega (Madison, WI) following the vendor's recommendations. Real-time PCR was performed using FastStart SYBR Green Master in Light Cycler 480 Instrument II (Roche, Mannheim, Germany) according to the manufacturer's protocol. Duplicate RT samples were used in each assay, data were normalized with β-actin housekeeping gene, and in a parallel way, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used. The comparative Ct method (ΔΔCt) was used to quantify gene expression, and relative quantification was calculated as 2− ΔΔCt for both housekeeping genes.
Disease-Free Survival
Disease-free survival (DFS) of the resulting patient groups was evaluated using the Kaplan-Meier method, and the statistical significance of survival differences was determined with the log-rank test. Multivariate analysis for confounding factors was performed with the Fisher exact test.
Results
Patient Characteristics
Relevant clinical information of 119 recruited patients in this study is shown in Table 1. The median age at diagnosis was 48 years (range 29 to 59 years). The majority of patients were diagnosed as IIB (63.8%) and IIIB stages (22.7%); 92.6% were squamous cell carcinomas, while 8.4% were adenocarcinoma histologic type. The main HPV types were 18 (18.5%) and 16 (37.7%); an important number of patients (27.7%) were infected with two or more HPV types. The median clinical follow-up was 24 months. Thirty-six (30.2%) patients had NR, while seventy-nine (66.4%) were diagnosed as complete responders (CR), and four patients (3.3%) withdrew from the protocol.
Gene Expression Profile from 89 Tumors
To identify genes differentially expressed in pretreatment biopsies of responders (CR) and non-responders (NR), we applied a supervised classification based on moderated t tests and significance statistics obtained by the empirical Bayes method. We obtained a list of 2133 genes with significant differential expression (P < .02). Figure 1 shows a two-dimensional hierarchical clustering using Pearson correlation distance and complete linkage clustering obtained from Genesis 2.1 software [22]. Dendrograms shown in Figure 1 represent the similarity between clinical samples based on gene expression profiles; the length and subdivision of the branches show the similarity between CC tumors (left) and the gene expression profiles (top). The four patients who withdrew from the protocol are represented in red in the left dendrogram. Two tumor groups are clearly observed; the dashed line indicates the subdivision of these two main groups. In the top group (cluster A), there are 56 tumor samples, from which 46 responded favorably (CR group), which represents 82.1% of tumors contained in cluster A. While the bottom group (cluster B) contains 33 samples, from which 14 (42.4%) had no response (NR group), this feature suggests that LACC tumors can be divided with respect to their clinical outcome based on the differential expression pattern of 2133 genes (fold change > 1.5 and P < .02). Information explaining clinical outcome and the relationship between both clusters to some relevant clinical features is shown in Table 2. To have a complete clinical perspective, we associated tumor gene expression profiles with informative clinical features: FIGO stage, tumor size, and co-infections by two or more HPV types. Of 68 tumors classified as IB2 to IIB stages, 42 were grouped in cluster A and 26 in cluster B; from 21 tumors classified IIIA to IIIB, 14 and 7 were grouped in clusters A and B, respectively. Tumor size is a clinical marker frequently used as a clinical response predictor [3,4]; from 54 biopsies with an initial tumor size greater than four centimeters, 29 (53.7%) belonged to cluster A and 25 (46.2%) were grouped in cluster B. Regarding HPV co-infection, of 23 samples with two or more HPV types, cluster A had 11 samples and the rest were grouped in cluster B; therefore, tumor samples with HPV co-infection grouped in cluster B represent 36.3% of the samples. The multivariate significance analysis of clinical characteristics summarized in Table 3 showed that only FIGO stage had a slight significant association to clinical response (P = .026).
Figure 1.
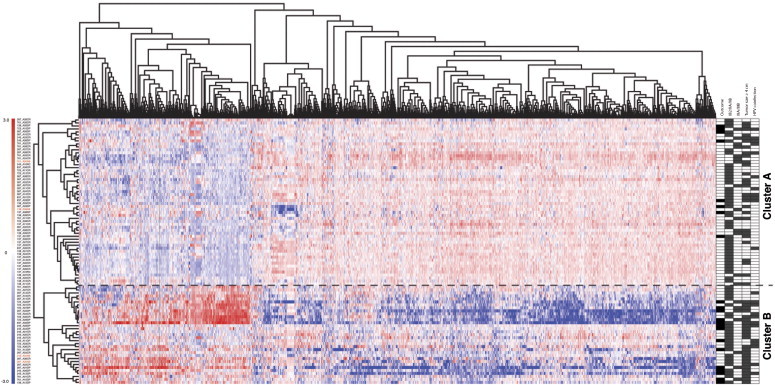
Supervised two-dimensional cluster analysis of 89 CC tumor profiles. Two-dimensional presentation of transcript ratios for 89 CC tumors. We selected 2133 genes with fold change > 1.5 and P value < .02. In the right panel, each individual’s response diagnosis status after 2.5-year follow-up period is indicated as black squares for NR and white squares for CR. Clinical characteristics such as stage (FIGO classification), tumor size > 4 cm, and co-infection by two of more HPV types are shown as gray squares.
Table 2.
Clinical Features Associated with Supervised Two-Dimensional Cluster Analysis of 89 CC Tumor Profiles
| N | Patients Who Withdrew | CR | NR | IB2 | IIA | IIB | IIIA | IIIB | Tumor Size ≥ 4 | HPV Co-Infection | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster A | 56 | 2 | 46 | 8 | 5 | 1 | 36 | 1 | 13 | 29 | 11 |
| Cluster B | 33 | 2 | 17 | 14 | 5 | 0 | 21 | 0 | 7 | 25 | 12 |
| Overall | 89 | 4 | 63 | 22 | 10 | 1 | 57 | 1 | 20 | 54 | 23 |
Table 3.
Multivariate Significance Analysis
| Clinical Feature | CR | NR | P Value | ||
|---|---|---|---|---|---|
| Multivariate analysis of cluster A with CR and NR (n = 56) | |||||
| FIGO stage | IB2/IIA/IIB | 35 | 7 | P = .664 | Not significantly |
| IIIA/IIIB | 13 | 1 | |||
| Tumor size | < 4 cm | 26 | 1 | P = .052 | Not significantly |
| ≥ 4 cm | 22 | 7 | |||
| HPV infection | Single | 40 | 5 | P = .181 | Not significantly |
| Co-infection | 8 | 3 | |||
| Multivariate analysis of cluster B with CR and NR (n = 33) | |||||
| FIGO stage | IB2/IIA/IIB | 18 | 8 | P = 0.026 | Significantly |
| IIIA/IIIB | 1 | 6 | |||
| Tumor size | < 4 cm | 7 | 1 | P ≤ .098 | Not significantly |
| ≥ 4 cm | 12 | 13 | |||
| HPV infection | Single | 12 | 9 | P = 1.000 | Not significantly |
| Co-infection | 7 | 5 | |||
P Values for stage, tumor size, and HPV co-infection were calculated by Fisher exact test.
Prediction Model for the Prognostic Profile in LACC
To identify genes with scores capable of discriminating between CR and NR clinical outcomes, we employed a supervised classification approach that has shown success in previous studies [20,21]. Genes ranked by significance of differential expression were used and a nearest mean classifier was trained in a leave-one-out cross-validation process to select genes with the best classifier performance. Using the predictive algorithm, a 27-gene signature was developed with maximum accuracy in predicting clinical response status (sensitivity: 74%, that is the capacity to predict patients with NR; specificity: 91.3%, that predict the patients with CR; and overall accuracy of 90%); Figure Supplementary S1 shows the classifier performance, while the description of the 27-gene signature is summarized in Supplementary Table S2. The expression pattern of genes present in this 27-gene signature panel is shown in Figure 2. The left panel shows the classifier itself, clinical outcome is represented with black circles for patients with NR and white circles for CR, and the score for each one is indicated in the x-axis. According to this score, patients were divided in two groups delimited by a red line: 17 of 21 (80%) patients with NR and 62 of 68 (91%) patients with CR were assigned the expected score (lying to the right and left of the red line, respectively) showing the high sensitivity and specificity of our predictor. Each patient’s 27-gene profile is displayed in the heat map at the right, where contrasting patterns can be observed at the top and bottom, suggesting an expression profile gradient between patients with the best (top) and worst (bottom) prognoses. Interestingly, the apparent threshold indicated by a horizontal black line corresponds to the actual disease outcome of the patients represented by black or white circles in the classifier at the left. Moreover, Kaplan-Meier analysis showed a statistically significant difference in DFS between the NR and CR groups (Figure 3). The NR group had a mean DFS of 16 months, whereas the CR had a median survival that had not yet been reached (log-rank P = 1 × 10− 16).
Figure 2.
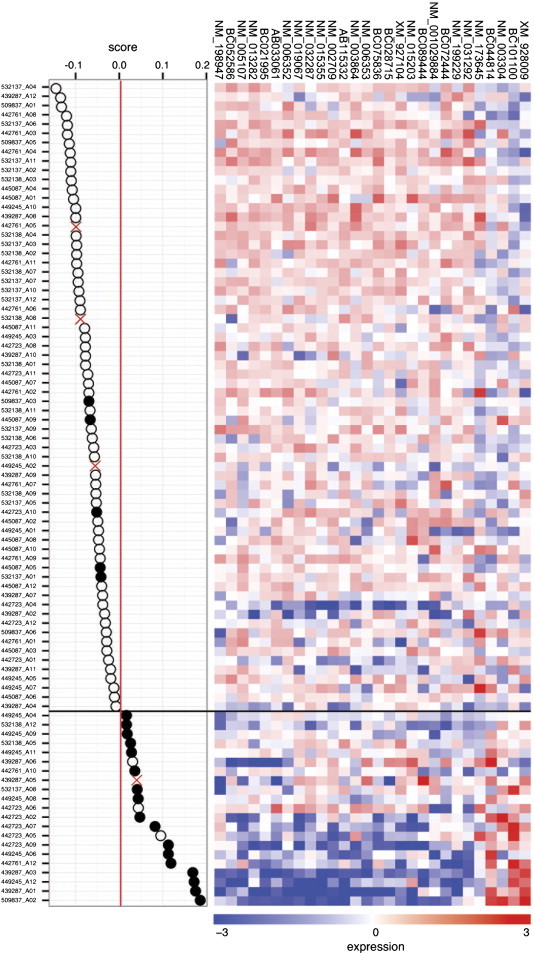
CC-CTRP. The predictor genes are clustered based on their similarities across the 89 tumors. The left panel shows tumors ordered according to their CC-CTRP score; the red line divides positive and negative values. White circles indicate CR tumors, and black circles indicate NR cases. The heat map (right) shows the expression of the 27-gene signature.
Figure 3.
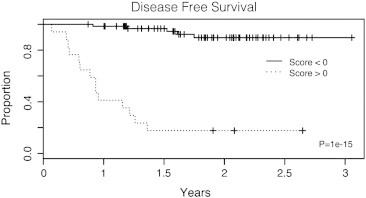
Kaplan-Meier DFS analysis based on 27-gene predictor stratification. The behavior of CC-CTRP for patients with NR to conventional treatment is shown by a dotted line and that for patients with CR is shown by a continuous line; both groups are clearly separated within the first months. Clinical response was assessed according to RECIST 1.1 criteria.
To identify genes with scores capable of discriminating between CR and NR clinical outcomes, we employed a supervised classification approach that has shown success in previous studies [20,21]. Genes ranked by significance of differential expression were used and a nearest mean classifier was trained in a leave-one-out cross-validation process to select genes with the best classifier performance. Using the predictive algorithm, a 27-gene signature was developed with maximum accuracy in predicting clinical response status (sensitivity: 74%, that is the capacity to predict patients with NR; specificity: 91.3%, that predict the patients with CR; and overall accuracy of 90%); Figure Supplementary S1 shows the classifier performance, while the description of the 27-gene signature is summarized in Supplementary Table S2. The expression pattern of genes present in this 27-gene signature panel is shown in Figure 2. The left panel shows the classifier itself, clinical outcome is represented with black circles for patients with NR and white circles for CR, and the score for each one is indicated in the x-axis. According to this score, patients were divided in two groups delimited by a red line: 17 of 21 (80%) patients with NR and 62 of 68 (91%) patients with CR were assigned the expected score (lying to the right and left of the red line, respectively) showing the high sensitivity and specificity of our predictor. Each patient’s 27-gene profile is displayed in the heat map at the right, where contrasting patterns can be observed at the top and bottom, suggesting an expression profile gradient between patients with the best (top) and worst (bottom) prognoses. Interestingly, the apparent threshold indicated by a horizontal black line corresponds to the actual disease outcome of the patients represented by black or white circles in the classifier at the left. Moreover, Kaplan-Meier analysis showed a statistically significant difference in DFS between the NR and CR groups (Figure 3). The NR group had a mean DFS of 16 months, whereas the CR had a median survival that had not yet been reached (log-rank P = 1 × 10− 16).
Validation by qRT-PCR
To confirm the discrimination capability of the Cervical Cancer Conventional Treatment Response Profile (CC-CTRP) gene signature, we searched for gene expression levels of all 27 genes that were validated by qRT-PCR in an independent group formed by 30 independent patients with LACC diagnosis. Total RNA was isolated from biopsies taken before treatment; diagnosis-wise 16 of these patients were classified as CR and 14 as NR. For the sake of clarity, we analyzed the qRT-PCR results as ΔΔCt(log2), which represents the fold change relative to the β-actin housekeeping gene (Figure 4A); in a parallel analysis, we used another housekeeping gene (GAPDH) to confirm the consistency of results (Figure 4B). Although GAPDH and β-actin housekeeping genes showed slight differences in expression levels (Supplementary Figure S2), the ability to discriminate both clinical responses (CR vs NR) does not show significant differences (Supplementary Figure S3, A and B). Hence, the RT-PCR results were evaluated as ΔΔCt(log2) grouped patients in accordance with the initial 27-gene classifier, regardless of the source of the data, qRT-PCR, or microarrays.
Figure 4.
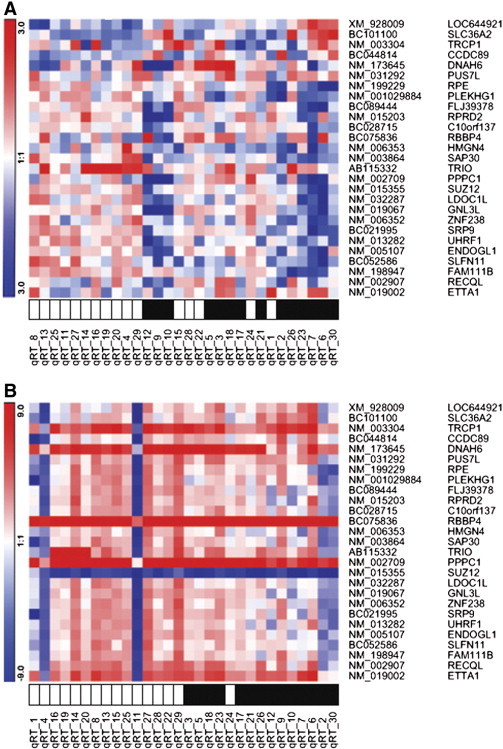
qRT-PCR validation. The cluster illustrates a comparison of data from real-time qRT-PCR of 30 patient analyses of each of the CC-CTRP signature. Clustering was obtained by means of their similarities (Pearson correlation and complete linkage clustering). Black squares represent non-responders; white squares represent complete responders. Gene expression levels were normalized to (A) β-actin and (B) GAPDH.
Discussion
Despite of the increase in early detection programs, CC still remains one of the principal neoplasms causing death of women throughout the world since most patients are diagnosed only when they arrive to health centers and the disease has often reached locally advanced stages. Thus far, no generally accepted molecular marker for CC has been reported [23] and clinical parameters are the only strategy currently used in the prediction of disease outcome.
In this work, we aimed to find a molecular signature associated with chemo-radioresistance of LACC; tumor biopsies were carefully selected to fulfill inclusion criteria, which included that each biopsy had more than 80% of tumor cells. We obtained the differentially expressed gene profiles and their correlation with the clinical outcome of 89 tumors and used these data to build a predictor algorithm to identify chemoradiotherapy-resistant individuals. The data were later validated by assessing 30 additional samples in an independent group, for a total of 119 LACC analyzed patients.
We identified a 27-gene molecular signature with high prognostic value, which proved to be more effective than tumor size in predicting disease outcome that is currently the main clinical approach used as predictive marker [3] (Table 3). Our predictor sorted correctly 17 of 22 patient diagnoses; this figure represents 80% of effectiveness. The Kaplan-Meier graph in Figure 3 shows that disease outcome can be identified as early as 4 months by using our approach.
Several works have used microarray technologies in the search for CC molecular signatures associated with different conditions such as radioresistance [24–26], early stage lymph node metastasis [27], and resistance to angiocidin and darapladib-based anti-tumoral and anti-inflammatory treatment [28]. However, despite assessing the same tumor type, these molecular signatures lack consistency, mainly because treatment selection is not based on NCI standards, which could affect the reported genes. In addition, this may result from the diverseness of the experimental designs or intrinsic bias of the different microarray platforms [23]. However, the populations that these authors have analyzed, while seemingly similar, are actually very diverging when observed from the perspective of the therapy. Thus, we find it reasonable to speculate that different treatments elicit variable but specific gene expression profiles. Moreover, intra-tumor source heterogeneity is an important but seldom considered point. Bachtiary and co-workers suggest that increasing sample size can serve as a remedial measure, based on variance-component analysis of the genetic properties of replicate cancer biopsies [29]. Currently, there is no previous report of a molecular signature associated with conventional treatment response. To our knowledge, this work is the first transcriptome-based molecular signature associated with conventional treatment based on chemoradiotherapy resistance and the largest LACC sample number assessed in such a study.
Conclusion
Patients diagnosed with LACC are submitted to conventional treatment, without certainty of a CR, due to tumor chemo-radiotherapy resistance. The CC-CTRP gene signature, obtained in this work, is a novel prognosis tool aimed at sorting patients with regard to their sensibility to conventional treatment; consequently, it would be possible to give those with a bad prognosis the opportunity to undertake alternative or complementary treatment without prior exposition to conventional treatment, avoiding unnecessary weakening and thus increasing their survival possibilities. However, more studies would be necessary to increase evidence about the utility of the current molecular signature.
The following are the supplementary data related to this article.
Primer Pairs Used for Relative Quantification by qRT-PCR
Genes Showing Different Expressions Between CR and NR Groups
Supplementary Figure S1. Predictor performance. A genomic average profile capable of discriminating favorable outcome or poor prognosis in LACC was obtained. The genomic average profile used 27 genes and performed with 74% sensitivity, 91.3% specificity, and 90% overall accuracy.
Supplementary Figure S2. Relative expression of housekeeping genes. Relative intensity of actin (ACT) and GAPDH housekeeping genes, tested in 89 biopsies of CC patients (top panel) by microarray analysis and 30 external samples by qRT-PCR (bottom panel).
Supplementary Figure S3 (A) qRT-PCR validation. Expression of 27 CC-CTRP genes in 30 independent LACC patients, using β-actin housekeeping gene as normalizer; 16 were CR, while 14 were NR. 2− ΔΔCt values were plotted in two sets of patients. The median values are shown as horizontal bars. P values for individual genes were calculated by Mann-Whitney test. (B) qRT-PCR validation. Expression of 27 CC-CTRP genes in 30 independent LACC patients; 2− ΔΔCt data were normalized with GAPDH housekeeping gene. Values were plotted in two sets of patients; 16 were CR, while 14 were NR. Median values are shown as horizontal bars. P values for individual genes were calculated by unpaired one-tailed test.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.tranon.2015.01.003.
Acknowledgements
This manuscript was submitted in partial fulfillment of J.F.-R.'s requirements for a PhD degree in Posgrado en Ciencias Biologicas, Universidad Nacional Autonoma de Mexico. We want to acknowledge the technical support of Abraham Pedroza for housekeeping gene experiments.
Footnotes
This work was supported in part by the Universidad Nacional Autonoma de Mexico (UNAM; grant IACOD-TB200111) and by Consejo Nacional de Ciencia y Tecnologia (CONACyT) (grants SALUD-2009-01-113948, SALUD-2010-01-141907 and CONACYT-Catedras-2425-Genomica Integral del Cancer) and National Cancer Institute of Mexico (INCAN) scientific research funds. J.F.-R. received a CONACyT fellowship (104639).
Conflicts of interest: The Cervical Cancer Conventional Treatment Response Profile (CC-CTRP) consisting in 27 genes has been protected by the Mexican Patent Office. The referral number is MX-E-2014-015826.
Author contributions: J.F.-R. and F.L.-G. contributed equally to this work. C.P.-P. planned, organized, and conceived the original idea. O.P.-Z., E.L.-U., J.C.-M., and D.C.L. monitored the patients who participated in this project. R.V.-R. and D.P.-M. did histopathologic analysis. J.F.-R. and F.L.-G. carried out the processing of microarrays, acquisition of data and analysis, and interpretation of data. J.F.-R., F.L.-G., E.L.-U., and C.P.-P. planned and draw all figures and discussed the manuscript. J.F.-R., F.L.-G., N.J.-H., N.R.-N., and C.P.-P. wrote and revised the manuscript.
This article refers to supplementary materials, which are designated by Supplementary Tables S1 and S2 and Supplementary Figures S1 to S3 and are available online at www.transonc.com.
References
- 1.Green JA, Kirwan JM, Tierney JF, Symonds P, Fresco L, Collingwood M. Survival and recurrence after concomitant chemotherapy and radiotherapy for cancer of the uterine cervix: a systematic review and meta-analysis. Lancet. 2001;358:781–786. doi: 10.1016/S0140-6736(01)05965-7. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Kapp KS, Poschauko J, Tauss J, Berghold A, Oechs A, Lahousen M. Analysis of the prognostic impact of tumor embolization before definitive radiotherapy for cervical carcinoma. Int J Radiat Oncol Biol Phys. 2005;62:1399–1404. doi: 10.1016/j.ijrobp.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 4.Kapp KS, Stuecklschweiger GF, Kapp DS, Poschauko J, Pickel H, Lahousen M. Prognostic factors in patients with carcinoma of the uterine cervix treated with external beam irradiation and IR-192 high-dose-rate brachytherapy. Int J Radiat Oncol Biol Phys. 1998;42:531–540. doi: 10.1016/s0360-3016(98)00255-7. [DOI] [PubMed] [Google Scholar]
- 5.Waggoner SE. Cervical cancer. Lancet. 2003;361:2217–2225. doi: 10.1016/S0140-6736(03)13778-6. [DOI] [PubMed] [Google Scholar]
- 6.National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology Cervical cancer. V. 1.2009. 2009. http://www.nccn.org
- 7.Kirwan JM, Symonds P, Green JA, Tierney J, Collingwood M, Williams CJ. A systematic review of acute and late toxicity of concomitant chemoradiation for cervical cancer. Radiother Oncol. 2003;68:217–226. doi: 10.1016/s0167-8140(03)00197-x. [DOI] [PubMed] [Google Scholar]
- 8.Pearcey R, Brundage M, Drouin P, Jeffrey J, Johnston D, Lukka H. Phase III trial comparing radical radiotherapy with and without cisplatin chemotherapy in patients with advanced squamous cell cancer of the cervix. J Clin Oncol. 2002;20:966–972. doi: 10.1200/JCO.2002.20.4.966. [DOI] [PubMed] [Google Scholar]
- 9.Chemoradiotherapy for Cervical Cancer Meta-Analysis Collaboration Reducing uncertainties about the effects of chemoradiotherapy for cervical cancer: a systematic review and meta-analysis of individual patient data from 18 randomized trials. J Clin Oncol. 2008;26:5802–5812. doi: 10.1200/JCO.2008.16.4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AAM, Voskuil DW. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 11.Yeoh E-J, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–143. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 12.Barrier A, Boelle P-Y, Roser F, Gregg J, Tse C, Brault D. Stage II colon cancer prognosis prediction by tumor gene expression profiling. J Clin Oncol. 2006;24:4685–4691. doi: 10.1200/JCO.2005.05.0229. [DOI] [PubMed] [Google Scholar]
- 13.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 14.Kittaneh M, Montero AJ, Glück S. Molecular profiling for breast cancer: a comprehensive review. Biomark Cancer. 2013;5:61–70. doi: 10.4137/BIC.S9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirschowitz L, Nucci M, Zaino RJ. Problematic issues in the staging of endometrial, cervical and vulval carcinomas. Histopathology. 2013;62:176–202. doi: 10.1111/his.12058. [DOI] [PubMed] [Google Scholar]
- 16.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 17.Sotlar K, Diemer D, Dethleffs A, Hack Y, Stubner A, Vollmer N. Detection and typing of human papillomavirus by e6 nested multiplex PCR. J Clin Microbiol. 2004;42:3176–3184. doi: 10.1128/JCM.42.7.3176-3184.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simon A, Biot E. ANAIS: analysis of NimbleGen arrays interface. Bioinformatics. 2010;26:2468–2469. doi: 10.1093/bioinformatics/btq410. [DOI] [PubMed] [Google Scholar]
- 19.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AAM, Mao M. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 21.Roepman P, Wessels LFA, Kettelarij N, Kemmeren P, Miles AJ, Lijnzaad P. An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet. 2005;37:182–186. doi: 10.1038/ng1502. [DOI] [PubMed] [Google Scholar]
- 22.Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18:207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- 23.Harima Y, Togashi A, Horikoshi K, Imamura M, Sougawa M, Sawada S. Prediction of outcome of advanced cervical cancer to thermoradiotherapy according to expression profiles of 35 genes selected by cDNA microarray analysis. Int J Radiat Oncol Biol Phys. 2004;60:237–248. doi: 10.1016/j.ijrobp.2004.02.047. [DOI] [PubMed] [Google Scholar]
- 24.Kitahara O, Katagiri T, Tsunoda T, Harima Y, Nakamura Y. Classification of sensitivity or resistance of cervical cancers to ionizing radiation according to expression profiles of 62 genes selected by cDNA microarray analysis. Neoplasia. 2002;4:295–303. doi: 10.1038/sj.neo.7900251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajkumar T, Vijayalakshmi N, Sabitha K, Shirley S, Selvaluxmy G, Bose MV. A 7 gene expression score predicts for radiation response in cancer cervix. BMC Cancer. 2009;9:365. doi: 10.1186/1471-2407-9-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong YF, Selvanayagam ZE, Wei N, Porter J, Vittal R, Hu R. Expression genomics of cervical cancer: molecular classification and prediction of radiotherapy response by DNA microarray. Clin Cancer Res. 2003;9:5486–5492. [PubMed] [Google Scholar]
- 27.Huang L, Zheng M, Zhou Q-M, Zhang M-Y, Jia W-H, Yun J-P. Identification of a gene-expression signature for predicting lymph node metastasis in patients with early stage cervical carcinoma. Cancer. 2011;117:3363–3373. doi: 10.1002/cncr.25870. [DOI] [PubMed] [Google Scholar]
- 28.Koch M, Wiese M. Gene expression signatures of angiocidin and darapladib treatment connect to therapy options in cervical cancer. J Cancer Res Clin Oncol. 2013;139:259–267. doi: 10.1007/s00432-012-1317-9. [DOI] [PubMed] [Google Scholar]
- 29.Bachtiary B, Boutros PC, Pintilie M, Shi W, Bastianutto C, Li J-H. Gene expression profiling in cervical cancer: an exploration of intratumor heterogeneity. Clin Cancer Res. 2006;12:5632–5640. doi: 10.1158/1078-0432.CCR-06-0357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer Pairs Used for Relative Quantification by qRT-PCR
Genes Showing Different Expressions Between CR and NR Groups
Supplementary Figure S1. Predictor performance. A genomic average profile capable of discriminating favorable outcome or poor prognosis in LACC was obtained. The genomic average profile used 27 genes and performed with 74% sensitivity, 91.3% specificity, and 90% overall accuracy.
Supplementary Figure S2. Relative expression of housekeeping genes. Relative intensity of actin (ACT) and GAPDH housekeeping genes, tested in 89 biopsies of CC patients (top panel) by microarray analysis and 30 external samples by qRT-PCR (bottom panel).
Supplementary Figure S3 (A) qRT-PCR validation. Expression of 27 CC-CTRP genes in 30 independent LACC patients, using β-actin housekeeping gene as normalizer; 16 were CR, while 14 were NR. 2− ΔΔCt values were plotted in two sets of patients. The median values are shown as horizontal bars. P values for individual genes were calculated by Mann-Whitney test. (B) qRT-PCR validation. Expression of 27 CC-CTRP genes in 30 independent LACC patients; 2− ΔΔCt data were normalized with GAPDH housekeeping gene. Values were plotted in two sets of patients; 16 were CR, while 14 were NR. Median values are shown as horizontal bars. P values for individual genes were calculated by unpaired one-tailed test.