

Abstract
The insulin-like growth factor 1 receptor (IGF-1R) has surfaced as a significant target in multiple solid cancers due to its fundamental roles in pro-survival and anti-apoptotic signaling. However, development of resistance to IGF-1R blockade represents a significant hindrance and limits treatment efficacy in the clinic. In this study, we identified acquired resistance to IGF-1R blockade with R1507, an antibody against IGF-1R, and with BMS-754807, a small molecular inhibitor of IGF-1R/insulin receptor (IR). We showed that treatment with an IGF-IR antibody, R1507, or an IR/IGF-IR kinase inhibitor, BMS-754807, was associated with increased activation of YES/SRC family tyrosine kinase (SFK) in rhabdomyosarcoma (RMS). Combining anti–IGF-1R agents with SFK inhibitors resulted in blockade of IGF-1R inhibition–induced activation of YES/SFK and displayed advantageous antitumor activity in vitro and in vivo. Our data provide evidence that IGF-1R blockade results in activation of the YES/SRC family kinase bypass resistance pathway in vitro and in vivo. This may be of particular clinical relevance since both Yes and IGF components are overexpressed in RMS. Increased YES/SFK activation might serve as a clinical biomarker for predicting tumor resistance to IGF-1R inhibition. Dual inhibition of IGF-1R and SFK may have a broader and enhanced clinical benefit for patients with RMS.
Introduction
Uncontrolled cell growth is central to carcinogenesis. Growth factors, as well as growth factor–initiated pathways that regulate cell growth control and modulate cell phenotype are undoubtedly important to the etiology of cancer. Studies aimed at deciphering the role of insulin/insulin-like growth factor (IGF) receptors and their cognate ligands in cancer biology have been ongoing for more than three decades due to their pivotal roles in regulating tumor cell growth, differentiation, tumor angiogenesis, metastasis, apoptosis, and resistance to chemotherapy [1]. To date, no cancer-specific mutations of the IGF 1 receptor (IGF-1R) or its ligands have been reported. The IGF signaling pathway is abnormally activated, either by loss of genomic imprinting or by abnormal stimulation by endocrine, autocrine, and paracrine mechanisms, in various pediatric sarcomas [2–4]. These changes lead to dysregulation of sarcoma growth and contribute to subsequent sarcoma development and progression. Hence, targeting of IGF signaling, specifically IGF-1R blockade, has emerged in recent years as a promising therapeutic approach in pediatric sarcomas. However, recent phase II clinical trials with several anti–IGF-1R monoclonal antibodies have shown relatively low response rates. For example, the Roche IGF–1R monoclonal antibody, R1507, showed modest therapeutic efficacy in 115 patients with recurrent or refractory Ewing sarcoma family of tumors, with an overall objective response rate of 10%. Furthermore, even in responding patients, most responses were transient, lasting less than 18 weeks. Thus, the majority of patients, even those with initial responses, do not have long-term benefit from IGF-IR blockade, indicating the presence of an innately resistant tumor mass or the recruitment of compensatory pathways allowing for continued growth [5]. We previously reported similar results in a rhabdomyosarcoma (RMS) xenograft model where tumors developed resistance after continuous treatment with IGF-1R antibody [6]. Therefore, there is clearly a need to identify resistance mechanisms to IGF-1R blockade and hopefully identify combination approaches that will improve on these early observations.
SRC family tyrosine kinases (SFKs) are non-receptor tyrosine kinases that are involved in multiple signaling pathways controlling cell growth, survival, differentiation, and migration, in response to the activation of cell surface receptors by growth factors, cytokines, and adhesion proteins [7,8]. SFKs consist of at least nine highly homologous family members: SRC, YES, FYN, LYN, LCK, HCK, FGR, BLK, and YRK, each composed of four SRC homology domains (SH1, SH2, SH3, and SH4) that share similar structure and function [9,10]. Although the SFKs are activated in various types of cancers, the exact mechanisms through which they contribute to the progression of individual tumors remain to be defined. As SFKs play important roles in the promotion of cell proliferation, adhesion, invasion, and motility, SFK has become a therapeutic target for drug development. Several SFK inhibitors, such as dasatinib, AZD0530, and SKI-606, are entering clinical trials as anticancer therapeutics [9]. Dasatinib is a multitargeted tyrosine kinase inhibitor that has been found to target BCR-ABL, SFKs (SRC, LCK, YES, and FYN), c-Kit, ephrin receptor A2, and PDGF receptor (PDGFR), which is capable of binding to these kinases, inhibiting their autophosphorylation and downstream phosphorylation of additional targets and thus blocking their oncogenic activities [11]. Dasatinib has been approved for the treatment of imatinib-resistant or imatinib-intolerant forms of chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia [12]. Previous studies have found that SFKs are involved in signaling from many receptor tyrosine kinases (RTKs) including PDGFR, epidermal growth factor receptor, fibroblast growth receptor, and IGF-1R [13]. SRC interacts with multiple RTKs through its SH2 domain and facilitates RTK-mediated signaling. We recently demonstrated that YES is highly expressed and functional in RMS [14]. These data led us to determine whether activation of YES might play a role in bypass resistance to IGF-IR inhibition.
In this study, we demonstrate that IGF-1R blockade results in an increase in Yes activation and is associated with resistance to IGF-1R blockade in RMS cell lines. On the basis of these findings, we tested IGF-1R blockade combined with SFK inhibition using the commercially available drug, dasatinib, as well as an independent SFK inhibitor, AZD0530. Dual blockade of IGF-1R and SFK pathways showed superior efficacy in vitro and in vivo. These data suggest that IGF-1R inhibition–induced activation of YES/SFK may act as a bypass pathway. Furthermore, these data suggest that dual IGF-1R and SFK kinase inhibition may lead to improved therapeutic outcomes.
Materials and Methods
Cell Lines and Cell Culture
Alveolar RMS cell lines Rh30 and Rh41 and embryonal RMS cell line RD have been previously described [15]. ARMS cell lines Rh5 and Rh28 and ERMS cell line Rh18 and TTC442 were obtained from Dr Javed Khan (NCI, Bethesda, MD). The cells were maintained in RPMI growth medium (Invitrogen, Carlsbad, CA) with 10% FBS (Sigma-Aldrich, St Louis, MO), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen), and 2 mM l-glutamine (Invitrogen) at 37°C in an atmosphere of 5% CO.
Compounds
AZD0530, dasatinib, and Gleevec were purchased from LC Laboratories (Woburn, MA). R1507 antibody was obtained from Roche Inc (Nultey, NJ), and h7C10 antibody was obtained from Merck Inc (Kenilworth, NJ). BMS-754807 was obtained from Bristol-Myers Squibb Inc (New York, NY).
Real-Time Polymerase Chain Reaction
Total RNA was isolated from each cell line, RD, Rh28, Rh30, and Rh41, with Qiagen RNeasy Mini Kit (74104). RNA (1 μg) was used for cDNA synthesis with Roche Transcriptor First-Strand cDNA Synthesis Kit (04379012001) using anchored oligo-dT. Normal human skeletal muscle RNA was purchased from Zyagen (San Diego, CA).
YES1 forward primer (ACAGCAAGACAAGGTGCAAA) and reverse primer (GTAAACCGACCATACAGTGCAG) and glyceraldehyde-3-phosphate dehydrogenase forward primer (AGGTCGGTGTGAACGGATTTG) and reverse primer (TGTAGACCATGTAGTTGAGGTCA) were used with Power SYBR Green PCR Master Mix (Applied Biosystems, Grand Island, NY) for quantitative polymerase chain reactions (PCRs) in a Bio-Rad C100 thermal cycler, and the result was analyzed with Bio-Rad CFX software. Annealing temperature was 55.7°C for both YES1 and GADPH primers.
Western Blot Analysis and Immunoprecipitation
Western blot analysis was done as published previously [16]. Antibodies to phospho-SFK (Tyr416), SRC, phospho-Akt (S473), cleaved caspase-3 (Asp175), and cleaved poly ADP ribose polymerase (PARP) (Asp214) were purchased from Cell Signaling Technology Inc (Beverly, MA). Anti-phosphotyrosine antibody was purchased from Millipore Inc. (Billerica, MA). Anti-YES used for immunoprecipitation (IP) was from Wako (Richmond, VA). Mouse anti-SRC antibodies for IP were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-actin antibody was purchased from Abcam Inc. (Cambridge, MA). IP assays were performed using the Universal Magnetic Co-IP kit from Active Motif Inc. (Carlsbad, CA) following the manufacturer’s instruction.
Cell Proliferation and Survival Assays
Cell proliferation and survival was determined using 3-(4,5-dimethylthiazolyl-2)-2, 5-diphenytetrazolium bromide (MTT) assay as described previously [17]. Cell proliferation kinetics were monitored and recorded with the IncuCyte System (Essen BioScience, Ann Arbor, MI).
In Vivo Study
Animal studies were performed in accordance with the guidelines of the National Institutes of Health Animal Care and Use Committee. Four- to 6-week-old female Fox Chase severe combined immunodeficiency-Beige mice were purchased from Charles River Laboratories (Wilmington, MA). Three million cells of Rh30 and RD were injected orthotopically into the gastrocnemius muscle in the left hind leg. Treatment with agents began when tumor was palpable, on day 14 in Rh30 tumor–bearing mice. In RD tumor–bearing mice, treatment began on day 11 after injection, before tumors were palpable. Dasatinib was given by oral gavage at 100 mg/kg daily × 5 days/week. R1507 was given IP at 6 mg/kg twice a week. Tumor volume was calculated by the following formula: V (mm3) = (D × d2)/6 × 3.14, where D is the longest tumor axis and d is the shortest tumor axis.
Statistical Analysis
Statistical analyses were performed in Prism version 4.0 (GraphPad Software) using a nonparametric t test. Statistical significance was defined as P < .05.
Results
IGF-2, IGF-1R, and YES Kinase are Highly Expressed in RMS
Our previous studies have demonstrated that IGF-2 is overexpressed and functions in an autocrine manner in RMS [16,18] and that YES kinase is overexpressed in RMS. [14] We also examined expression of other members of SFKs in addition to YES, IGF-1, IGF-2, and IGF-1R, in cell lines, xenografts, and human tumor samples of RMS by cDNA microarray (Oncogenomic Database, http://home.ccr.cancer.gov/oncology/oncogenomics/). As shown in Figure 1A, IGF-2 and IGF-1R are both highly expressed in cell lines, xenografts, and human tumor samples of RMS. We further confirmed that YES is highly expressed in almost all cell lines, xenografts, and human tumor samples of RMS compared to expression of other SFKs and non-RMS tumors. In contrast, both SRC and LYN are minimally expressed in RMS cell lines and tumors (Figure 1B). Data from quantitative reverse transcription–PCR further demonstrate that YES is highly expressed in RMS cell lines compared to normal human skeletal muscle (Figure 1C). These results suggest that YES is the predominantly expressed SFK in RMS. In addition, Western blot analysis revealed that YES protein is expressed, while phospho-SFK is expressed at modest levels in seven RMS cell lines (Figure 1D).
Figure 1.

(A and B) cDNA expression profile shows expression levels of IGF-1, IGF-2, IGF-1R, and SFKs in human RMS (ERMS, embryonal RMS, ARMS, and alveolar RMS) cell lines [C], xenograft tumors of human cell lines [X], and human tumor tissue [T]. For comparison, non-RMS tumors and normal human skeletal muscle are shown. The Affy ID refers to specific probes used in U133 plus 2.0 Arrays and are designed to provide multiple probes for the same gene. The scale is median centered (in log2) with a saturation threshold of + 2 (red; four-fold higher) to − 2 (green; four-fold lower). (C) By quantitative PCR, YES1 RNA expression in RMS cell lines was higher compared to expression in normal human skeletal muscle (NSM). YES and GADPH SQ values were generated with Bio-RAD CFX software and normalized to GADPH. (D) YES expression and SFK phosphorylation were analyzed by Western blot analysis in RMS cell lines.
IGF-1R Inhibition Induces Activation of YES Kinase
Since IGFs and YES are both highly expressed in RMS cell lines and tumor samples, we examined the effect of anti–IGF-1R agents on the phosphorylation of SFK and downstream targets involved in the IGF-1R signaling pathway. As shown in Figure 2A, Western blot analysis identified that single-agent treatment with either of the IGF-1R inhibitors, R1507 or BMS-754807, or both agents in combination for 24 hours resulted in an increase in phosphorylation of SFK (Tyr416). This appeared to be further enhanced with combination IGF-1R inhibition in both Rh30 and RD cell lines. Treatment of cells with dual anti–IGF-1R and SFK inhibitors resulted in blockade of IGF-1R inhibition–induced activation of SFK. Phosphorylation of Akt was downregulated in the face of both single-agent IGF-1R or SFK inhibition and dual IGF-1R and SFK blockade. Further, rapid up-regulation of SFK activation was observed within 4 hours of IGF-1R blockade in RMS cell lines (Figure 2B). To determine if IGF-1R inhibition–induced up-regulation of SFK activation is specifically associated with YES activation, we performed IP assays with SRC and YES-specific antibodies followed by immunoblot analysis with an anti-phosphotyrosine antibody. A significant increase in YES tyrosine phosphorylation was detected following 4 hours of R1507 treatment in Rh30 and 24 hours of R1507 treatment in RD, compared to a minimal increase in phosphorylation of SRC (Figure 2, C and D). These results suggest that IGF-1R inhibition induces YES activation. To evaluate whether dual blockade of IGF-1R and SFK can augment cellular apoptotic response, we determined the levels of cleaved caspase-3 and cleaved PARP, key biochemical markers of apoptosis. Significantly increased cleaved caspase-3 and cleaved PARP were observed after 24 hours of treatment with anti–IGF-1R agents in combination with the SFK inhibitor dasatinib, when compared to treatment with each agent alone (Figure 2E). These results suggest that IGF-1R inhibition–induced activation of YES kinase may act as a bypass resistance pathway.
Figure 2.

IGF-1R inhibition is associated with a compensatory increase in YES/SFK activation. (A and B) Cells were treated with single-agent R1507, BMS-754807, or dasatinib or in combination using dasatinib with R1507/BMS-754807 at 24 (A) or 4 hours (B). Both single-agent R1507 and BMS-754807 induced an increase of SFK phosphorylation at 24-hour treatment (A) and 4-hour treatment (B) by Western blot analysis. (C and D) IGF-1R inhibition–induced up-regulation of SFK activation is specifically associated with YES. Protein lysates from Rh30 and RD cells with or without R1507 treatment at indicated times were immunoprecipitated with normal IgG, SRC, and YES antibodies overnight. These blots were probed with mouse anti-phosphotyrosine, SRC, or YES antibodies (C). (D) The phosphotyrosine intensities from C were scanned and analyzed by ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA). (E) The levels of cleaved caspase-3 and PARP were significantly increased with combination IGF-1R and SFK inhibition. Apoptosis was determined by cleaved caspase-3 and PARP. Western blot analysis demonstrates a significant increase in cleaved caspase-3 and PARP at 24 hours of combination treatment compared to single-agent treatment.
Effects of Dual Inhibition of IGF-1R and SFK on Cell Growth Inhibition of RMS In Vitro
Since both IGF signaling and SFK pathways play important roles in cell growth and survival, we tested the rationale for combined inhibition of IGF and SFK in RMS cell lines in vitro. Inhibition of IGF-1R and SFK with a combination of R1507 and the SFK inhibitor AZD0530 resulted in more potent inhibition of both RD and Rh30 cell growth than did either agent alone or other combined treatments including R1507 plus Gleevec (Figure 3A). Since Gleevec (also known as imatinib) is a tyrosine kinase inhibitor with activity against Bcr-Abl, c-Kit, and PDGFR that lacks activity against SFK, the lack of enhanced activity with the combination of Gleevec plus IGF-IR blockade in this assay further points to the activation of YES as a potential bypass resistance pathway induced by IGF-IR blockade. Similar results were observed with combined treatment using h7c10, another IGF-1R antibody that has been used previously [17] (Figure 3B). We also tested the combination of R1507 and dasatinib. Both RD and Rh30 cells were treated with dasatinib (10 nM to 10 μM) alone or in combination with R1507 for 72 hours. The combination of dasatinib (10 nM to 10 μM) and R1507 resulted in additive negative effects on cell growth (Figure 3C). In addition, combined treatment of RMS cells with BMS-754807 (1 μM), a small molecule inhibitor of the IGF-1R/IR and dasatinib (1 μM), led to more effective inhibition of RMS cell growth than each agent alone, as shown in Figure 3D.
Figure 3.

Effects of dual inhibition of IGF-1R and SFK on RMS cell growth in vitro. (A and B) Both RD and Rh30 cells were plated onto a 96-well plate (5000 cells per well) in regular growth medium. After 16 hours of incubation, cells were treated with single agent or combined agents as indicated in the figures for 48 hours (*P < .0001; 10 μM AZD0530 + R1507 vs R1507 or 10 μM AZD0530 alone in both Rh30 and RD cell lines) (A) or 72 hours (*P < .0001; 10 μM AZD0530 + h7C10/R1507 vs 10 μM AZD0530 or R1507 alone in Rh30 cell line; **P < .0005; 1 μM AZD0530 + h7C10/R1507 vs 1 μM AZD0530 or R1507 alone in Rh30 cell line; ***P < .05-.001; 1 μM AZD0530/10 μM AZD0530 + h7C10/R1507 vs 1 μM AZD0530/10 μM AZD0530 alone in RD cell line) (B). (C) Cells were treated with dasatinib alone at a range of concentrations from 0.01 to 10 μM or in combination with R1507 at a fixed concentration (100 nM) for 72 hours. Cell growth was measured by MTT (*P < .0002-.0001; 10 nM-10 μM dasatinib + R1507 vs 10 nM-10 μM dasatinib alone in both Rh30 and RD cell lines. (D) The combination of dasatinib and BMS-754807, a small-molecule inhibitor of IGF-1R/IR, results in more potent inhibition of RMS cell growth than does either agent alone. Cells were treated with single agent alone or combined agents for 72 hours as indicated. Images were taken 4 hours after incubation with MTT.
We next examined the effect of dual inhibition of IGF-1R and YES/SFK on additional ARMS cell lines Rh5 and Rh28 and ERMS cell line TTC442. The combination of dasatinib (100 nM) and R1507 or BMS-754807 significantly inhibited cell growth and survival compared to single agent alone in all tested cell lines (Figure 4). These data suggest that dual blockade of IGF-1R and SFKs has efficacy in RMS in vitro.
Figure 4.

Effects of dual inhibition of IGF-1R and SFK on cell growth were examined in additional RMS cell in vitro. Rh5, Rh28, and TTC442 cells were plated onto a 96-well plate and treated with single-agent R1507 (100 nM), BMS-754807 (1 μM), or dasatinib (100 nM) or in combination using dasatinib (100 nM) with R1507 (100 nM)/BMS-754807 (1 μM). These cells were monitored and recorded with the IncuCyte System. Arrow indicates beginning of treatment.
Effect of Combination of R1507 and Dasatinib on Growth of RMS Xenografts In Vivo
We next sought to determine whether the effect of dual IGF-1R and SFK inhibition observed in vitro would translate to the in vivo setting. The in vivo antitumor efficacy of R1507 and dasatinib alone and in combination was tested in xenograft models of RMS. We treated RMS xenograft–bearing mice with R1507 and dasatinib alone or in combination beginning when tumor was palpable (day 14) in Rh30 tumor–bearing mice and 11 days after tumor injection for RD tumor–bearing mice. As shown in Figure 5, R1507 alone significantly inhibited xenograft growth compared to the control group after 31 days of treatment for Rh30 xenografts (Figure 5B) and 60 days of treatment for RD xenografts (Figure 5E; P < .0001 for R1507 group vs vehicle group in both models); dasatinib alone had less effect on tumor growth inhibition compared to R1507 alone (P = .0795 in Rh30 model and P = .041 in RD model for R1507 group vs vehicle group; Figure 5, B and E). In the Rh30 model, all mice in both of the R1507 and dasatinib groups developed resistance to single-agent treatment by 55 days of treatment (P = .117 for R1507 group and P = .1114 for dasatinib group; Figure 5C). In the RD model, most of the mice in both the R1507 and dasatinib groups developed resistance to single-agent treatment by 79 days of treatment (P = .4309 for R1507 group and P = .7167 for dasatinib group; Figure 5F). Combining R1507 and dasatinib led to more significant inhibition of tumor growth (P < .0001 in both models), and some tumors did not become resistant even after 55 or 79 days of treatment (Figure 5, C and F).
Figure 5.
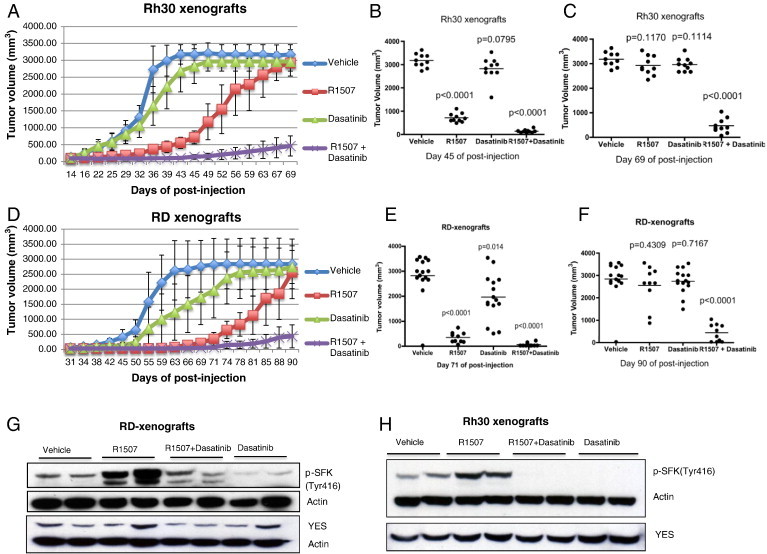
Dual IGF-1R and SFK blockade inhibit RMS xenograft growth. (A and D) Mice were injected with 3 × 106 RD cells per mouse into the gastrocnemius muscle of the left hind leg. Treatment with agents began when tumor was palpable (day 14) in Rh30 tumor–bearing mice (A) and 11 days following injection of cells in RD tumor–bearing mice (B). Dasatinib was given by oral gavage at 100 mg/kg daily × 5. R1507 was given IP at 6 mg/kg twice a week. Tumor growth was measured twice per week, and tumor volume was determined using the formula (D × d2/6 × 3.14). (B and C) Measured tumor volume of each of the individual mice bearing Rh30 xenografts shown on day 45 (B) and day 69 (C) post-injection. (E and F) Measured tumor volume of each of the individual mice bearing RD xenografts shown on day 71 (B) and day 90 (C) post-injection. (G and H) SFK phosphorylation was analyzed by Western blot in RD (G) and Rh30 (H) xenograft tumor samples.
IGF-1R Inhibition Induces Activation of YES/SFK In Vivo
To confirm whether IGF-1R inhibition induced activation of SFK in vivo, xenograft tumor lysates from each treatment group were subjected to Western blot analysis. As seen in Figure 5, G and H, treatment with R1507 increased SFK phosphorylation in both RD and Rh30 xenografts, whereas R1507 in combination with dasatinib abrogated this increase in SFK activation. These data further demonstrate that IGF-1R inhibition induces YES/SFK activation in vivo.
Discussion
Disruption of the IGF-1R signaling pathway has proven to be a promising therapeutic strategy in a variety of human cancers due to its role in cancer cell proliferation, survival, tumor angiogenesis, and metastasis [1]. We recently completed a phase II study targeting the IGF-1R signaling pathway in refractory Ewing’s and other sarcomas. We demonstrated an objective response rate of 10%, but most responses were transient lasting less than 18 weeks. The majority of patients, even those with initial responses, do not have long-term benefit from IGF-1R blockade, indicating the presence of both an innately resistant tumor mass as well as acquired resistance to the anti–IGF-1R antibody R1507 [5]. Thus, it is important to identify the mechanisms of resistance to IGF-1R blockade so that rational therapeutic strategies can be identified to overcome resistance. The aim of this study was to identify the mechanisms of acquired resistance to IGF-1R inhibition. In the present study, we identified activation of YES/SFK as a consequence of IGF-IR blockade in RMS cell lines and xenografts. On the basis of these observations, we then demonstrated that combined treatment with IGF-1R antibody plus SFK kinase inhibition enhanced growth inhibitory activity both in vitro and in vivo and led to prolonged responses in vivo.
Accumulating studies have reported that multiple mechanisms of acquired resistance are involved with IGF-1R inhibition. heat shock protein 90 has been reported to stabilize IGF-1R in some cancers and to increase following IGF-1R inhibition [19]. Overexpression of IGF binding proteins 3 and 6 might alter ligand stability and lead to resistance to IGF-1R inhibitors [20]. PDGFR-A was found to confer resistance to IGF-1R inhibitor in a drug-selected RMS cell line [21]. A recent report found that formation of IGF-1R and Her2 heterodimers is one of the mechanisms of rapidly developing resistance to IGF-1R inhibitors in primary mouse RMS cells [22]. It is not surprising that multiple mechanisms of resistance may operate depending on the cellular context as well as other variables. Furthermore, since IGF signaling is such a central biologic process, multiple redundant pathways should be anticipated in reaction to IGF blockade.
SFKs are activated in various types of cancer, but the family member(s) that contribute to the progression of individual tumors have not yet been defined. Because currently available SFK inhibitors are all dual SRC/Abl inhibitors and target multiple members of the SFKs, we cannot conclude that YES is the only target of dual inhibition experiments. We recently identified CRKL/YES as critical interrelated pathways necessary for RMS growth and survival by a loss-of-function screen using an inducible small hairpin RNA library [14]. Further, knockdown of YES with lentivirus small hairpin RNA dramatically inhibited RMS cell growth. These data suggest that YES seems to play a more important role in the regulation of RMS cell growth and survival.
IGF-1R/IR hybrid receptors are activated by IGF-1 and IGF-2 with similar function to IGF-1R in cancer [23,24]. IR has been reported to contribute to resistance to IGF-1R blockade [25]. It has therefore been suggested that IGF-1R/IR kinase inhibitors may prove to be more effective than IGF-1R antibody therapy by blocking both IR and IGF-1R. However, at least in our study, we found that targeting both IR and IGF-1R with the kinase inhibitor BMS-754807 also resulted in activation of YES. Our study therefore suggests that both IGF-1R blockade by anti-IGF-1R antibody and IGF-1R blockade by IGF-1R/IR small molecule inhibitors are associated with increased activation of YES. This finding may be of particular relevance clinically, since both YES and IGF components are overexpressed in RMS tumors (Figure 1). Increased YES/SFK activation might serve as a biomarker for predicting tumor resistance to IGF-1R inhibition clinically. In addition to IGF-1R inhibition, increased SFK activation has also been implicated in epidermal growth factor receptor inhibitor–mediated resistance mechanisms in different types of cancer [26,27]. Therefore, activation of SFK may confer a broader role for RTK blockade–mediated resistance.
The mechanism for IGF-1R inhibition–induced SFK activation is not clear. Shin et al. reported that increased SRC activation occurring with IGF-1R blockade in human head and neck squamous cell carcinoma and non–small cell lung cancer cell lines is associated with integrin β3 [28]. To determine whether integrin β3 is a potential mediator for increased YES/SFK activation on IGF-1R blockade, we co-treated RMS cells with anti–integrin β3 and anti-R1507 antibodies. However, anti–integrin β3 antibody did not affect IGF-1R inhibition–induced YES/SFK activation (data not shown). Thus, in RMS, IGF-1R blockade–induced YES/SFK activation is independent of integrin β3 signaling. It is possible that the compensatory activation of the YES/SFK pathway represents an important adaptive survival response in which cancer cells become resistant to IGF-1R–targeted therapeutics. Since IGF-1R lacks mutations and/or amplifications in cancer cells, induction of alternate compensatory pathways may be the more expected resistance mechanism. Therefore, combined targeting of IGF-IR and SFK may provide a new impetus for more effective applications of this targeted therapy.
In summary, we identified a mechanism of IGF-1R blockade–induced Yes/SFK activation responsible for acquired resistance to IGF-1R blockade in RMS. Due to the heterogeneous nature of tumors and alternative survival pathways developed during treatment, it is likely that additional resistance mechanisms may coexist or develop. It is clear even from our experiments that not all mice were cured with dual IGF-1R/YES blockade, and additional pathways became critical. These additional pathways remain to be studied in the future. Nonetheless, our data clearly suggest potential therapeutic strategies that could be employed now for potential patient benefit.
Acknowledgements
We are thankful to Roche Inc for providing us with R1507, Merck Inc for providing us with h7C10 antibody, and Bristol-Myers Squibb Inc for providing BMS-754807. We thank Javed Khan for providing the cDNA microarray database.
Footnotes
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. Disclosure of potential conflicts of interest: none.
Contributor Information
Xiaolin Wan, Email: xiaolinw@mail.nih.gov.
Lee J. Helman, Email: helmanl@nih.gov.
References
- 1.Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505–518. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- 2.Merlino G, Helman L. Rhabdomyosarcoma—working out the pathways. Oncogene. 1999;18:5340–5348. doi: 10.1038/sj.onc.1203038. [DOI] [PubMed] [Google Scholar]
- 3.Scrable H, Cavenee W, Ghavimi F, Lovell M, Morgan K, Sapienza C. A model for embryonal rhabdomyosarcoma tumorigenesis that involves genome imprinting. Proc Natl Acad Sci U S A. 1989;86:7480–7484. doi: 10.1073/pnas.86.19.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim SY, Toretsky JA, Scher D, Helman LJ. The role of IGF-1R in pediatric malignancies. Oncologist. 2009;14:83–91. doi: 10.1634/theoncologist.2008-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pappo AS, Patel SR, Crowley J, Reinke DK, Kuenkele KP, Chawla SP, Toner GC, Maki RG, Meyers PA, Chugh R. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol. 2011;29:4541–4547. doi: 10.1200/JCO.2010.34.0000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao L, Yu Y, Darko I, Currier D, Mayeenuddin LH, Wan X, Khanna C, Helman LJ. Addiction to elevated insulin-like growth factor I receptor and initial modulation of the AKT pathway define the responsiveness of rhabdomyosarcoma to the targeting antibody. Cancer Res. 2008;68:8039–8048. doi: 10.1158/0008-5472.CAN-08-1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown MT, Cooper JA. Regulation, substrates and functions of Src. Biochim Biophys Acta. 1996;1287:121–149. doi: 10.1016/0304-419x(96)00003-0. [DOI] [PubMed] [Google Scholar]
- 8.Bolen JB. Nonreceptor tyrosine protein kinases. Oncogene. 1993;8:2025–2031. [PubMed] [Google Scholar]
- 9.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470–480. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 10.Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6:587–595. doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- 11.Steinberg M. Dasatinib: a tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Clin Ther. 2007;11:2289–2308. doi: 10.1016/j.clinthera.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O'Brien S, Nicaise C, Bleickwood E. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 13.Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004;23:7957–7968. doi: 10.1038/sj.onc.1208079. [DOI] [PubMed] [Google Scholar]
- 14.Yeung CL, Ngo VN, Grohar PJ, Arnaldez FI, Asante A, Wan X, Khan J, Hewitt SM, Khanna C, Staudt LM. Loss-of-function screen in rhabdomyosarcoma identifies CRKL-YES as a critical signal for tumor growth. Oncogene. 2013;32:5429–5438. doi: 10.1038/onc.2012.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wan X, Yeung C, Kim SY, Dolan JG, Ngo VN, Burkett S, Khan J, Staudt LM, Helman LJ. Identification of FoxM1/Bub1b signaling pathway as a required component for growth and survival of rhabdomyosarcoma. Cancer Res. 2012;72:5889–5899. doi: 10.1158/0008-5472.CAN-12-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wan X, Helman LJ. Levels of PTEN protein modulate Akt phosphorylation on serine 473, but not on threonine 308, in IGF-II-overexpressing rhabdomyosarcomas cells. Oncogene. 2003;22:8205–8211. doi: 10.1038/sj.onc.1206878. [DOI] [PubMed] [Google Scholar]
- 17.Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 18.Wan X, Helman LJ. Effect of insulin-like growth factor II on protecting myoblast cells against cisplatin-induced apoptosis through p70 S6 kinase pathway. Neoplasia. 2002;4:400–408. doi: 10.1038/sj.neo.7900242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martins AS, Ordonez JL, Garcia-Sanchez A, Herrero D, Sevillano V, Osuna D, Mackintosh C, Caballero G, Otero AP, Poremba C. A pivotal role for heat shock protein 90 in Ewing sarcoma resistance to anti-insulin-like growth factor 1 receptor treatment: in vitro and in vivo study. Cancer Res. 2008;68:6260–6270. doi: 10.1158/0008-5472.CAN-07-3074. [DOI] [PubMed] [Google Scholar]
- 20.Huang F, Greer A, Hurlburt W, Han X, Hafezi R, Wittenberg GM, Reeves K, Chen J, Robinson D, Li A. The mechanisms of differential sensitivity to an insulin-like growth factor-1 receptor inhibitor (BMS-536924) and rationale for combining with EGFR/HER2 inhibitors. Cancer Res. 2009;69:161–170. doi: 10.1158/0008-5472.CAN-08-0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang F, Hurlburt W, Greer A, Reeves KA, Hillerman S, Chang H, Fargnoli J, Graf Finckenstein F, Gottardis MM, Carboni JM. Differential mechanisms of acquired resistance to insulin-like growth factor-I receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 2010;70:7221–7231. doi: 10.1158/0008-5472.CAN-10-0391. [DOI] [PubMed] [Google Scholar]
- 22.Abraham J, Prajapati SI, Nishijo K, Schaffer BS, Taniguchi E, Kilcoyne A, McCleish AT, Nelon LD, Gilles FG, Efstratiadis A. Evasion mechanisms to Igf1r inhibition in rhabdomyosarcoma. Mol Cancer Ther. 2011;10:697–707. doi: 10.1158/1535-7163.MCT-10-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrione A, Valentinis B, Xu SQ, Yumet G, Louvi A, Efstratiadis R, Baserga R. Insulin-like growth factor II stimulates cell proliferation through the insulin receptor. Proc Natl Acad Sci U S A. 1997;94:3777–3782. doi: 10.1073/pnas.94.8.3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-Yamaguchi Y, Siddle K, Goldfine ID, Belfiore A. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin Cancer Res. 1999;5:1935–1944. [PubMed] [Google Scholar]
- 25.Hendrickson AW, Haluska P. Resistance pathways relevant to insulin-like growth factor-1 receptor-targeted therapy. Curr Opin Investig Drugs. 2009;10:1032–1040. [PubMed] [Google Scholar]
- 26.Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011;17:461–469. doi: 10.1038/nm.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Girotti MR, Pedersen M, Sanchez-Laorden B, Viros A, Turajlic S, Niculescu-Duvaz D, Niculescu-Duvaz D, Zambon A, Sinclair J, Hayes A. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013;3:158–167. doi: 10.1158/2159-8290.CD-12-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin DH, Lee HJ, Min HY, Choi SP, Lee MS, Lee JW, Johnson FM, Mehta K, Lippman SM, Glisson BS. Combating resistance to anti-IGFR antibody by targeting the integrin β3-Src pathway. J Natl Cancer Inst. 2013;105:1558–1570. doi: 10.1093/jnci/djt263. [DOI] [PMC free article] [PubMed] [Google Scholar]