

Abstract
Breast tumors in (FVB × BALB-NeuT) F1 mice have characteristic loss of chromosome 4 and sporadic loss or gain of other chromosomes. We employed the Illumina GoldenGate genotyping platform to quantitate loss of heterozygosity (LOH) across the genome of primary tumors, revealing strong biases favoring chromosome 4 alleles from the FVB parent. While allelic bias was not observed on other chromosomes, many tumors showed concerted LOH (C-LOH) of all alleles of one or the other parent on sporadic chromosomes, a pattern consistent with cytogenetic observations. Surprisingly, comparison of LOH in tumor samples relative to normal unaffected tissues from these animals revealed significant variegated (stochastic) deviations from heterozygosity (V-LOH) in every tumor genome. Sequence analysis showed expected changes in the allelic frequency of single nucleotide polymorphisms (SNPs) in cases of C-LOH. However, no evidence of LOH due to mutations, small deletions, or gene conversion at the affected SNPs or surrounding DNA was found at loci with V-LOH. Postulating an epigenetic mechanism contributing to V-LOH, we tested whether methylation of template DNA impacts allele detection efficiency using synthetic oligonucleotide templates in an assay mimicking the GoldenGate genotyping format. Methylated templates were systematically over-scored, suggesting that the observed patterns of V-LOH may represent extensive epigenetic DNA modifications across the tumor genomes. As most of the SNPs queried do not contain standard (CpG) methylation targets, we propose that widespread, non-canonical DNA modifications occur during Her2/neuT-driven tumorigenesis.
Abbreviations: SNP, single nucleotide polymorphism; LOH, loss of heterozygosity; ASO, allele-specific oligonucleotide probe; LSO, locus-specific oligonucleotide probe
Introduction
Genetic instability is a key characteristic of most advanced cancers as they progress toward increasingly malignant phenotypes [1]. Genetic instability also complicates the long-term success of conventional chemotherapies, target-specific therapies, as well as newer cancer vaccine strategies, as genetic diversity within tumor cell populations allows for multiple mutant phenotypes to be acquired and exist in dynamic genetic and epigenetic landscapes [2,3].
Mouse models of cancer provide important insights into the biology of spontaneous cancer development [4,5]. Using inbred animals, stages of tumor development can be followed under reproducible, controlled circumstances allowing dissection of regulatory checkpoints that are overcome as tumors emerge. These models also provide experimental systems for evaluating strategies for cancer therapy [6–8]. Whereas tumors are sometimes cured in mice, translation of the same strategies to human patients has been less effective [9–11].
One limitation of inbred experimental tumor models is their inability to account for genetic events that generate populations of phenotypically distinct cells on the assortment of alleles among mitotic progeny. Loss of heterozygosity (LOH) is a common characteristic of human cancers [12–14], but the importance of mechanisms leading to LOH in tumor evolution in humans is primarily investigated once tumors are already established [15,16]. Mechanisms leading to tumor development influenced by LOH are underappreciated in most mouse models as LOH is masked in inbred animals.
Nonetheless, several studies have used F1 intercrosses between two inbred mouse strains to demonstrate patterns of LOH. Early studies were interpreted as evidence for selective loss of tumor suppressor genes [17–19]. Most of these studies used low-resolution mapping of tumor genotypes with microsatellite and single nucleotide polymorphism (SNP) typing and, thus, provided a limited view of genetic instability in the emerging tumors. Major genetic instability on mouse chromosome 4 has been observed repeatedly using these methods, as have minor patterns of LOH on other chromosomes [20,21].
In the present study, we used a medium density genotyping bead array to evaluate the assortment of alleles in spontaneous breast tumors emerging in genetically identical FVB × BALB-NeuT F1 female mice. The array is capable of determining the genotypes of up to 32 DNA samples simultaneously; parental alleles differ at 553 polymorphic SNPs, representing every chromosome of the mouse genome. Using an internally matched set of normal samples as reference, we quantified LOH for each heterozygous SNP in each tumor sample. Two patterns of LOH were revealed, one in which the same parental allele was poorly represented for most or all SNPs along a given chromosome. This concerted LOH (C-LOH) was validated by sequencing genomic regions around several SNPs using DNA from individual tumors. A second, unexpected pattern of LOH (variegated) was also revealed. These regions of the tumor genomes were found to have remained heterozygous (i.e., mirroring the germline). As genotyping interrogates regions of naked DNA flanking each SNP, we present data to suggest that genome-wide epigenetic events were detected by this assay in the F1 tumor genomes. As such, this experimental approach provides a new method for evaluating genetic and environmental variables regulating somatic genetic changes in evolving tumors.
Materials and Methods
Mice
Hemizygous BALB/c-neuT mice were originally acquired from Dr Guido Forni [7,22] and were maintained by intercross with transgene negative BALB/c female littermates such that all transgene-positive animals contained a single copy of the mouse mammary tumor virus promoter–driven neuT transgene. The presence of the transgene was monitored using DNA primers (gtaacacaggcagatgtagga and actggtgatgtcggcgatat) in a standard polymerase chain reaction (PCR) assay. F1 hybrid mice were from matings of neuT transgene-positive BALB/c males with wild-type FVB/J females. Only female progeny were used in this study. Low-fat diet (LFD) and high-fat diet (HFD) were from Research Diets (New Brunswick, NJ) (D12450H and D12451) and introduced at weaning.
Tumor Incidence and Recovery of Tumors and Other Tissues
Animals were monitored weekly for the appearance of palpable tumors. All animals developed multiple tumors (≥ 5 of 10 glands affected). Tumors were excised from each of the F1 mice when any one tumor exceeded 100 mm2 (width × length). Other tissues (tail, ear, liver, lung, and kidney) were free of visible tumor nodules and were excised as sources of reference DNAs.
DNA Preparation and Analysis
DNA was extracted from the tumors and reference tissues using DNAeasy (Qiagen, Venlo, Netherlands) according to the manufacturer’s procedures.
Genotyping and Statistical Analyses
DNA samples from normal tissue (ear, liver, kidney, and tail) and breast tumors of FVB × BALB-NeuT F1 mice were used for genotyping using the GoldenGate Bead Array by the Mayo Clinic Genotyping Core of the Medical Genomics Facility according to the manufacturer’s recommendations (Illumina, Inc, San Diego, CA). The mouse MD linkage panel used contains ~ 1600 mouse SNPs, ~ 550 of which were found empirically to be heterozygous in normal tissue from our F1 mice. There was some difference in the reported genotypes for FVB and BALB/c relative to the animals used in our study, and we excluded SNPs from the analysis when one allele was not detected with at least 33% intensity of the other allele in the reference tissues. Raw data files were processed using GenomeStudio software (Illumina, Inc). From these data, we extracted measurements of LOH by first computing z-scores ([value − meannormal]/standard deviationnormal) indicating the deviation from the average value from normal tissue for each of the genotyped SNPs. We tested for significant differences in these z-scores between normal and tumor samples using two-sample rank-sum tests for individual SNPs across the entire genome, and also for each chromosome for each tumor by calculating the average z-score on a given chromosome. Z-scores were compared between the 21 tumor samples and the seven normal tissues used in the analysis (see Supplemental Tables 1 and 2). DNA from one tumor sample (a 22nd) was excluded for technical reasons following DNA isolation, and an eighth normal tissue sample was excluded in some cases on the basis of being a significant outlier (more than 5 SD outside the tight cluster of the other samples). Heat maps in Figure 2 summarize analyses both including and excluding this outlier normal sample. Two DNA samples, one normal reference and one tumor, were measured as replicates to assess the reproducibility of measurements on this platform. The concordance correlation coefficients were 0.706 and 0.943, respectively. Parallel statistical assessments including this outlier yielded the same overall conclusions, but the variance of the normal samples was substantially distorted by including this sample.
Figure 2.

Extensive LOH in primary F1 breast tumors. Allelic ratios for each SNP in each tumor sample were converted to z-scores based on the degree of deviation from the average allele intensities for the normal tissue DNAs. SNPs are shown color-coded across each of the chromosomes for tumors (N = 21) if the SNP z-score is > 2 SDs outside of heterozygous measurements of normal tissues (white, apparent loss of FVB SNP, z-score > 2; black, apparent loss of BALB/c SNP, z-score < − 2; gray, retained heterozygosity). (A) Calculations based on analysis including an apparent outlier normal sample (> 5 SDs outside the other samples) from tumor-bearing mice; (B) calculations based on data omitting that outlier. While the overall results are similar, this approach reveals additional positions of LOH throughout the genomes of the tumors.
PCR Amplification and Sequence Validation of Tumor Genotype Calls
We used the BALB genome to guide the design of oligonucleotide primers that would amplify approximately 2 kb of genomic DNA flanking SNP gnf04.123.467 on chromosome 4 (forward primer: 5′-TGGACACTTTGCCCCTTCTTAGAAT-3′ and reverse primer: 5′-TTCCATTTTCCATTTCATAAATGAGG-3′) and CEL-15_9687257 on chromosome 15 (forward primer: 5′-CACTGTGCTGCCTTTGACAAGGATTC-3′ and reverse primer: 5′-CTTTGGCAGATAAAGTTTGCACGACC-3′). Genomic DNA used previously for genotyping assay was amplified with Phusion Hot Start DNA Polymerase according to the manufacturer’s recommendations (New England BioLabs, Ipswich, MA). PCR products were resolved by agarose gel electrophoresis, purified, and subjected to Sanger sequencing.
In Vitro SNP Detection and Impact of Modified DNA Templates
A “GG-in-a-tube” assay was developed on the basis of the workflow diagrams of the GoldenGate Genotyping Assay and known genomic sequences around SNP CEL-15_9687257 on mouse chromosome 15. Synthetic FVB.0 template (5′-GATATACATGCATACTGAGACTCAGTGGACAGAGAAAGCAGAAGCTTTCTAGC-3′), and BALB.0 template (5′-GATATACATGCATACTGAGACTCAGTAGAC*AGAGAAAGC*AGAAGCTTTCTAGC-3′), wild-type or internally substituted (at the *) 5-methyl- or 5-hydroxymethyl-deoxycytosine templates, were obtained from Integrated DNA Technologies Inc (Coralville, IA). Allele-specific and locus-specific primers with added tags for post-annealing amplification were designed based on the manufacturer’s (Illumina, Inc) application notes as follows: FVB–allele-specific oligonucleotide (ASO) 5′-ACTTCGTCAGTAACGGACGCTAGAAAGCTTCTGCTTTCTCTGTCC-3′; BALB-ASO 5′-ACTTCGTCAGTAACGGACGCTAGAAAGCTTCTGCTTTCTCTGTCT-3′; locus-specific oligonucleotide (LSO) 5′-CTGAGTCTCAGTATGCATGTATATCAGTCCGAACCTGCCTATGATTCGGTCTGCCTATAGTGAGTC-3′; universal P1 primer 5′-ACTTCGTCAGTAACGGAC-3′; universal P3 primer 5′-GACTCACTATAGGCAGAC-3′. Regions shown underlined correspond to genomic sequences around the SNP. FVB.0 (5 ng) and BALB.0 (25 ng) or modified BALB templates were mixed together in thin-walled PCR tubes with a standardized mixture of FVB-ASO (10 pmol), BALB-ASO (100 pmol), and LSO (10 pmol) primers in Quick Solution buffer, 2.5 U PfuUltra polymerase (Stratagene, LaJolla, CA), and deoxynucleoside triphosphates (Roche, Mannheim, Germany). The reactions were placed in a thermocycler programmed for one cycle: 95°C for 1 minute, 55°C for 1 minute, and 68°C for 15 minutes to anneal and extend the primers. After quick clean-up (Qiagen), the reaction products were ligated using T4 ligase according to the rapid ligation protocol (Invitrogen, Carlsbad, CA) and then used directly for 20 cycles of PCR amplification with P1/P3 primers and Hi-Fidelity polymerase (Roche). Amplification products were resolved by agarose gel electrophoresis, excised, purified (Qiagen), and sequenced using the P3 primer.
Results
Spontaneous Breast Tumors Emerging in Heterozygotes Have Patterns of Cytogenetic Diversity and Extensive LOH in the F1 Tumor Genomes
Female heterozygous FVB × BALB-NeuT F1 mice expressing activated ErbB2 (neu) oncogene under transcriptional control of the mouse mammary tumor virus promoter were monitored weekly for spontaneous tumor development. All animals developed multiple tumors that were palpable by approximately 13 weeks of age. Tumors for study were harvested when any one tumor reached 100 mm2. Spectral karyotyping (SKY) showed changes in chromosome number, deletions, and translocations (Supplemental Figure 1). Loss of chromosome 4 or other aberrations involving the fourth chromosome was a common feature in these tumors (100/100 mitotic figures from 10 different animals). No chromosome 4 alterations were observed in mitotic spreads of normal heterozygous fibroblasts.
The genomes of F1 tumors were surveyed for LOH using genomic DNA purified from F1 tumors and F1 normal tissues (GoldenGate Genotyping Assay; Illumina, Inc). There are 553 SNPs represented on the mouse medium density array that distinguish FVB/J from BALB/c mice. An overview of SNP genotype allele frequencies for 8 normal and 21 tumor genomes is shown in Figure 1B. For nearly every individual SNP across the genome, the allelic ratios of the samples were clustered at 0.5 as expected for an F1 genetic cross. However, closer inspection of the data revealed that the tumor allele frequencies were much more varied compared to the allele frequencies of the normal tissue DNA samples from the same animals.
Figure 1.
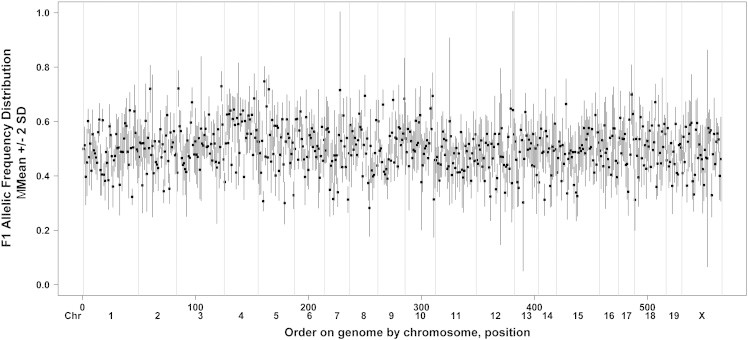
DNA samples from F1 tumors (N = 21) and F1 normal tissues (N = 7) were assessed for heterozygosity using the GoldenGate Assay (Illumina, Inc). The expected frequency of alleles is 0.5. The range of 2 SDs around the mean z-score at each heterozygous SNP is shown chromosome by chromosome. SNPs with a median empirical allele frequency score between 0.75 and 0.25 were selected for further analysis (see below).
To evaluate the tumor allele frequencies more closely, a heterozygosity score was defined operationally for each assay using the mean score empirically established for each SNP using DNA of normal tissue isolated from the F1 animals. Deviation from this mean value was then calculated using a z-score ([value − meannormal]/standard deviationnormal) for each tumor and normal DNA sample (summarized in Supplemental Table 1). Negative z-scores were defined to signify under-representation of the BALB/c allele and positive scores were defined to signify under-representation of the FVB allele. Support for directional loss of LOH was only evident on chromosome 4 (Supplemental Table 2). While the genotyping calls for individual SNPs on chromosomes 8, 11, 12, and 18 were suggestive of skewing toward one allele or the other, this trend was not supported in a follow-up study. However, the strong bias toward loss of BALB/c alleles on chromosome 4 was consistently observed.
The degree of SNP heterozygosity was visualized using the calculated z-scores. A threshold of 2.0 in absolute value was used to score LOH, and the results of the individual SNP calls were displayed for each tumor and normal sample in a heat map (Figure 2; gray, heterozygous; white, apparent loss of FVB allele; black, apparent loss of BALB/c allele). Two versions of this heat map are shown, as genotyping data from one of the normal tissue samples displayed many allelic ratios outside the otherwise tight cluster of normal. Similar results were found with or without this outlier in the analysis. These data revealed extensive LOH throughout the genomes of the tumor samples; 96% of the SNPs shown had undergone LOH in at least 1 of the 21 tumor samples studied. There was concerted loss of the same allele (concerted or “C-LOH”) at adjacent SNPs on chromosome 4 for nearly all tumors. Most often, the C-LOH on chromosome 4 was a loss of BALB alleles, suggesting an allelic bias in whatever mechanism results in loss of chromosome 4. Some tumors also showed C-LOH elsewhere in the genome. No skewing relative to centromeres was noted, indicating that somatic crossing over was not likely a major contributor to tumorigenesis in this model. A portion of the observed patterns of LOH is consistent with the deletions and duplications of chromosomes seen in our cytogenetic analysis. However, many of the measured events in the array analysis did not fit this pattern but instead displayed a variegated pattern of LOH (“V-LOH”) in which one locus exhibited loss of one parental allele, whereas a linked SNP had loss of the opposite allele. A second independent analysis of 16 additional tumors (including analysis of two tumors from the same mouse) and 6 normal tissues revealed the same patterns (not shown). Independent tumors recovered from the same animals (upper left vs lower right quadrants) displayed unrelated patterns of LOH (not shown).
LOH at Chromosome 4 Represents Chromosome Loss
The allelic skewing of the genotypes on chromosome 4 suggested that gross gains or losses of genetic information had occurred at loci across this chromosome, a hypothesis supported by cytogenetic analysis (Figure 1). To verify that genotyping in this manner can be used to visualize a true loss of one of the parental alleles, we identified genomic sequences for both parent strains around one of the SNPs on chromosome 4 (gnf04_123.367; Figure 3A) and used PCR to amplify approximately 2 kb of genome from the same tumor and normal DNA used for genotyping. Sanger sequencing revealed allelic ratios in genomic DNA (Figure 3). Both BALB and FVB alleles were present in DNA sequenced (Figure 3B) from tumor samples scored as heterozygous by genotyping (gray in Figure 3A; absolute z-score < 2 in Figure 3C) such as tumor 23. In contrast, in DNA from samples such as tumor 8 (white in Figure 3A; z-score ~ 14, Figure 3C) and tumor 16 (black in Figure 3A; z-score ~ − 37, Figure 3C), one of the alleles was dominant by both sequencing and genotyping. A similar analysis of SNPs on chromosome 12 validated that the genotyping approach was also sensitive to copy number changes in individual tumors displaying C-LOH at a second genomic location (not shown).
Figure 3.
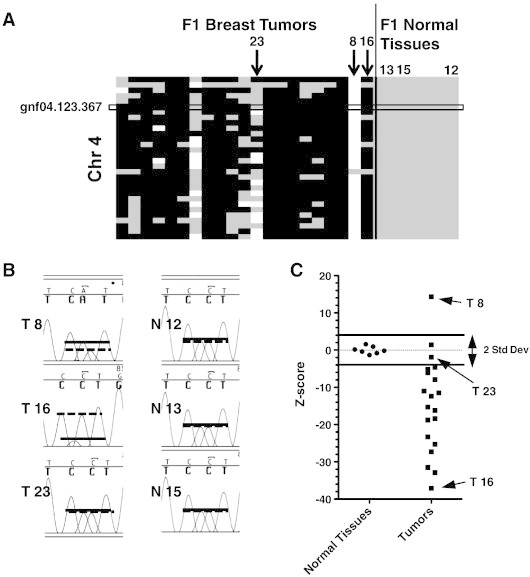
C-LOH on chromosome 4 validated by sequence analysis. (A) Enlargement of heat map from Figure 2, box indicates SNP gnf04.123.367 used for Sanger sequence analysis. Only samples shown in B and C are numbered; arrows point to tumors. (B) Sequencing chromatograms focusing on nucleotide polymorphism and shown for three tumors and three normal tissues from (BALB/c-neuT × FVB) F1 mice. Lines represent allelic measurements (A, solid line—BALB allele; C, dashed line—FVB allele). (C) Allelic ratios from genotype array for gnf04.123.367, represented as z-scores for normal and tumor DNA samples (negative z-score represents loss of BALB allele). Arrows indicate samples whose sequence chromatograms are shown in B.
Detection of a Second Pattern of LOH Indicates Another Mechanism(s) at Play
The V-LOH pattern observed by genotyping individual tumors was surprising in that it was widespread yet seemingly distributed stochastically in any given tumor. To determine whether the V-LOH was also caused by physical loss or gain of genomic DNA, we repeated the PCR and sequencing approach, choosing SNPs at locations where both C-LOH and V-LOH were apparent among different tumor samples (Figure 4). For example, tumor 13 (arrow in Figure 4A) displayed C-LOH across chromosome 15 and analysis of SNP CEL-15_9687257 showed allelic skewing compared to normal tissue (Figure 4B). In contrast, sequence analysis of tumor 16 displaying a strong V-LOH measurement found SNP to be heterozygous, indistinguishable from the normal unaffected tissue. This finding was consistent, even in cases where the genotyping call of the V-LOH sample was quantitatively more extreme than the same SNP for another tumor in the same set displaying C-LOH (genotype quantitations expressed as z-scores; Figure 4C). Moreover, sequence analysis revealed no evidence for additional mutations, rearrangements, or deletions in the vicinity of the SNPs analyzed in these tumors.
Figure 4.
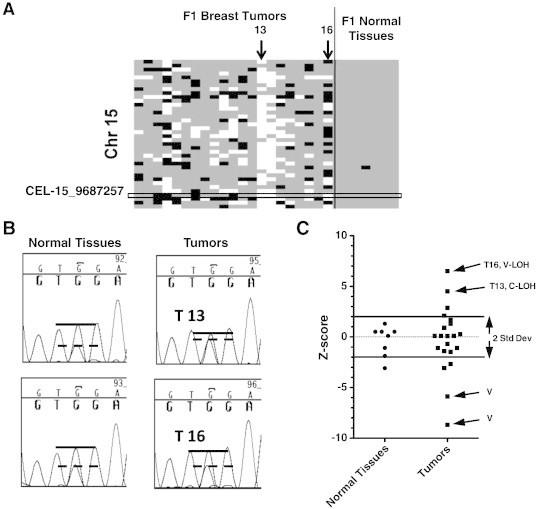
C-LOH but not V-LOH displays change in allelic ratios. (A) Enlargement of heat map from Figure 2, box indicates SNP CEL-15_9687257 on chromosome 15 used for Sanger sequence analysis. Sample numbers and arrows indicate tumors with C-LOH and V-LOH compared in B and C. (B) Chromatograms focusing on the nucleotide polymorphism (A—BALB allele; G—FVB allele; note that the empirical measure of heterozygosity for this SNP is not of equal intensities of the A and G nucleotide peaks). (C) Allelic ratios from genotype array for CEL-15_9687257, represented as z-scores for normal and tumor DNA samples (positive z-score represents loss of FVB or gain of BALB allele). Arrows indicate samples whose sequence chromatograms are shown in B and additional samples having aberrant genotype calls in patterns of V-LOH that were also heterozygous by sequence analysis (not shown).
The V-LOH appears to be fundamentally different from the C-LOH, indicating two different underlying mechanisms. This conclusion is supported by quantitation of the allelic ratios in the sequence analysis and comparing those ratios to the genotyping quantitation (z-scores) of tumor and normal samples. Figure 5 shows the tumor-to-normal ratios, calculated from analyses of six different SNPs using DNA from 26 independent tumors plotted versus the z-scores for the same samples. The data for C-LOH and V-LOH are displayed separately and show the clear correlation of allelic ratios detected by sequence analysis and bead array in cases of C-LOH (top panel; R2 = 0.6783; P = .0016). Despite multiple examples of SNPs exhibiting high deviation in allelic ratios by bead array genotyping, the sampled examples of V-LOH showed no correlation between genotype ratios and allelic ratios determined by direct sequencing (bottom panel; R2 = 0.0057; P = .73).
Figure 5.
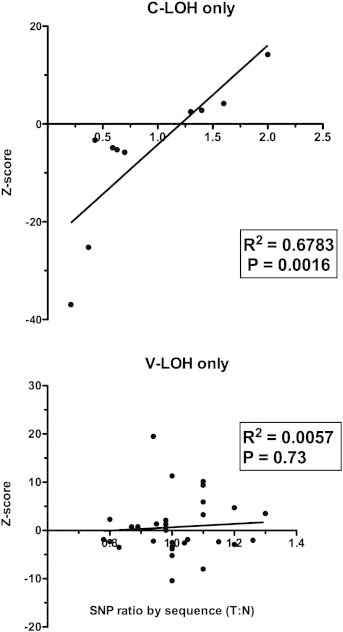
C-LOH but not V-LOH correlates with quantitative genotyping. Peak height ratios were calculated from Sanger sequence chromatograms for tumor (n = 26) and normal (n = 10) DNA samples. SNP peak height ratios for tumor samples were normalized to the average peak height ratio for normal tissue DNA and are shown as a function of genotype array z-scores for those tumor samples. Top panel: Combined analysis of four different SNPs found in patterns of C-LOH. Bottom panel: Combined analysis of four different SNPs found in patterns of V-LOH.
Influence of Dietary Fat on Developing Breast Cancer and V-LOH
A major advantage for using genotyping to study genomic LOH events in tumors is that a variety of preventative and therapeutic interventions can be easily assessed on individual animal tumors simultaneously. As a first test, we investigated whether dietary modification in breast cancer–prone F1 mice altered tumor LOH patterns. Animals were fed either defined low-fat or high-fat chow beginning at weaning. Tumor development was monitored until all mice developed breast tumors. In mice fed the LFD, tumor onset was delayed by 1 week, resulting in a commensurate 1-week difference in the timing of sacrifice due to tumor burden (median survival: HFD, 16 weeks; LFD, 17 weeks; hazard ratio = 2.62; P = .0023 Mantel-Cox; n = 46 per group). Tumors from mice receiving HFD (n = 11) and LFD (n = 8) were genotyped as before, along with normal tissue samples from the same animals. Diet appeared to have little influence on loss of chromosome 4, the bias toward preferential loss of the BALB/c chromosome, or other gross chromosomal changes as visualized by C-LOH (not shown). However, lowering the fat content of the diet decreased the percentage of SNPs in a V-LOH context by approximately half. To test for any quantitative effect on the genotyping assay, we compared the absolute z-scores for SNP CEL-15_9687257 in tumor DNA from individual mice fed with HFD or LFD (Figure 6). The tumors from HFD-fed mice had significantly higher median deviations from normal (z-scores) than did tumors from LFD-fed mice (P = .028, Kruskal-Wallis test; HFD vs LFD, P < .05, Dunn’s multiple comparison test). Whether the delay in tumor development is related to the drop in V-LOH is not known. However, because C-LOH appears similar in tumors from the two diet groups, this possibility is attractive and needs further investigation.
Figure 6.
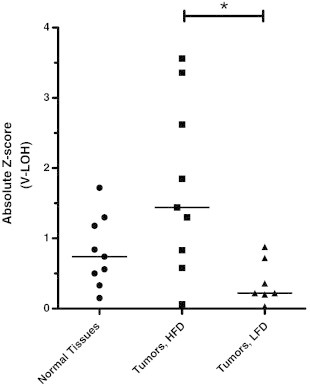
V-LOH in tumor genotype influenced by dietary fat in FVB × BALB-NeuT F1 mice. Heterozygous animals were assigned to an HFD or LFD at weaning. Genotyping data from breast tumor DNA samples were analyzed as before. The absolute z-scores are shown for SNP CEL-15_9687257 for individual tumors where V-LOH on chromosome 15 was observed. Absolute z-scores for normal tissues did not differ by diet group and are shown combined. Lines indicate the median values for each group.
Methyl- and Hydroxymethylcytosines Near the SNP Can Disrupt Template-Assisted Ligation-Based Genotyping
Several factors point to an epigenetic etiology of the V-LOH pattern in these primary breast tumors: the retention of allelic frequencies where V-LOH is observed, the absence of mutations, the widespread distribution of V-LOH throughout the tumor genomes, and the finding that environmental changes can influence the pattern. Epigenetic modifications to DNA play key regulatory roles in developmental processes, and altered patterns of CpG methylation have been described in a variety of cancer settings [2]. We reasoned that epigenetic DNA modifications in tumors compared to normal tissue DNA might interfere with the mechanics of the GoldenGate genotyping assay. In this approach to SNP detection, approximately 50 nt region of native DNA around the polymorphism is interrogated by allele-specific probes. These probes compete for annealing to the targeted area. The annealed product is extended and ligated to an adjacent locus-identifying probe; the final product is amplified for detection. The output is thus a measure of the efficiency of these annealing, extension, and ligation reactions.
To test the hypothesis that modifications to the target DNA might influence SNP detection efficiency, we developed an in vitro version of the assay (outlined in Figure 7A). Instead of genomic DNA, we substituted synthetic, methylation-free or methylcytosine- or hydroxymethylcytosine-containing templates corresponding to SNP CEL-15_9687257 (studied in Figure 4). Unmodified templates containing either the BALB (A) or FVB (G) allele were synthesized. Figure 7B shows that BALB/FVB heterozygosity can be measured using a mixture of control templates (top chromatogram). The BALB:FVB allelic ratio was increased when the BALB target DNA contained modified cytosines (P < .0001). As the presence of methylcytosine can increase the local Tm of DNA [23], we infer that our estimates of heterozygosity are substantially altered when one allelic template is modified. As the SNPs we examined in our experiments are not found in or near CpG islands and as 75% to 80% of the genomic sequences adjacent to the SNP loci probed by the genotyping assay contain no CG dinucleotides, we propose that non-canonical epigenetic modifications may be more widespread in cancers than previously shown.
Figure 7.
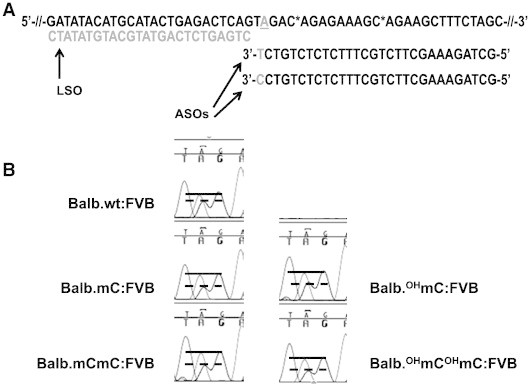
Efficiency of SNP genotyping assay affected by the presence of non-canonical DNA methylation. (A) Synthetic single-stranded templates were used in an in vitro assay involving competitive annealing of ASO probes (3′ T or C colored gray) followed by extension and ligation to an LSO probe. A wild-type FVB template was used for all reactions and mixed with an unmodified BALB template (shown) or templates synthesized to contain one or two 5-methylcytosines or 5-hydroxymethylcytosines (locations indicated by C*). The reactions create a new strand that is amplified and sequenced (B) to reveal how template strand modification affected ASO-dependent allelic ratios. Results shown are from one of three independent experiments; statistics are for the pooled data.
Discussion
Understanding the mechanisms contributing to the genetic heterogeneity of cancers remains a key to our ability to prevent and treat this multifaceted disease [24]. Animal models remain an important tool for cancer researchers as they often provide insights into fundamental properties of cellular transformation and malignant evolution [4]. We have developed a new approach using F1 heterozygous progeny from two inbred strains and quantitative genotyping measurements to simultaneously measure gross chromosomal changes and epigenetic modification of the cancer genome. This approach allows evaluation of contributive factors and provides a platform for testing intervention strategies in cancerous tissues.
In the Her2/neu oncogene model used in this study, deletions or losses of chromosome 4 is strongly associated with tumorigenesis, as all the tumors examined had this phenotype in both the inbred (not shown) and heterozygous F1 mouse lines. Cytogenetic analysis showed no example of trisomy at chromosome 4 and a loss of chromosome 4 was favored. Loss of chromosome 4 is one of the most common events in mouse tumor models [17–19,25]. However, the reasons for this are still unknown. Our genotyping approach readily detected a strong preference for the loss of the BALB/c fourth chromosome in FVB × BALB-NeuT F1 mice, similar to that observed in other studies of F1 studies with neuT animals [18]. In our study, sporadic loss of the FVB chromosome was observed in roughly 10% of the analyzed tumors. Whether this pervasive monoploidy is a fundamental step in tumor development or is consequential to an inherent instability of chromosome 4 remains to be determined.
Our unexpected finding that genetic regions marked by SNPs throughout the genome are modified at a high frequency in breast cancers could be an important clue for understanding how this cancer develops. We raise the possibility that a dysregulation of DNA-modifying machinery is a critical step in promoting tumor growth. Stepwise changes in DNA methylation have been shown to be linked to breast epithelial cell transformation in vitro [26], and differences in methylation may define human breast cancer subtypes [27,28]. Epigenetic modifications to DNA can alter DNA repair mechanisms as well as change gene expression programs leading to further gains or losses of encoded traits [29–31]. In heterozygous cells where allelic diversity in gene function is pervasive, silencing or activation of cellular functions can occur through an epigenetic mechanism with a single hit changing expression of only one of the parental genes. While most of the events measured in our study appear to be silent with respect to allelic preference (no evidence of selection), the sheer number of these events increases the likelihood that relevant genes will be affected, providing an opportunity for the evolution of gene expression profiles favoring uncontrolled growth.
An unexpected finding was the extensive LOH in 2 of 14 normal F1 reference samples (one of which can be seen in Figure 2). Each tissue sample was free of tumor by visual inspection. In fact, in these studies where no evidence of metastasis to any organ in the mice studied, these normal tissues also retained heterozygous signatures of SNPs on chromosome 4. It appears, therefore, that DNA from these cancer-prone animals contains some level of epigenetic modification, perhaps providing fertile ground for subsequent transformational or metastatic events. The events driving LOH in normal tissues remain to be elucidated.
The plasticity of genome in cancer has implications for the design of effective strategies to treat tumors. Most cancer models use inbred mice. As humans are an outbred population characterized by genome-wide heterozygosity, the behavior of cancers might be modeled more closely by heterozygous animals. We have shown that tumors driven by the Her2/neu oncogene contain both chromosomal losses and other patterns of allelic imbalances that appear to be epigenetically driven. Reducing the fat in the diets of the animals led to a measurable delay in tumor onset and mortality in the hybrid mice and an apparent decrease in V-LOH throughout the genome. These observations suggest that genetic instability might be modifiable and that F1 models may be advantageous for evaluating certain interventions. Therapeutic interventions, especially those designed to target specific cellular molecules, likely kill cells within a heterogeneous tumor with varying effectiveness. Testing and linking the assortment of functional traits to allele-specific events using F1 animals has the potential to elucidate mechanisms of tumor resistance under therapeutic pressure. This would be particularly true when a therapy targets a single allele.
The chemical nature of the epigenetic changes visualized in our study is not known. A substantial portion of the SNPs we assayed is devoid of CpG dinucleotides, suggesting that another epigenetic mechanism(s) is in play in our F1 breast tumors. Cytosine methylation is the predominant modification to eukaryotic genomic DNA and, until recently, thought to be restricted to CpG sequence contexts [32,33]. The presence of non-CpG cytosine methylation has been documented in embryonic and multipotent stem cells [30]. More recently, Guo et al. [34] showed that non-CG methylation regulates neuronal function in a region of the adult mouse brain associated with regeneration potential, suggesting that this form of DNA modification correlates with cellular plasticity. How pervasive other modification patterns are throughout the tumor genomes remains to be tested, but certainly our experiments show that methylated cytosine can contribute to some of the LOH signals. However, canonical CpG methylation cannot be the whole story. How DNA modifications function during tumor evolution, and to what extent they can be controlled by conventional therapies, by new targeted therapies or by lifestyle choices remains to be determined in future studies.
Acknowledgements
SKY and cytogenetic analyses were provided by Darlene Knutson and Patricia T. Greipp in the Mayo Cytogenetics Core. Additional technical support from the Genotyping and Molecular Biology cores (supported by the Mayo Clinic Cancer Center CA15083) is appreciated.
R.G.V. and L.R.P. designed the study. V.P.V.K., M.J.H., M.P.B., S.J.F., K.A., and A.A.B. performed the experiments. J.M.C., T.L.H., V.S.P., S.J.F., and L.R.P. analyzed and interpreted the data. S.J.F., V.P.V.K., and L.R.P. wrote the manuscript.
Footnotes
This article refers to supplementary materials, which are designated by Supplemental Tables 1 and 2 and Supplemental Figure 1 and are available online at www.neoplasia.com.
This work was supported by grant funding from NIH NCI P50 CA116201. Conflicts of interest: The authors disclose no potential conflicts of interest.
Appendix A. Supplementary Materials
Supplemental Figure 1. Genomic instability of spontaneous breast tumors in heterozygous mice. (A) Chromosomes of primary cells from FVB × BALB-NeuT F1 tumors were examined using SKY. The complement of chromosome 4 was altered in 100 of 100 tumor cells examined. Additional gains, losses, and translocations were observed at other chromosomes, primarily at chromosomes 1 and 12.
Supplemental Table 1. Significant LOH on Each Chromosome in Spontaneous Breast Tumors. Z-Scores Were Calculated from the Raw Allele Intensities of the Genotyping Data, and Absolute Values of SNP Probes Are Summarized Chromosome by Chromosome for F1 Tumor and Normal Samples
Supplemental Table 2. Analyzing Genotype Data for Selection, i.e., a Quantitative Measurement toward One Allele Reveals Significant Loss of BALB/c Alleles on Chromosome 4. Locus-by-Locus Analysis Used the Positive (Gain of BALB/c) or Negative (Loss of BALB/c) Z-Scores Calculated Before. Only Probes on Chromosome 4 Were Significantly Skewed in Three of Three Arrays
References
- 1.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gillies R.J., Verduzco D., Gatenby R.A. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12:487–493. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andrechek E.R., Nevins J.R. Mouse models of cancers: opportunities to address heterogeneity of human cancer and evaluate therapeutic strategies. J Mol Med. 2010;88:1095–1100. doi: 10.1007/s00109-010-0644-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ursini-Siegel J., Schade B., Cardiff R.D., Muller W.J. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007;7:389–397. doi: 10.1038/nrc2127. [DOI] [PubMed] [Google Scholar]
- 6.Gendler S.J., Mukherjee P. Spontaneous adenocarcinoma mouse models for immunotherapy. Trends Mol Med. 2001;7:471–475. doi: 10.1016/s1471-4914(01)02124-4. [DOI] [PubMed] [Google Scholar]
- 7.Nava-Parada P., Forni G., Knutson K.L., Pease L.R., Celis E. Peptide vaccine given with a Toll-like receptor agonist is effective for the treatment and prevention of spontaneous breast tumors. Cancer Res. 2007;67:1326–1334. doi: 10.1158/0008-5472.CAN-06-3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varghese S., Rabkin S.D., Nielsen G.P., MacGarvey U., Liu R., Martuza R.L. Systemic therapy of spontaneous prostate cancer in transgenic mice with oncolytic herpes simplex viruses. Cancer Res. 2007;67:9371–9379. doi: 10.1158/0008-5472.CAN-07-0674. [DOI] [PubMed] [Google Scholar]
- 9.Chi M., Dudek A.Z. Vaccine therapy for metastatic melanoma: systematic review and meta-analysis of clinical trials. Melanoma Res. 2011;21:165–174. doi: 10.1097/CMR.0b013e328346554d. [DOI] [PubMed] [Google Scholar]
- 10.Draube A., Klein-Gonzalez N., Mattheus S., Brillant C., Hellmich M., Engert A. Dendritic cell based tumor vaccination in prostate and renal cell cancer: a systematic review and meta-analysis. PLoS One. 2011;6:e18801. doi: 10.1371/journal.pone.0018801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Florescu A., Amir E., Bouganim N., Clemons M. Immune therapy for breast cancer in 2010-hype or hope? Curr Oncol. 2011;18:e9–e18. doi: 10.3747/co.v18i1.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellsworth R.E., Ellsworth D.L., Patney H.L., Deyarmin B., Love B., Hooke J.A. Amplification of HER2 is a marker for global genomic instability. BMC Cancer. 2008;8:297. doi: 10.1186/1471-2407-8-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bacolod M.D., Schemmann G.S., Giardina S.F., Paty P., Notterman D.A., Barany F. Emerging paradigms in cancer genetics: some important findings from high-density single nucleotide polymorphism array studies. Cancer Res. 2009;69:723–727. doi: 10.1158/0008-5472.CAN-08-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dutt A., Beroukhim R. Single nucleotide polymorphism array analysis of cancer. Curr Opin Oncol. 2007;19:43–49. doi: 10.1097/CCO.0b013e328011a8c1. [DOI] [PubMed] [Google Scholar]
- 15.Alexandrov L.B., Nik-Zainal S., Wedge D.C., Campbell P.J., Stratton M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246–259. doi: 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nik-Zainal S., Van Loo P., Wedge D.C., Alexandrov L.B., Greenman C.D., Lau K.W., Raine K., Jones D., Marshall J., Ramakrishna M. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ritland S.R., Rowse G.J., Chang Y., Gendler S.J. Loss of heterozygosity analysis in primary mammary tumors and lung metastases of MMTV-MTAg and MMTV-neu transgenic mice. Cancer Res. 1997;57:3520–3525. [PubMed] [Google Scholar]
- 18.Cool M., Jolicoeur P. Elevated frequency of loss of heterozygosity in mammary tumors arising in mouse mammary tumor virus/neu transgenic mice. Cancer Res. 1999;59:2438–2444. [PubMed] [Google Scholar]
- 19.Cool M., Depault F., Jolicoeur P. Fine allelotyping of Erbb2-induced mammary tumors in mice reveals multiple discontinuous candidate regions of tumor-suppressor loci. Genes Chromosomes Cancer. 2006;45:191–202. doi: 10.1002/gcc.20276. [DOI] [PubMed] [Google Scholar]
- 20.Montagna C., Andrechek E.R., Padilla-Nash H., Muller W.J., Ried T. Centrosome abnormalities, recurring deletions of chromosome 4, and genomic amplification of HER2/neu define mouse mammary gland adenocarcinomas induced by mutant HER2/neu. Oncogene. 2002;21:890–898. doi: 10.1038/sj.onc.1205146. [DOI] [PubMed] [Google Scholar]
- 21.Weaver Z.A., McCormack S.J., Liyanage M., du Manoir S., Coleman A., Schrock E., Dickson R.B., Ried T. A recurring pattern of chromosomal aberrations in mammary gland tumors of MMTV-cmyc transgenic mice. Genes Chromosomes Cancer. 1999;25:251–260. [PubMed] [Google Scholar]
- 22.Quaglino E., Mastini C., Forni G., Cavallo F. ErbB2 transgenic mice: a tool for investigation of the immune prevention and treatment of mammary carcinomas. Curr Protoc Immunol. 2008 doi: 10.1002/0471142735.im2009s82. Chapter 20: Unit 20.9.1–20.9-10. [DOI] [PubMed] [Google Scholar]
- 23.Burrell R.A., McGranahan N., Bartek J., Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 24.Padilla-Nash H.M., Hathcock K., McNeil N.E., Mack D., Hoeppner D., Ravin R., Knutsen T., Yonescu R., Wangsa D., Dorritie K. Spontaneous transformation of murine epithelial cells requires the early acquisition of specific chromosomal aneuploidies and genomic imbalances. Genes Chromosomes Cancer. 2012;51:353–374. doi: 10.1002/gcc.21921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Novak P., Jensen T.J., Garbe J.C., Stampfer M.R., Futscher B.W. Stepwise DNA methylation changes are linked to escape from defined proliferation barriers and mammary epithelial cell immortalization. Cancer Res. 2009;69:5251–5258. doi: 10.1158/0008-5472.CAN-08-4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bediaga N.G., Acha-Sagredo A., Guerra I., Viguri A., Albaina C., Ruiz Diaz I., Rezola R., Alberdi M.J., Dopazo J., Montaner D. DNA methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res. 2010;12:R77. doi: 10.1186/bcr2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novak P., Stampfer M.R., Munoz-Rodriguez J.L., Garbe J.C., Ehrich M., Futscher B.W., Jensen T.J. Cell-type specific DNA methylation patterns define human breast cellular identity. PLoS One. 2012;7:e52299. doi: 10.1371/journal.pone.0052299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin B., Ernst J., Tiedemann R.L., Xu H., Sureshchandra S., Kellis M., Dalton S., Liu C., Choi J.H., Robertson K.D. Linking DNA methyltransferases to epigenetic marks and nucleosome structure genome-wide in human tumor cells. Cell Rep. 2012;2:1411–1424. doi: 10.1016/j.celrep.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lister R., Pelizzola M., Dowen R.H., Hawkins R.D., Hon G., Tonti-Filippini J., Nery J.R., Lee L., Ye Z., Ngo Q.M. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nowsheen S., Aziz K., Tran P.T., Gorgoulis V.G., Yang E.S., Georgakilas A.G. Epigenetic inactivation of DNA repair in breast cancer. Cancer Lett. 2014;342:213–222. doi: 10.1016/j.canlet.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 31.Stroud H., Do T., Du J., Zhong X., Feng S., Johnson L., Patel D.J., Jacobsen S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol. 2014;21:64–72. doi: 10.1038/nsmb.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ziller M.J., Gu H., Muller F., Donaghey J., Tsai L.T., Kohlbacher O. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo J.U., Su Y., Shin J.H., Shin J., Li H., Xie B., Zhong C., Hu S., Le T., Fan G. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014;17:215–222. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodríguez López C.M., Guzmán Asenjo B., Lloyd A.J., Wilkinson M.J. Direct detection and quantification of methylation in nucleic acid sequences using high-resolution melting analysis. Anal Chem. 2010;82:9100–9108. doi: 10.1021/ac1024057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Genomic instability of spontaneous breast tumors in heterozygous mice. (A) Chromosomes of primary cells from FVB × BALB-NeuT F1 tumors were examined using SKY. The complement of chromosome 4 was altered in 100 of 100 tumor cells examined. Additional gains, losses, and translocations were observed at other chromosomes, primarily at chromosomes 1 and 12.
Supplemental Table 1. Significant LOH on Each Chromosome in Spontaneous Breast Tumors. Z-Scores Were Calculated from the Raw Allele Intensities of the Genotyping Data, and Absolute Values of SNP Probes Are Summarized Chromosome by Chromosome for F1 Tumor and Normal Samples
Supplemental Table 2. Analyzing Genotype Data for Selection, i.e., a Quantitative Measurement toward One Allele Reveals Significant Loss of BALB/c Alleles on Chromosome 4. Locus-by-Locus Analysis Used the Positive (Gain of BALB/c) or Negative (Loss of BALB/c) Z-Scores Calculated Before. Only Probes on Chromosome 4 Were Significantly Skewed in Three of Three Arrays