

Abstract
Sepsis is an aggressive inflammatory syndrome and a global health burden estimated to kill 7.3 million people annually. Single-target molecular therapies have not addressed the multiple disease pathways triggered by septic injury. Cell therapies might offer a broader set of mechanisms of action that benefit complex, multifocal disease processes. We describe a population of immune-specialized myofibroblasts derived from lymph node tissue, termed fibroblastic reticular cells (FRCs). Because FRCs have an immunoregulatory function in lymph nodes, we hypothesized that ex vivo–expanded FRCs would control inflammation when administered therapeutically. Indeed, a single injection of ex vivo–expanded allogeneic FRCs reduced mortality in mouse models of sepsis when administered at early or late time points after septic onset. Mice treated with FRCs exhibited lower local and systemic concentrations of proinflammatory cytokines and reduced bacteremia. When administered 4 hours after induction of lipopolysaccharide endotoxemia, or cecal ligation and puncture (CLP) sepsis in mice, FRCs reduced deaths by at least 70%. When administered late in disease (16 hours after CLP), FRCs still conveyed a robust survival advantage (44% survival compared to 0% for controls). FRC therapy was dependent on the metabolic activity of nitric oxide synthase 2 (NOS2) as the primary molecular mechanism of drug action in the mice. Together, these data describe a new anti-inflammatory cell type and provide preclinical evidence for therapeutic efficacy in severe sepsis that warrants further translational study.
INTRODUCTION
Sepsis is a life-threatening systemic inflammatory response estimated to kill more than 140,000 people globally each week—with a mortality incidence greater than that of lung, breast, and colorectal cancers combined (1–5). Septic shock occurs when the immune system detects blood-borne microbes and induces a systemic cascade of inflammation and hypotension, impairing oxygenation of vital organs, particularly the lungs, liver, intestine, and kidneys (2). Most studies estimate mortality at 30 to 40%, despite administration of antibiotics and supportive care (1, 2, 6, 7).
Sepsis pathology is driven primarily by a cooperative response between the innate immune system and the endothelium—a layer of endothelial cells that lines the interior surface of blood and lymphatic vessels. Microbial products or other unknown stimuli in the bloodstream induce Toll-like receptor (TLR) signaling in macrophages and neutrophils, which activates the cells and triggers the release of successive waves of inflammatory cytokines, beginning with tumor necrosis factor–α (TNFα) (which peaks within 90 min after exposure) followed by interleukin-1 (IL-1) and IL-6 (8, 9). These proinflammatory cytokines together with downstream mediators induce a hyperdynamic state characterized by abnormal vasodilation, fever, systemic endothelial permeability, and tissue edema. This in turn leads to a hypodynamic state, with severe hypotension, lymphocyte apoptosis, organ hypoxia, disseminated intravascular coagulation, and frequently organ failure (10, 11). Potential therapeutic options for sepsis have failed to translate to clinical efficacy. Reasons for this likely include the complex inflammatory cytokine cascade, which contains enormous redundancy of action and, therefore, cannot be disabled through targeting a single pathway; the unsuitability of in vitro systems for testing human therapies; and the swiftness with which a treatment must be administered and function. Beyond antibiotics, there are few pharmacological options for reduction of mortality. Anti-TNFα antibody therapy has not been successful in human sepsis (1, 12), and activated protein C, initially thought to increase survival, has since been proven ineffective (13).
The foundation of modern drug development, single-molecule targeted therapeutics, has been tested in sepsis clinical trials with little success (6, 14, 15). In contrast, cell therapies target multiple molecules and processes and are responsive to a patient’s disease state; thus, cellular therapeutics may offer a broader-spectrum approach to treating disease. This is particularly pertinent to sepsis, wherein the pathophysiological response is not controlled by a single inflammatory mediator or pathway (15). Cells administered to a patient sense and respond to the host’s environment, for example, through manipulation of multiple cytokine levels (16, 17). Thus, the right cell, if well tolerated and appropriately administered, could target multiple molecules and pathways, in effect providing a multifaceted pharmacological intervention with a single therapeutic agent (17, 18).
Lymphoid tissue myofibroblasts known as fibroblastic reticular cells (FRCs) are found in T cell zones of secondary lymphoid organs, which have evolved to regulate the immune response at many levels (19–25). FRCs follow a well-charted differentiation pathway generated by stromal organizer cells in the lymphoid organ anlagen during ontogeny, developing via myofibroblastic precursors and requiring signals through the lymphotoxin β receptor (LTβR) for full immunological maturation (25, 26). This ontogeny is distinct from other fibroblastic cell types, such as bone marrow–derived mesenchymal stromal cells (MSCs). Accordingly, FRCs display transcriptomes that differ broadly from other fibroblastic populations, with a notable degree of immunological specialization, including significant enrichment in cytokine signatures (23). During infection, draining secondary lymphoid organs are bathed in a complex inflammatory milieu, forming important crossroads for chemical or physical communication between various leukocyte cell types (19–22). FRCs have evolved to respond to these immunological cues, actively play a role in regulating immune responses (23), and are hypothesized to react strongly to the presence of bacteria in vivo. FRCs express TLRs (23, 27) and, within 12 hours of exposure to microbial lipopolysaccharide (LPS; also called endotoxin), produce a robust interferon (IFN)–TLR4 and acute-phase protein response (23).
Here, we describe a new cell therapeutic composed of murine FRCs and show that, when expanded ex vivo and administered to septic or endotoxemic mice, FRCs modulate a systemic response to infection. When used in allogeneic cell therapy, the FRCs significantly increased survival under clinically relevant conditions. FRCs reduced bacterial progression, increased splenic mass, and defused the sepsis-associated cytokine storm. Murine FRCs required the activity of inducible nitric oxide synthase 2 (NOS2) for a therapeutic effect, reducing mortality even when administered 16 hours after disease induction.
RESULTS
We hypothesized that the immunoregulatory potential of FRCs can be maintained after transplantation for the purpose of modulating an uncontrolled, cytokine-dominated immune response to infection. After 10 days of expansion in culture (Fig. 1A), purified FRCs isolated from pooled cutaneous and mesenteric lymph nodes showed a fibroblastic phenotype characterized by expression of podoplanin (gp38), CD140a (PDGFRα), CD44, CD90 (Thy1), CD105 (endoglin), CD106 (VCAM-1), and Sca-1; the hematopoietic determinant CD45; endothelial identifiers CD31, CD34, and Lyve-1; and follicular dendritic cell markers CD35 (CR1) and FDC-M1 (fig. S1). Cultured FRCs also expressed immunomodulatory molecules PD-L1, CD40, and CD80 while lacking expression of the MSC marker CD73 (28, 29) (fig. S1). A timeline summarizing the preparation, in vivo use, and endpoint analysis of FRC treatment is shown in Fig. 1A.
Fig. 1. Therapeutically administered FRCs impart a survival benefit after LPS endotoxemia or CLP sepsis induction.

In all experiments, stated groups received a single intraperitoneal dose of FRCs ex vivo expanded from sex-matched mice or saline (vehicle alone control). All CLP mice received standard of care, that is, antibiotics administered from the time of FRC administration and maintained throughout the experiment, and fluid resuscitation at the time of surgery. (A) Schematic timeline of experimental setup and analyses for LPS and CLP sepsis studies. LN, lymph node. (B) C57BL6 mice aged 4 to 6 weeks received 350 μg of LPS in saline intraperitoneally. Four hours later, mice received 1 × 106 syngeneic C57BL6-derived FRCs (n = 5), bone marrow MSCs (n = 7), or vehicle (saline, n = 5) intraperitoneally and were monitored for survival. (C) Aged C57BL6 mice (18 to 24 months age-matched between groups) received 150 μg of LPS intraperitoneally, and 4 hours later, this was followed by a single intraperitoneal injection of 1 × 106 syngeneic FRCs derived from 4- to 6-week-old C57BL6 mice (n = 6) or vehicle (saline, n = 10). Mice were monitored for survival. (D) CLP sepsis was induced in C57BL6 mice aged 4 to 6 weeks. Four hours later, mice received a single dose of 1 × 106 autologous FRCs (n = 5) or vehicle (saline, n = 13) intraperitoneally. (E) CLP sepsis was induced in BALB/c mice aged 4 to 6 weeks. Mice received a single dose of allogeneic C57BL6-derived FRCs (n = 18), bone marrow MSCs (n = 7), or vehicle (saline, n = 21) intraperitoneally at 4 hours and were monitored for survival. (F) BALB/c mice aged 4 to 6 weeks received a single dose of 1 × 106 allogeneic C57BL6-derived FRCs (n = 9) or saline (n = 10) intraperitoneally 16 hours after CLP surgery. Mice were monitored for survival. All data depict a minimum of two independent experiments. The log-rank (Mantel-Cox) test was used to test for significance. Arrows depict the time of cell administration.
The primary endpoint of FRC therapy for the treatment of septic shock was measured by survival analysis in two widely studied mouse models (7, 11, 30). In a model of endotoxemia, C57BL6/J mice received a lethal dose of LPS, which plays a dominant role in the pathogenesis of gram-negative bacterial sepsis (9). Young (4- to 6-week-old) C57BL6/J mice were given a single intraperitoneal dose of autologous FRCs that had been expanded in culture (1 × 106 cells) or saline (vehicle-only control) 4 hours after LPS treatment. To assess the specificity of FRC therapeutic results, we administered, at the same dose and by the same route, bone marrow–derived MSCs as a stromal cell–based control. Four hours was chosen as a therapeutically relevant time point, after which TNFα and IL-6 concentrations have reached maximal levels in both septic mice and humans (31–33). FRC-treated mice showed a significant increase in survival compared to mice given saline alone (Fig. 1B). MSCs also showed a therapeutic response compared to saline treatment [P = 0.016, log-rank (Mantel-Cox) test]; however, the FRC therapeutic response was found to be superior to that of MSCs [P = 0.0071, log-rank (Mantel-Cox) test].
Given that young C57BL6/J mice are relatively resistant to both LPS endotoxemia and sepsis (34–36), and because the rate of sepsis hospitalization is more than 10-fold higher in people older than 65 years relative to younger subjects (4), we also tested LPS-treated aged (18- to 24-month-old) C57BL6/J mice, which show greater sensitivity and susceptibility to disease in general. Aged mice were also protected from endotoxemia-related mortality when an intraperitoneal dose of autologous FRCs was administered 4 hours after LPS compared to saline-treated controls (Fig. 1C). These results suggested therapeutic potential for FRCs in murine sepsis.
Therefore, we explored the breadth of FRC therapy using a severe model of sepsis, the cecal ligation and puncture (CLP) model, which imposes physical trauma to the gut and exposure of gut contents to the peritoneum so as to induce a heavy, progressive, polymicrobial infection, with LPS detected in serum within 60 min after surgery (32). Because any clinical application of FRCs would need to complement and improve on current standards of care for sepsis, we administered all septic mice (FRC- and saline-treated controls) with antibiotics and fluid resuscitation as supportive care. C57BL6/J mice aged 4 to 6 weeks were given CLP sepsis, followed by administration of antibiotics as standard of care. Autologous FRCs or saline were infused 4 hours later. A reproducible therapeutic effect on survival was observed in the CLP model (Fig. 1D), with an efficacy similar to the FRC effect in LPS endotoxemia. Thus, FRCs improved survival of mice in two high-mortality models of systemic shock.
Modeling sepsis in the clinic
Our next set of studies focused on testing of FRCs in clinically relevant conditions to meet the demands of sepsis patients. Sepsis is a swiftly progressive medical emergency, and treatment must be administrated with minimal delay. The ability to use stored, “off-the-shelf” allogeneic cells is therefore essential for any future clinical applications. We tested the efficacy, in BALB/c mice 4 hours after CLP, of a single intraperitoneal infusion (1 × 106 cells) of allogeneic FRCs that had been derived from C57BL6/J mice and expanded in culture. The FRC-treated BALB/c mice also showed a robust survival advantage (Fig. 1E) relative to saline-treated mice. Allogeneic MSCs failed to protect at the 4-hour therapeutic time point (Fig. 1E), a finding that corroborates a previous study that showed a lack of efficacy when MSCs were administered to mice more than 60 min after CLP (30).
Diagnosis of sepsis is often made by exclusion, and thus, diagnosis may not be definitive. The delay in sepsis diagnosis requires a therapeutic to meet the severity of injury well after disease onset. Although administration of FRCs 4 hours after disease onset is a therapeutically relevant window, we further challenged the benefit of FRCs to improve survival from sepsis very late in the disease course. To this end, mice were treated with allogeneic FRCs 16 hours after CLP, a time at which mice typically approach euthanasia endpoints—a robust test of efficacy. Survival remained highly significant compared to saline-treated controls (44% compared to 0%, P = 0.007) (Fig. 1F). As expected, a higher proportion of FRC-infused mice died when treated 16 hours after CLP compared to those treated 4 hours after CLP (P = 0.007, log-rank test). The survival rate of mice treated with FRCs at 16 hours was 44% compared with 0% for saline-treated mice. The survival rate of mice treated with FRCs after 4 hours was 89% compared with 14.2% for saline-treated mice (P < 0.001, log-rank test).
The bacterial load of mice induced with CLP sepsis in peritoneal and blood compartments was surveyed after treatment to determine whether there was an effect of FRCs on bacterial clearance in the presence of broad-spectrum antibiotic therapy. Mice treated with FRCs 4 hours after CLP showed no difference in bacterial load in the peritoneum at 16 hours after CLP (Fig. 2A). Yet, we detected a significant difference in bacteremia: 78% of mice (seven of nine) treated with FRCs 4 hours after CLP had no detectable bacteria in the blood 16 hours after surgery (Fig. 2B). The observation of improved survival (Fig. 1E) when FRCs were administered to mice at a time point when bacterial load was high (16 hours, Fig. 2A) showed that FRC therapy is systemically effective in mice with established high-load bacteremia. As a mechanism, we first hypothesized a direct microbicidal effect of FRCs. In a commonly used Transwell coculture system (37, 38), FRCs inhibited Escherichia coli growth (fig. S2); however, post hoc analysis of these data in the context of this study’s findings suggested that this in vitro effect was subject to artifacts (see Discussion); thus, the assay was deemed unsuitable for further investigation of the FRC-mediated reduction in bacteremia observed in vivo. We therefore considered alternate approaches for deciphering the compartmental-specific effects of FRC therapy.
Fig. 2. FRC treatment of CLP sepsis inhibits bacteremia in vivo.
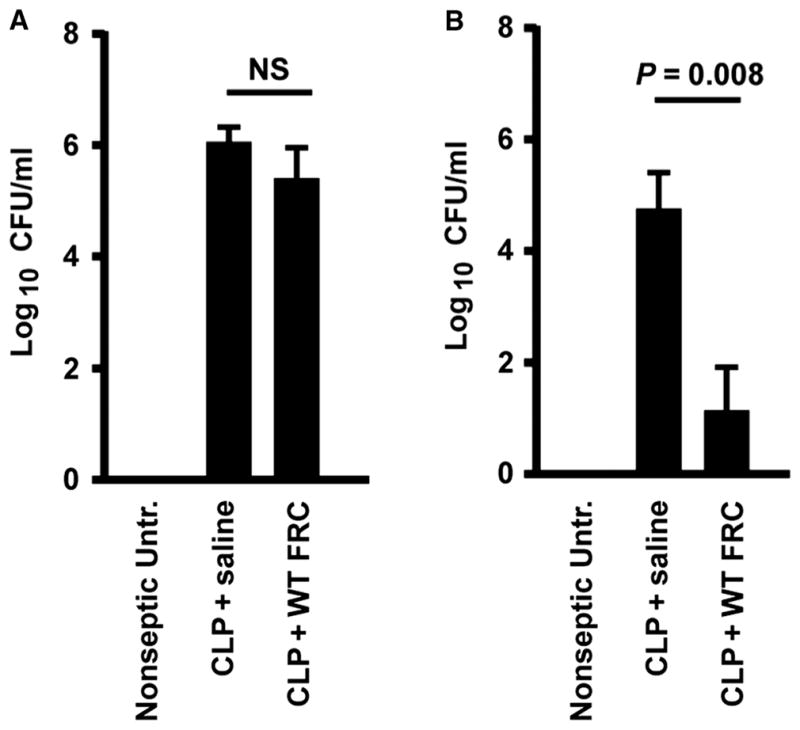
BALB/c mice aged 4 to 6 weeks received CLP sepsis, followed 4 hours later by intraperitoneal injection of a single dose (1 × 106) of allogeneic C57BL6-derived FRCs or saline (vehicle alone control). Nonseptic untreated mice are also shown. Septic mice received standard of care, including antibiotics administered from the time of FRC treatment and fluid resuscitation after surgery. (A and B) Peritoneal lavage (A) and blood samples (B) were taken 16 hours after surgery. Bacterial colony-forming units (CFUs) were calculated from serially diluted samples of (A) peritoneal lavage fluid and (B) blood. Mean ± SEM is shown. Peritoneal samples: n = 4 untreated, n = 5 saline-treated, n = 5 FRC-treated. Blood samples: n = 5 untreated, n = 6 saline-treated, n = 10 FRC-treated. Data depict three to five different experiments. Statistical analysis was performed using the Mann-Whitney U test, comparing saline- versus FRC-treated groups. NS, not statistically significant.
Because FRC treatment reduced bacteremia, we next hypothesized that FRCs may be trafficking to other parts of the body from the peritoneal cavity. Bioavailability of the cell transplant in the local peritoneal cavity of LPS-injured mice was first measured using noninvasive bioluminescent imaging of FRCs isolated from luciferase-engineered knock-in mice. FRCs were administered 4 hours after LPS injection and were present in the peritoneum of LPS-injured mice for ~48 hours (Fig. 3A), with signal detectable and declining for up to a week after cell transplantation (Fig. 3, A and B). Because the detection capability of bioluminescence decreases significantly in deep tissues, flow cytometric profiling of nonengineered allogeneic FRC peritoneal transplants was used to assess the presence of infused FRCs in the blood and spleen over time. The donor FRCs were detected using anti-podoplanin and anti-CD90 as donor cell markers and were found at low but detectable numbers in the blood and spleen at 8 hours (Fig. 3, C and D). FRCs in the periphery accounted for <1% of the initial injection (Fig. 3, C and D). These data support the notion that FRCs were predominantly retained in the peritoneum with rather insignificant numbers observed in circulation or engrafted in the spleen.
Fig. 3. FRCs are retained in the peritoneum.

(A) Mice received 350 μg of LPS 4 hours before intraperitoneal injection of 1 × 106 allogeneic FRCs. Mice then received 4.5 mg of luciferin intraperitoneally and were imaged every 5 min for 45 min or until peak luminescence was reached. The maximum luminescence for each mouse over the imaging period was then recorded. The same maximum and minimum acquisition settings were used for all time points. Images depict FRC luminescence and localization in anesthetized recipient mice over time. Two mice most closely representing the group average are depicted (n = 5 mice per group). (B) Maximum luminescence over the imaging period was recorded for each mouse at each time point. Bars represent mean ± SEM (n = 3 mice per experiment). (C and D) Mice received 350 μg of LPS 4 hours before intraperitoneal injection of 1 × 106 allogeneic antibody-labeled FRCs or saline (no stroma control). Blood (C) and spleen (D) were harvested after 1 h, 8 h, or 24 h. Donor FRCs were identified using flow cytometry. n = 2 (no stroma), n = 3 (1 h time point), n = 3 (8 h time point), n = 3 (24 h time point). Bars represent mean ± SEM.
Because FRCs were concentrated in the peritoneum, we next tested whether the transplanted cells acted locally to contain the polymicrobial infection within the peritoneum, systemically to hinder bacterial infection of the blood, or both. Mining of mRNA microarray data of FRCs isolated from pooled cutaneous and mesenteric lymph nodes showed high levels of chemokine expression that were consistent with the hypothesis that FRCs were acting locally (Fig. 4A). However, despite expression of these chemokines (at the mRNA transcript level in vitro), when mice were given an FRC transplant 4 hours after CLP and assessed 16 hours after surgery, the transplanted FRCs did not modulate local sepsis-induced leukocyte influx into the peritoneum, because these changes were entirely sepsis-dependent (compare treated to untreated mice) and were not affected by FRC administration (Fig. 4, B to E). The increase in the total number of viable, erythrocyte-depleted cells in the mouse peritoneum was a result of the CLP sepsis-inducing injury (Fig. 4B), and sepsis similarly drove an increase in granulocyte cell types that occurred concomitantly to a loss of F4/80-expressing macrophages, T cells, and B cells (Fig. 4, C to E, and fig. S3). None of these changes were altered by FRC administration. We next tested whether FRCs enhanced phagocytic activity of immune cells in vitro, despite the comparable numbers of cells found in the septic peritoneal cavity with and without FRC treatment. The ability of CD11b+ splenocytes to phagocytose bacterial components was unaffected by coculture with FRCs (Fig. 4F). Collectively, analysis of the local peritoneal cavity did not reveal a major change in the composition or effector function of immune cells.
Fig. 4. FRCs do not alter peritoneal immune cell migration or effector function.

(A) RNA was extracted from two independent purified FRC cultures and sent for cDNA microarray analysis. The heat map measures selected chemokines, depicted as log2 of the mRNA expression value (EV). Log2EV of 6.9 represents the post-normalization threshold for expression. Dark blue represents nonexpression, whereas light blue, pink, and red depict increasing levels of expression. (B) BALB/c mice aged 4 to 6 weeks received CLP sepsis, followed 4 hours later by intraperitoneal injection of a single dose (1 × 106) of allogeneic C57BL6-derived FRCs or saline (vehicle alone control). Results with nonseptic untreated (healthy) mice are also shown. Septic mice received standard of care, including antibiotics administered from the time of FRC treatment and fluid resuscitation after surgery. Peritoneal lavage samples were taken 16 hours after surgery. Peritoneal lavage leukocyte counts are shown along with means (bars) ± SEM (n = 4 untreated, n = 6 saline-treated, and n = 7 FRC-treated mice). (C and D) Two myeloid cell subsets (%) in peritoneal lavage fluid are shown [n = 9 mice for untreated (black), n = 9 mice for saline-treated (light gray), and n = 10 mice for FRC-treated (dark gray) groups]; results are from three to four independent experiments. (E) B and T lymphocyte subsets (%) from peritoneal lavage fluid are shown (n = 9 for untreated, n = 9 for saline-treated, and n = 10 for FRC-treated mice; results are from three to four independent experiments). **P < 0.01, ***P < 0.001, ****P < 0.0001, untreated compared to FRC-treated mice, using the Mann-Whitney U test. (F) Splenocytes and purified mouse FRCs were cocultured with pHrodo Red–labeled E. coli conjugates for 30 min at 37°C (gray) or on ice (white). Cells were analyzed using flow cytometry. Histograms depict the percentage of CD11b+ cells that are pHrodo Red–positive. Histograms represent two independent experiments. NS, not significantly different.
Similar to the peritoneal cavity assessments, in the peripheral blood of CLP mice, we observed sepsis-specific changes in leukocyte composition that were not changed by FRC therapy (Fig. 5, A to C, and fig. S4). CLP significantly reduced the proportion of peripheral blood B cells while boosting the T cell pool (P < 0.0001 and P = 0.027, two-tailed Mann-Whitney U test).
Fig. 5. FRC treatment prevents sepsis-associated reduction in splenic leukocyte populations.

BALB/c mice aged 4 to 6 weeks received CLP sepsis, followed 4 hours later by a single dose (1 × 106) of allogeneic C57BL6-derived FRCs or saline (vehicle alone control). Results for nonseptic untreated (healthy) mice are also shown. Septic mice received standard of care, including antibiotics administered from the time of FRC treatment and fluid resuscitation after surgery. Sixteen hours after CLP, mice were humanely killed, blood and spleens were harvested for flow cytometric assessment, and results were compared to untreated (nonseptic) controls. (A) Percentage of CD11c+ and F4/80+ myeloid subsets from blood [n = 4 mice for untreated (black) and saline-treated (light gray) groups; n = 5 mice for FRC-treated (dark gray) group]. (B) Expression of Gr1 and CD11b on F4/80−CD11c− leukocytes isolated from blood (n = 9 mice for the untreated, saline-treated, and FRC-treated groups). (C) Percentage of T and B cells in blood (n = 9 untreated, n = 8 saline-treated, and n = 10 FRC-treated groups). (D) Total number of spleen cells (n = 6 mice for untreated and n = 7 mice for saline- and FRC-treated groups). (E) Percentage of apoptotic (annexin V+PI−) splenocytes (n = 5 mice for untreated, saline-treated, and FRC-treated groups). (F) Expression of Gr1 and CD11b on F4/80−CD11c− leukocytes isolated from spleens (n = 4 mice for untreated and saline-treated groups and n = 5 mice for the FRC-treated group). Bars depict means ± SEM and represent at least two independent experiments. Statistical significance was assessed using a two-tailed Mann-Whitney U test for nonparametric data. NS, not statistically significant.
A reduction in leukocyte numbers in the spleen is a hallmark of mouse and human sepsis (39–41). Therefore, at 16 hours after CLP in mice, we measured leukocyte numbers in spleens isolated from mice that were either untreated or treated 4 hours after surgery with an allogeneic FRC transplantation. We observed significant changes in leukocyte numbers in the spleens of FRC-treated septic mice compared to saline-treated septic controls (Fig. 5, D to F, and fig. S5). Specifically, FRC-treated mice showed a significant increase in overall spleen cellularity 16 hours after CLP (Fig. 5D; P = 0.006, two-tailed Mann-Whitney U test) accompanied by a reduction in leukocyte apoptosis (Fig. 5E; P = 0.008, two-tailed Mann-Whitney U test). Compared to saline-treated septic controls, FRC-treated mice also had significantly higher numbers of B cells, CD4+ and CD8+ T cells, and myeloid cell subsets in the spleen (Fig. 5F and fig. S5E). Although no changes in activated T cell numbers were seen in the spleen, FRC-treated septic mice showed significantly higher numbers of naïve T cells compared to saline-treated septic controls, suggesting a protective effect of FRC administration on preservation of naïve T cells (fig. S5, M and O; P = 0.016, two-tailed Mann-Whitney U test). Together, these data show that by 16 hours after CLP, FRCs mediated significant protective effects on leukocyte subsets distal to the initial infection site. The splenic protection observed was in lieu of the large fraction of the FRC graft residing in the peritoneal compartment and led us to consider an intermediary signaling mechanism that was transmitted into the bloodstream from the FRCs.
Distal effects of FRCs
Murine FRCs exert immunosuppressive effects in lymph nodes and ex vivo through targeted NOS2-dependent release of nitric oxide (NO) in situ (42–44). Therefore, we hypothesized that FRC transplants release NO as a direct or indirect molecular mediator of FRC therapy in mice. Nitrite, a by-product of NO metabolism, was detectable in the peritoneum and blood as early as 60 min after allogeneic FRCs were transplanted into LPS-injured mice, with a peak burst at 4 hours after transplantation (fig. S6). In vitro studies confirmed that FRCs did not constitutively produce NO (fig. S7A), nor was secretion induced by LPS treatment alone (fig. S7B). Secretion of NO was observed when FRCs were cocultured with CD3/28-activated splenocytes (fig. S7B). The TNFα/TNFRI (TNF receptor type I) axis—known to be important in inducing NO production by activated FRCs in vitro (42)—was not critical to the observed efficacy of FRC therapy, because a similar level of survival was observed with wild-type allogeneic FRCs and FRCs in which TNFRI was blocked by neutralizing antibodies when the cells were administered 4 hours after mice were subjected to CLP (fig. S8).
With the knowledge that the FRCs were viable after transplantation and NOS2 activity was measurable (indicated by nitrite production), we tested FRC transplants derived from NOS2−/− mice for protection in CLP sepsis. FRCs isolated from NOS2−/− mice failed to protect BALB/c mice from sepsis-mediated mortality (Fig. 6A), with survival kinetics and rates similar to treatment with saline or MSCs (compare with Fig. 1E). These data suggest NOS2 as a primary molecular mediator of protection by FRCs in mouse models. Accordingly, mice treated with NOS2−/−FRCs at 4 hours after CLP showed a similar degree of bacteremia 16 hours after CLP (Fig. 6B), and splenic apoptosis rates were significantly higher in mice treated with NOS2−/−FRCs at 4 hours after CLP than in mice treated with wild-type FRCs (Fig. 6C). Therefore, the major hallmarks of FRC therapy appear to be NOS2-dependent.
Fig. 6. Survival effects of FRC therapy are dependent on NOS2.
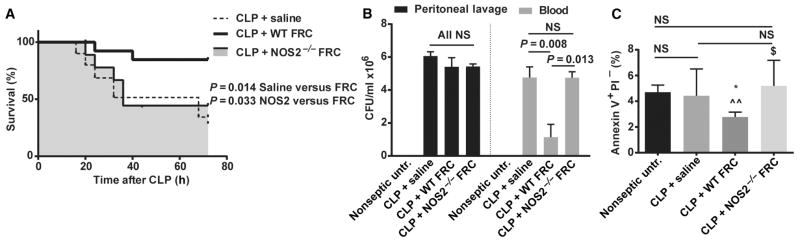
BALB/c mice aged 4 to 6 weeks received CLP sepsis, followed 4 hours later by intraperitoneal injection of a single dose (1 × 106) of allogeneic C57BL6-derived FRCs (n = 18), NOS2−/− FRCs (n = 9), or saline (vehicle alone control, n = 10). Results with untreated (nonseptic) mice are shown for comparison. Septic mice received standard of care, including antibiotics administered from the time of FRC treatment and fluid resuscitation after surgery. (A) Mice were monitored for survival, and the groups were compared for statistically significant differences using the log-rank (Mantel-Cox) test. Data represent a minimum of two independent experiments. (B) Serum and peritoneal lavage samples were taken 16 hours after CLP surgery. Bacterial CFUs were calculated from serially diluted samples of blood and peritoneal lavage. Means ± SEM are shown. Peritoneal samples: n = 4 mice for the untreated group, n = 5 mice for the saline-treated group, n = 5 mice for the FRC-treated group, and n = 4 mice for the NOS2−/− FRC–treated group. Blood samples: n = 5 mice for the untreated group, n = 6 mice for the saline-treated group, n = 10 mice for the FRC-treated group, and n = 4 mice for the NOS2−/− FRC–treated group. Data depict results from two to five experiments. Statistical analysis was performed using the Mann-Whitney U test, comparing saline- versus FRC-treated groups. (C) The percentage of apoptotic splenocytes was calculated from flow cytometric staining of samples taken 16 hours after CLP. *P < 0.05 for FRC-treated mice compared to untreated; ^^P < 0.01 for FRC-treated mice compared to saline-treated mice; $P < 0.05 for NOS2−/− FRC–treated mice compared to FRC-treated mice, all calculated using the Mann-Whitney U test. n = 5 mice for the untreated, saline-treated, and FRC-treated groups; n = 4 mice for the NOS2−/−FRC–treated group. NS, not statistically significant. Data depicted are from two independent experiments. For clarity of comparison, CLP-saline and CLP-FRC data points in this figure are also depicted in Figs. 1E, 2, and 5D. WT, wild type.
The biological response to FRC therapy and the role of NOS2 activity were assessed on the host proinflammatory cytokine response, both as a key driver of sepsis-related pathology and as a factor common to both the endotoxin and CLP models. At 16 hours after CLP was induced (12 hours after treatment with antibiotics and either FRCs or saline), FRC-treated mice displayed significantly lower levels of peritoneal TNFα relative to saline-treated control septic mice (Fig. 7A; P = 0.02, one-tailed Mann-Whitney U test) and other proinflammatory cytokines (IL-1α and IL-1β) (Fig. 7, B and C; P < 0.05 in all comparators, one-tailed Mann-Whitney U test). Peritoneal IL-17 and IFN-γ were not reduced to statistically significant levels by FRC treatment (Fig. 7, D and E). No significant differences were observed in peritoneal levels of the anti-inflammatory cytokine IL-10 (Fig. 7F), suggesting that the boosting of IL-10, as reported for MSC therapy (30), was not a primary molecular mechanism for increasing survival by FRC treatment.
Fig. 7. FRCs reduce expression of proinflammatory cytokines in the peritoneum and blood in a NOS2-dependent manner.

BALB/c mice aged 4 to 6 weeks received CLP sepsis, followed 4 hours later by an intraperitoneal injection of a single dose (1 × 106) of allogeneic C57BL6-derived FRCs, NOS2−/− FRCs, or saline (vehicle alone control). Results for untreated (healthy nonseptic) mice are shown for comparison. Septic mice received standard of care, including antibiotics administered from the time of FRC treatment and fluid resuscitation after surgery. (A to L) Peritoneal lavage (PL) and blood samples were obtained 16 hours after CLP and analyzed using the Luminex multiplex platform, examining concentrations of (A) PL TNFα, (B) PL IL-1α, (C) PL IL-1β, (D) IFN-γ, (E) IL-17, (F) IL-10, (G) blood TNFα, (H) blood IL-17, (I) blood IFN-γ, (J) blood IL-6, (K) blood MCP-1, and (L) blood IL-10. Bars represent means ± SEM. PL, all groups: n = 5 mice. Blood: n = 4 mice for untreated, saline-treated, and NOS2−/− FRC–treated groups; n = 5 mice for FRC-treated groups. The data shown are compiled from two independent experiments. Groups were compared using a one-tailed Mann-Whitney U test for nonparametric data. NS, not significantly different.
Moreover, the prototypical septic cytokine storm in mouse serum was strongly regulated by FRC therapy in CLP-treated mice. Serum TNFα, IFN-γ, and IL-17 levels were completely reduced to near wild-type levels (Fig. 7, G to I), whereas IL-6 and MCP-1 levels were attenuated from their pathogenic values (Fig. 7, J and K) in FRC-treated relative to saline-treated control mice. As in the peritoneum, serum IL-10 concentrations were not substantially altered by FRC treatment and did not correlate with the antiseptic therapeutic benefit of FRCs, unlike MSC therapy, in which boosting of IL-10 concentrations was key (30) (Fig. 7L).
To rule out a permanent anergizing effect of FRCs, we isolated and reactivated leukocytes from the peritoneums or spleens of septic animals 16 hours after CLP, and found that reactivated cells remained capable of producing normal levels of TNFα (fig. S9) in vitro. Hence, FRC therapy reduced the cytokine storm without impairing natural immunosurveillance mechanisms of immune cells and permanently impairing their capacity to produce cytokines. Overall, the biological response to FRC therapy was driven by a major reversal in cytokine pathways that required NOS2 production by FRCs.
Together, our data pinpoint FRCs as a potential new immunomodulatory cell therapy candidate for the treatment of murine models of inflammation and sepsis.
DISCUSSION
Stromal cell–based therapies have risen in prominence in recent years. MSCs, in particular, have proven to be well tolerated and are being heavily tested in clinical trials to control immune responses and promote tissue regeneration across a range of diseases and pathologies for which current treatments are inadequate (16–18). In 2012, allogeneic bone marrow–derived MSCs were approved for use in New Zealand and Canada to treat steroid-refractory pediatric graft-versus-host disease; in South Korea, allogeneic umbilical cord blood–derived MSCs were approved to treat degenerative arthritis, whereas autologous adipose-derived MSCs were approved to treat anal fistulas caused by Crohn’s disease (45).
However, MSCs have not shown marked therapeutic promise in preclinical sepsis models. Multiple studies report improved survival and anti-inflammatory properties when MSCs propagated from various tissues were administered to rodents at the time (within 60 min) of septic insult (30, 46–49) but not at therapeutically relevant time points. One opposing study reported a survival benefit 6 hours after CLP (50). Compared to MSCs, FRCs are a mesenchymal cell type of unique ontogeny, phenotype, and site-specific specialization. Their immune-specialized capabilities, in particular, suggest that FRCs might yield a distinct or enhanced therapeutic profile in septic patients compared with other stromal cell therapies currently being tested in clinical trials. Like other stromal cell therapies, FRCs share desirable characteristics for a cell therapy candidate: ease of isolation and ex vivo expansion to clinical cell masses, allogeneic efficacy, and high viability in frozen storage for point-of-care use. Our data demonstrate that murine FRCs effectively reduced death from sepsis, inhibited key proinflammatory cytokines in blood and peritoneum, and reduced bacterial load.
The inspiration for using FRC transplants as a therapy for inflammatory disease such as sepsis was based on the in situ immunoregulatory functions of FRCs, which have become better understood over the past decade. FRCs are predominantly stromal cell population in lymph nodes and spleen and have historically been studied as important structural cells that secrete extracellular matrix components as well as cytokines CCL19, CCL21, and IL-7 to create a three-dimensional environment that permits T cells, B cells, and dendritic cells to interact and initiate an immune response (19, 23–25, 51). Recently, a new paradigm of peripheral tolerance was described, mediated by FRCs in situ (27, 42–44, 52, 53). FRCs can impose both deletional and regulatory tolerance; they can present autoantigens and activate autoreactive T cells in a manner that induces their loss from the T cell pool (27, 52, 53), as well as suppress the proliferation of T cells that are activated by other nearby cells or stimuli (42–44). Accordingly, FRCs have been shown to reduce T cell proliferation within lymph nodes after exposure to IFN-γ and TNFα, with signaling through the IFN-γ receptor (IFNGR) and TNFRI (42). TNFRI was dispensable for FRC therapy in our CLP studies, although we cannot rule out a role for other signaling molecules such as IFN-γ. A recently published transcriptome of FRCs suggests a strong capacity to modulate the inflammatory response through extensive expression of cytokine receptors, chemokines, and growth factors (23).
A key finding in our model, unlikely to be coincidental, is that FRCs show the ability to defuse the sepsis-associated “cytokine storm” with a concomitant reduction in serum concentrations of TNFα, IL-6, MCP-1, IL-17, and, to a lesser extent, IFN-γ. Serum concentrations of IL-10 [which is a commonly reported player in MSC-mediated protection against cytokine-induced pathology, including in sepsis (30)] were not changed in our studies, and MSCs did not improve survival when administered to septic mice at time points late in the course of the disease. On the other hand, we observed a reduction in serum concentrations of IL-17 in FRC-treated septic mice, a finding that agrees with those of another study in which neutralizing IL-17 antibodies yielded a significant survival benefit in mice that had undergone CLP even at late administration times (54). Previous studies have not reported a reduction in serum IL-17 concentrations when mouse MSCs are transplanted into mice intravenously (55, 56); this is another indication, aside from a superior survival benefit, that the biological response to FRCs differs from that of other MSCs. Therefore, in our studies, the main effect of FRC administration was to reduce the concentration of key proinflammatory cytokines in both blood and peritoneum.
Reduction of splenic cellularity is a noted effect in human sepsis, with increases in both leukocyte apoptosis and a likely efflux of leukocytes to blood, other tissues, and the original infection site. Saline-treated CLP mice did not show an increase in spleen cell apoptosis compared to untreated mice at the time point tested (16 hours after CLP induction), but FRC-treated mice exhibited a significant reduction in apoptotic (annexin V+PI−) splenocytes compared to saline-treated CLP mice, suggesting a degree of protection. FRCs express a variety of molecules known to function in the maintenance, expansion, migration, and maturation of myeloid cells, including Flt3L (supports and expands dendritic cells), CSF-1 (colony-stimulating factor 1), IL-34 (a macrophage colony-stimulating factor receptor ligand), and CXCL14 (a monocyte and dendritic cell chemoattractant) (23). Accordingly, we observed significant FRC-mediated protection of myeloid cell subsets, notably F4/80+CD11c− macrophages and F4/80− granulocytes that were CD11b+ and with high, low, or negative Gr1 expression. The loss of B cells and T cells (both CD4+ and CD8+) seen in saline-treated CLP mice was also ameliorated by FRC treatment. FRCs also exerted a protective effect on naïve T cells, correlating with a known function of FRCs (19, 51, 57).
The bacterial load in the bloodstream was markedly reduced by FRC therapy, which we speculate to be the result of increased splenic numbers with unimpaired antibacterial activity (58). FRCs inhibited E. coli growth in vitro, although this is likely an artifact of the assay, because bacterial inhibition was not specific to FRCs and was not NOS2-dependent; these finding suggest that the in vitro E. coli cell growth inhibition model is irrelevant to FRC sepsis therapy. Bacteria levels in the peritoneum did not differ among untreated and FRC-treated septic mice, thereby implying that the infection source was still present in all groups. The cellular composition of the peritoneum and blood was generally unchanged by FRC treatment but showed sepsis-specific changes such as the predominant influx of CD11b+Ly6G+ granulocytes in the peritoneum and a loss of peripheral blood B cells. On the other hand, leukocyte populations from the spleen were markedly altered, and in vitro results showed that FRCs did not impair phagocytosis activity of splenocytes or their ability to secrete TNFα once isolated and stimulated ex vivo after CLP sepsis. Together, our results suggest that a greater splenic mass was preserved by FRC treatment and could likely reduce bacteremia through natural phagocytosis and sequestration.
We observed that the prosurvival effects of FRC therapy were lost when NOS2-deficient FRCs were administered to mice, implying a role for NO in survival. NO is a highly pleiotropic and immunomodulatory cell signaling molecule that can stably react with blood components to reach distal organs (59), and its breadth of action could explain the observed effects of FRC therapy on cytokines, leukocytes, and bacterial levels in septic mice (59–62). Alternatively, NO’s effects could involve interactions with other cellular and molecular partners, which in turn mediate the observed outcomes. Directly or indirectly, NO is capable of shutting down an immune response at multiple levels (59–63).
The biochemistry of NO function is complex and depends on both concentration and the local oxidative microenvironment, but major mechanisms of action involve binding to “metal centers” such as heme components of soluble guanylyl cyclase and S-nitrosylation of proteins and lipids (64). NO has a short half-life and was originally thought to act when released in close proximity to target cells. Yet, we now know that NO can form stable compounds in circulation through S-nitrosylation or other reactions and thus can reach distal organs in a bioactive state (15, 59) or can create a tissue-wide antimicrobial milieu (63). NO has antiapoptotic effects; reduces both neutrophil adhesion to blood vessels and extravasation to tissues (64); increases antibacterial capabilities of macrophages; directly inhibits bacterial DNA replication, protein synthesis, and platelet aggregation; and increases smooth muscle relaxation and metabolism (64). Manipulation of NO metabolism may represent a therapeutic option for sepsis. Selected NO donors can target hypoxic, acidified tissues for increased micro-circulation without negatively affecting macrocirculatory vascular function (15, 65, 66). Treatment of animal models with NO donors reduces organ failure and, in many cases, mortality (15). Administration of the NO donor nitroglycerin to humans with septic shock decreased mortality (67), whereas treatment of septic patients in a phase 3 trial with NOS inhibitor 546C88 increased mortality (68).
Our studies support the notion that NOS2 is essential for the benefits observed after FRC transplantation. FRCs are conceivably a major source of NO, as estimated by in vitro studies showing that FRCs contribute 110-fold more NO on a per-cell basis compared to activated splenocytes (fig. S7). However, we cannot rule out the possibility that other mediators are involved in our observed survival outcomes. NOS2 might also participate in immunoevasion of an allogeneic cell transplant reaction, allowing the cells to persist long enough to impart an immunosuppressive effect before clearance. Our results showed that FRCs were confidently detectable in mice for up to 50 hours after transplantation.
Human FRCs are poorly studied, although they show immunoregulatory properties in culture that are similar to those of mouse FRCs in our study (69). Although their therapeutic potential has not been tested preclinically, human FRCs can be isolated from lymph node biopsies or palatine tonsil tissues and expanded ex vivo (70, 71) to provide possible source for future cell formulations. To date, the therapeutic capacity of mouse or human FRCs to dampen pathogenic immune responses has not been tested. It is notable that although murine MSCs use NO as a primary mechanism for immunosuppression in several models of T cell–mediated diseases, human MSCs yield similar effects but without dependence on NO release, primarily using indoleamine 2,3-dioxygenase, cyclooxygenase enzymes, and prostaglandin E2 (17, 55, 72, 73). The lack of suitable in vitro and in vivo models for testing human FRC efficacy in sepsis prevents direct tests of its efficacy at this point, particularly because human FRCs do not conduct effective immunological crosstalk with mouse parenchymal and hematopoietic cells, hindering the use of xenograft models.
Rodent models of sepsis have come under scrutiny because of a lack of effective sepsis drugs arising from such research. Microarray data suggest that rodents receiving low doses of LPS do not recapitulate the transcriptional changes that humans exhibit in response to low doses of LPS (74). However, severe models of murine sepsis and endotoxemia do exhibit many key features of the pathological immune response long reported in humans, including an uncontrolled cytokine storm led by high systemic production of TNFα, IL-6, and IL-1. These cytokines activate platelets and induce disseminated intravascular coagulation and systemic loss of endothelial cell integrity, which in turn induces loss of blood pressure, followed by hypoxic organ damage and high mortality. Among other criticisms [recently presented by (75)], none of these hallmarks are induced by the low doses of LPS used in (74), rendering interpretation of the model difficult. Despite important and acknowledged differences between mice and humans, preclinical testing in rodent models remains critical to sepsis research because of the immense difficulties inherent in recapitulating the complex systemic response to sepsis in vitro. Testing in multiple models and mouse strains and use of clinically relevant treatment regimens are prudent if we hope to increase translation of therapies to large animal models and human subjects.
Deaths from sepsis are reaching crisis point: the mortality rate is high, and overall incidence is increasing as a result of the current growth in aging populations and increasing rates of surgical intervention, type II diabetes, and antibiotic resistance (76–78). Our results support the notion that FRC-based therapy warrants further study and may represent a new, multifactorial intervention for sepsis.
MATERIALS AND METHODS
Study design
The aim of this study was to evaluate FRC treatment in models of sepsis and endotoxemia in mice. FRC therapy was administered to injured mice that received standard of care in clinically relevant studies, with primary endpoints being survival and secondary endpoints as serology, cytometry, and bacteriology analysis of animals at an interim time point. All studies were powered to achieve a >25% benefit in the endpoints stated. Subjects were randomly assigned to experimental groups, and external testing of samples collected from the studies was analyzed by blinded core facility technicians. All studies had several internal replicates of each experimental group and were replicated through at least two independent trials as described in the figure legends. FRCs provided a significant improvement in primary and many secondary endpoints in these efficacy studies.
Mice
Female C57BL/6 and BALB/c mice aged 4 to 6 weeks were obtained from The Jackson Laboratory or bred in-house at Monash University Animal Research Laboratories; 18- to 24-month female C57BL/6 mice were aged in-house. Nos2−/− mice backcrossed to a C57BL/6 background (strain name: B6.129S2-Nos2tm1MrI N12) and wild-type littermates were from Taconic. All mice were specific pathogen–free and cared for in accordance with institutional and National Institutes of Health (NIH) guidelines. Mice were acclimated to new housing conditions for a minimum of 5 days before experimentation. Experiments were conducted with approval of the Research Animal Care subcommittees at Massachusetts General Hospital or Dana-Farber Cancer Institute, or the Monash University Animal Welfare Committee.
FRC isolation and ex vivo expansion
Mouse lymph nodes (inguinal, axillary, brachial, cervical, and mesenteric) were pooled and digested as described (21), using 5 ml of enzyme mix containing collagenase P (0.2 mg/ml) (Roche), deoxyribonuclease I (0.1 mg/ml) (Invitrogen), and Dispase (0.8 mg/ml) (Roche) at 37°C for 15 min. Tissues were agitated, medium was collected, and the digestion mix was replaced. This proceeded for 50 to 60 min, when lymph nodes were completely digested. Single-cell suspensions were recovered by centrifugation and cultured in α-MEM (minimum essential medium) with 10% batch-selected fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cultures were washed after 48 hours and allowed to expand for one to two passages before use. Cells were washed with phosphate-buffered saline (PBS) to remove residual protein and harvested using 0.2% trypsin with 5 mM EDTA in PBS. FRCs were purified by MACS-depleting endothelial and hematopoietic cells as described to >95% purity (21). Where stated, FRCs were preincubated for 20 min with either anti-TNFRI blocking antibody (BioLegend, clone 55R-170) or isotype control (BioLegend, clone HTK888). Cells were washed three times in sterile PBS before injection into mice.
MSC isolation and expansion
Bone marrow was isolated from tibias and femurs and plated for MSC expansion according to standard methods (79). Briefly, marrows were gently dissociated in sterile α-MEM with 10% batch-selected FBS and 1% penicillin/streptomycin before plating at a density of 2.6 × 104 cells/mm2. Cells were grown until 80 to 90% confluent, then harvested using 0.2% trypsin with 5 mM EDTA in PBS, and subcultured for further expansion. Before use, MSCs were purified by MACS-depleting endothelial and hematopoietic cells as described for FRCs (21), according to the manufacturer’s instructions (Miltenyi Biotec). MSCs were used at passage 2.
Endotoxemia and sepsis induction
Endotoxemia was induced by administering LPS (O111:B4, Sigma) intraperitoneally at a dose of 350 μg in 100 μl of saline for 4- to 6-week-old mice weighing about 20 g (80), or 150 μg in 100 μl of saline for aged mice weighing about 40 g. Sepsis was also induced using a modified CLP protocol with fluid resuscitation, as described (11). Briefly, mice were anesthetized using ketamine/xylazine, given sterile eye drops to protect vision, and incised 2 cm vertically along the abdominal midline. Eighty percent of the cecum distal to the ileocecal valve was ligated using 4.0 sutures. The cecum was punctured twice with a 27-gauge needle, and a drop of fecal matter was exuded before reinstating the cecum to the peritoneal cavity and suturing the muscle and skin closed. Mice were given 1 ml of saline subcutaneously. To mimic the clinical standard of care, all CLP-treated mice also received 1.5 mg of ampicillin (Sigma) in saline subcutaneously once every 12 hours, beginning at the time of first FRC treatment, and maintained until the end of the experiment. Mice were monitored at four hourly intervals through critical stages of disease and euthanized at objective, predefined endpoints: loss of circulation to tail or feet, loss of responsiveness to stimuli, or with a breathing rate less than 120 breaths per minute. Survivors were monitored intensively for 72 hours after surgery and euthanized 96 hours after surgery. Where stated, mice were euthanized 16 hours after CLP for immunological analysis, histopathology, serology, or bacteriology.
FRC and MSC treatment of septic and endotoxemic mice
Ex vivo–expanded FRCs were washed twice in sterile PBS and resuspended at 1 × 107 cells/ml. Mice received 1 × 106 cells in 100 μl of saline intraperitoneally at stated time points (either 4 or 16 hours after sepsis or endotoxemia induction).
Bacteriology
Peritoneal lavage fluid, or blood collected in 20 mM EDTA to prevent clotting, was serially diluted 10-fold in sterile saline, and a lawn culture was made on nutrient agar. Three to four dilutions were plated per mouse in triplicate, and plates were incubated at 37°C. Colonies were counted after 16 hours, and average CFUs per milliliter of blood or lavage fluid were calculated per mouse.
Quantification of cytokines
TNFα, IL-6, MCP-1, IL-17, IFN-γ, IL-1α IL-1β, and IL-10 were quantified from serum and peritoneal lavage samples using the Milliplex MAP Mouse Cytokine/Chemokine Panel (Millipore) and read on a Luminex MAGPIX instrument, and data were analyzed with Luminex xPONENT software. The detection limits for each cytokine were 1.0, 1.8, 5.3, 0.5, 0.9, 5.1, 2.0, and 3.3 pg/ml, respectively. Measurements for all cytokines were performed according to protocols provided by the manufacturer.
Flow cytometry
Peritoneal lavage was made using 3 ml of sterile PBS injected into the peritoneal cavity, mixed well by palpitation, and then withdrawn using a syringe. Spleens were dissociated using 70-μm mesh and the plunger of a syringe. Blood was collected in 20 mM EDTA in PBS, with cells pelleted by centrifugation. Red blood cells for all cell preparations were lysed for 60 s using ACK lysis buffer (Fisher). Cells were then counted and stained for flow cytometry. Cells were stained for 20 min with antibodies against extracellular determinants at predetermined titrations and diluted in PBS with 1% heat-inactivated fetal calf serum and 2.5 mM EDTA. For restimulation of cells from septic mice, cells were plated in 96-well flat-bottom tissue culture plates for 5 hours in the presence of LPS (1 μg/ml) and GolgiStop (BD Biosciences) before being stained with extracellular antibodies, then fixed and permeabilized using the BD Biosciences Fix/Perm kit according to the vendor’s instructions, and then stained with anti-TNFα for 30 min. Antibodies used were as follows: CD11c-PE (phycoerythrin) (clone HL-3, BD Biosciences), F4/80-APC (allophycocyanin) (clone BM8, Invitrogen), Ly6G-PECy7 (clone RB6-8C5, BioLegend), CD11b-FITC (fluorescein isothiocyanate) (clone M1/70, eBioscience), TCRb-APC (clone H57-597, BD Biosciences), CD19-PE (clone 1D3, eBioscience), CD8a-FITC (clone 53-6.7, BD Biosciences), CD62L-PECy7 (clone MEL-14, BioLegend), and anti-TNFα–PE (clone MP6-XT22, eBioscience). Annexin V and propidium iodide (PI) staining was made using the Annexin V Staining kit (BD Biosciences) according to supplied instructions. Data were acquired using a FACSAria IIU or FACSCalibur (both BD Biosciences) and analyzed using FlowJo (Tree Star).
Tracking FRCs
For tracking via bioluminescent imaging, 1 × 106 purified FRCs were intraperitoneally injected into mice 4 hours after LPS injury. Mice received an intraperitoneal injection of 4.5 mg of luciferase substrate (Molecular Imaging Products) solution before imaging. The bioluminescent signal was measured in anesthetized mice on an IVIS 100 imaging system (Caliper Life Sciences) until a peak signal was reached. Data are expressed as photons/second per cm2. For flow cytometric tracking, 1 × 106 purified FRCs were labeled with anti-gp38 (BioLegend, clone 8.1.1) and anti-CD90 (BD Biosciences, clone 30. H12), washed three times, and then intraperitoneally injected into mice 4 hours after LPS injury. Mice were humanely killed at stated time points after cell transfer. Blood was collected in 0.5 M EDTA to prevent clotting. Spleen was dissociated using the frosted ends of glass slides. Cell preparations were stained with CD45 (BD Biosciences, clone 30. F11) to exclude hematopoietic cells and reduce signal-to-noise ratio, acquired on a FACSCalibur (BD Biosciences), and analyzed using FlowJo (Tree Star).
Phagocytosis assay
FRCs (1 × 105) were cultured in a 96-well flat-bottom culture plate overnight to adhere. Splenocytes (1 × 106) were added, with pHrodo Red–labeled E. coli conjugate (Molecular Probes) in the provided buffer, according to the manufacturer’s instructions, for 30 min at 37°C or on ice. Cells were analyzed using flow cytometry. Histograms are gated on CD11b+ cells and represent two independent experiments.
Microarray analysis
Lymph node stromal cells were isolated from 4- to 6-week-old C57BL6/J mice and then ex vivo–expanded and purified as described above. Purified FRCs were recultured for 2 days to recover. FRCs from two independent cultures were harvested and lysed, RNA was purified, and Affymetrix MoGene 1.0 ST arrays were used to generate expression profiles as described (23). Results were robust multiarray average (RMA)–normalized, log2-transformed, and analyzed using GenePattern software as described (23). A post-normalized expression value of 120 was used to indicate gene expression.
Statistical analyses
Survival data were compared using the log-rank (Mantel-Cox) test. Normal distribution was not assumed, and flow cytometry, bacteriology, and protein analysis data were compared using a nonparametric two-tailed Mann-Whitney U test. As outlined in the figure legends, a one-tailed Mann-Whitney U test was performed when the directionality of a difference between groups was predicted on the basis of previous supported hypotheses. For all tests, an α threshold of 0.05 was used. Variance is depicted as either mean ± SD or mean ± SEM as described in the figure legends. Statistical analysis was performed using GraphPad Prism version 6.01 for Windows.
Supplementary Material
Acknowledgments
We thank the Center for Systems Biology for technical assistance with pharmacological analysis of FRCs, and M. Armant, R. Harding, and S. Bouch for helpful discussions.
Funding: Supported in part by grants R01EB012521 (B.P.), K01DK087770 (B.P.), R01 DK074500 (S.J.T.), and P01 AI045757 (S.J.T.) from the U.S. NIH and by the Shriners Hospitals for Children (B.P.). A.L.F. was the recipient of an Early Career Fellowship (546259) from the National Health and Medical Research Council of Australia; a training grant from Monash University, Australia; and a Birmingham Fellowship, University of Birmingham, UK. T.S.P.H. was supported by an Australian Postdoctoral (Industry) Fellowship from the Australian Research Council (LP110201169).
Footnotes
www.sciencetranslationalmedicine.org/cgi/content/full/6/249/249ra109/DC1
Fig. S1. Phenotype of ex vivo–expanded FRCs.
Fig. S2. FRCs inhibit growth of E. coli in vitro in a NOS2-independent manner.
Fig. S3. Flow cytometric analysis of leukocyte subsets in peritoneal lavage fluid after FRC therapy.
Fig. S4. Flow cytometric analysis of leukocyte subsets in blood after FRC therapy.
Fig. S5. Flow cytometric analysis of leukocyte subsets in spleen after FRC therapy.
Fig. S6. Nitrite measurement in serum after FRC therapy.
Fig. S7. FRCs do not secrete NO in vitro in response to LPS.
Fig. S8. TNFRI does not mediate the survival benefit seen in FRC-treated mice with CLP sepsis.
Fig. S9. TNFα production by restimulated peritoneal and splenic leukocytes.
Table S1. GenBank accession number for microarray analyses of cultured FRCs.
Competing interests: B.P. has an equity interest in Sentien Biotechnologies Inc., which develops cell therapeutics and has an option to license relevant patents generated by B.P. that may lead to financial benefits. A.L.F., S.J.T., and B.P. are inventors on a patent application related to this work.
Data and materials availability: GenBank accession codes are provided in table S1.
Author contributions: A.L.F., S.J.T., and B.P. conceived, designed, and financed the study; interpreted the data; and wrote the paper. R.L.B. interpreted the data and provided financial support. A.L.F., J.S.E., J.A., R.M., N.S., K.K., F.D.B., F.A.S., J.M.N., T.S.P.H., and J.D.R. performed the experiments.
REFERENCES AND NOTES
- 1.Carney DE, Matsushima K, Frankel HL. Treatment of sepsis in the surgical intensive care unit. Isr Med Assoc J. 2011;13:694–699. [PubMed] [Google Scholar]
- 2.Daniels R. Surviving the first hours in sepsis: Getting the basics right (an intensivist’s perspective) J Antimicrob Chemother. 2011;66(Suppl 2):ii11–ii23. doi: 10.1093/jac/dkq515. [DOI] [PubMed] [Google Scholar]
- 3.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Hall MJ, Williams SN, DeFrances CJ, Golosinskiy MS. Inpatient Care for Septicemia or Sepsis: A Challenge for Patients and Hospitals. National Center for Health Statistics; Hyattsville, MD: 2011. [PubMed] [Google Scholar]
- 5.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. GLOBOCAN 2008 v.2.0. Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 10. http://globocan.iarc.fr.
- 6.Vincent JL. Acute kidney injury, acute lung injury and septic shock: How does mortality compare? Contrib Nephrol. 2011;174:71–77. doi: 10.1159/000329238. [DOI] [PubMed] [Google Scholar]
- 7.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: Setting the stage. Nat Rev Drug Discov. 2005;4:854–865. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 8.Rodríguez-Gaspar M, Santolaria F, Jarque-López A, González-Reimers E, Milena A, de la Vega MJ, Rodríguez-Rodríguez E, Gó-Sirvent JL. Prognostic value of cytokines in SIRS general medical patients. Cytokine. 2001;15:232–236. doi: 10.1006/cyto.2001.0932. [DOI] [PubMed] [Google Scholar]
- 9.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 10.London NR, Zhu W, Bozza FA, Smith MC, Greif DM, Sorensen LK, Chen L, Kaminoh Y, Chan AC, Passi SF, Day CW, Barnard DL, Zimmerman GA, Krasnow MA, Li DY. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med. 2010;2:23ra19. doi: 10.1126/scitranslmed.3000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest. 2009;119:2868–2878. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Carr B, Goyal M, Gaieski DF. Sepsis: The inflammatory foundation of pathophysiology and therapy. Hosp Pract. 2011;39:99–112. doi: 10.3810/hp.2011.08.585. [DOI] [PubMed] [Google Scholar]
- 13.Martí-Carvajal AJ, Solà I, Lathyris D, Cardona AF. Human recombinant activated protein C for severe sepsis. Cochrane Database Syst Rev. 2012;3:CD004388. doi: 10.1002/14651858.CD004388.pub5. [DOI] [PubMed] [Google Scholar]
- 14.Cohen J. Adjunctive therapy in sepsis: A critical analysis of the clinical trial programme. Br Med Bull. 1999;55:212–225. doi: 10.1258/0007142991902222. [DOI] [PubMed] [Google Scholar]
- 15.Cauwels A, Brouckaert P. Nitrite regulation of shock. Cardiovasc Res. 2011;89:553–559. doi: 10.1093/cvr/cvq317. [DOI] [PubMed] [Google Scholar]
- 16.Le Blanc K, Pittenger M. Mesenchymal stem cells: Progress toward promise. Cytotherapy. 2005;7:36–45. doi: 10.1080/14653240510018118. [DOI] [PubMed] [Google Scholar]
- 17.Pittenger M. Sleuthing the source of regeneration by MSCs. Cell Stem Cell. 2009;5:8–10. doi: 10.1016/j.stem.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 18.De Miguel MP, Fuentes-Julián S, Blázquez-Martínez A, Pascual CY, Aller MA, Arias J, Arnalich-Montiel F. Immunosuppressive properties of mesenchymal stem cells: Advances and applications. Curr Mol Med. 2012;12:574–591. doi: 10.2174/156652412800619950. [DOI] [PubMed] [Google Scholar]
- 19.Bajénoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, Germain RN. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25:989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mueller SN, Matloubian M, Clemens DM, Sharpe AH, Freeman GJ, Gangappa S, Larsen CP, Ahmed R. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc Natl Acad Sci USA. 2007;104:15430–15435. doi: 10.1073/pnas.0702579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fletcher AL, Malhotra D, Turley SJ. Lymph node stroma broaden the peripheral tolerance paradigm. Trends Immunol. 2011;32:12–18. doi: 10.1016/j.it.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luther SA, Vogt TK, Siegert S. Guiding blind T cells and dendritic cells: A closer look at fibroblastic reticular cells found within lymph node T zones. Immunol Lett. 2011;138:9–11. doi: 10.1016/j.imlet.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Malhotra D, Fletcher AL, Astarita J, Lukacs-Kornek V, Tayalia P, Gonzalez SF, Elpek KG, Chang SK, Knoblich K, Hemler ME, Brenner MB, Carroll MC, Mooney DJ, Turley SJ Immunological Genome Project Consortium. Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat Immunol. 2012;13:499–510. doi: 10.1038/ni.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Acton SE, Astarita JL, Malhotra D, Lukacs-Kornek V, Franz B, Hess PR, Jakus Z, Kuligowski M, Fletcher AL, Elpek KG, Bellemare-Pelletier A, Sceats L, Reynoso ED, Gonzalez SF, Graham DB, Chang J, Peters A, Woodruff M, Kim YA, Swat W, Morita T, Kuchroo V, Carroll MC, Kahn ML, Wucherpfennig KW, Turley SJ. Podoplanin-rich stromal networks induce dendritic cell motility via activation of the C-type lectin receptor CLEC-2. Immunity. 2012;37:276–289. doi: 10.1016/j.immuni.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chai Q, Onder L, Scandella E, Gil-Cruz C, Perez-Shibayama C, Cupovic J, Danuser R, Sparwasser T, Luther SA, Thiel V, Rülicke T, Stein JV, Hehlgans T, Ludewig B. Maturation of lymph node fibroblastic reticular cells from myofibroblastic precursors is critical for antiviral immunity. Immunity. 2013;38:1013–1024. doi: 10.1016/j.immuni.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–674. doi: 10.1038/nri2832. [DOI] [PubMed] [Google Scholar]
- 27.Fletcher AL, Lukacs-Kornek V, Reynoso ED, Pinner SE, Bellemare-Pelletier A, Curry MS, Collier AR, Boyd RL, Turley SJ. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J Exp Med. 2010;207:689–697. doi: 10.1084/jem.20092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 29.Katebi M, Soleimani M, Cronstein BN. Adenosine A2A receptors play an active role in mouse bone marrow-derived mesenchymal stem cell development. J Leukoc Biol. 2009;85:438–444. doi: 10.1189/jlb.0908520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Németh K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, Mezey E. Bone marrow stromal cells attenuate sepsis via prostaglandin E2–dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neilson D, Kavanagh JP, Rao PN. Kinetics of circulating TNF-α and TNF soluble receptors following surgery in a clinical model of sepsis. Cytokine. 1996;8:938–943. doi: 10.1006/cyto.1996.0126. [DOI] [PubMed] [Google Scholar]
- 32.Villa P, Sartor G, Angelini M, Sironi M, Conni M, Gnocchi P, Isetta AM, Grau G, Buurman W, van Tits LJ, Ghezzi P. Pattern of cytokines and pharmacomodulation in sepsis induced by cecal ligation and puncture compared with that induced by endotoxin. Clin Diagn Lab Immunol. 1995;2:549–553. doi: 10.1128/cdli.2.5.549-553.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Møller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem. 2008;15:1697–1705. doi: 10.2174/092986708784872393. [DOI] [PubMed] [Google Scholar]
- 34.Saito H, Sherwood ER, Varma TK, Evers BM. Effects of aging on mortality, hypothermia, and cytokine induction in mice with endotoxemia or sepsis. Mech Ageing Dev. 2003;124:1047–1058. doi: 10.1016/j.mad.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Turnbull IR, Wlzorek JJ, Osborne D, Hotchkiss RS, Coopersmith CM, Buchman TG. Effects of age on mortality and antibiotic efficacy in cecal ligation and puncture. Shock. 2003;19:310–313. doi: 10.1097/00024382-200304000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Maddens B, Vandendriessche B, Demon D, Vanholder R, Chiers K, Cauwels A, Meyer E. Severity of sepsis-induced acute kidney injury in a novel mouse model is age dependent. Crit Care Med. 2012;40:2638–2646. doi: 10.1097/CCM.0b013e3182591ebe. [DOI] [PubMed] [Google Scholar]
- 37.Krasnodembskaya A, Song Y, Fang X, Gupta N, Serikov V, Lee JW, Matthay MA. Antibacterial effect of human mesenchymal stem cells is mediated in part from secretion of the antimicrobial peptide LL-37. Stem Cells. 2010;28:2229–2238. doi: 10.1002/stem.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 39.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 40.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci USA. 1999;96:14541–14546. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharron M, Hoptay CE, Wiles AA, Garvin LM, Geha M, Benton AS, Nagaraju K, Freishtat RJ. Platelets induce apoptosis during sepsis in a contact-dependent manner that is inhibited by GPIIb/IIIa blockade. PLOS One. 2012;7:e41549. doi: 10.1371/journal.pone.0041549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lukacs-Kornek V, Malhotra D, Fletcher AL, Acton SE, Elpek KG, Tayalia P, Collier AR, Turley SJ. Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat Immunol. 2011;12:1096–1104. doi: 10.1038/ni.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siegert S, Huang HY, Yang CY, Scarpellino L, Carrie L, Essex S, Nelson PJ, Heikenwalder M, Acha-Orbea H, Buckley CD, Marsland BJ, Zehn D, Luther SA. Fibroblastic reticular cells from lymph nodes attenuate T cell expansion by producing nitric oxide. PLOS One. 2011;6:e27618. doi: 10.1371/journal.pone.0027618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khan O, Headley M, Gerard A, Wei W, Liu L, Krummel MF. Regulation of T cell priming by lymphoid stroma. PLOS One. 2011;6:e26138. doi: 10.1371/journal.pone.0026138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Syed BA, Evans JB. Stem cell therapy market. Nat Rev Drug Discov. 2013;12:185–185. doi: 10.1038/nrd3953. [DOI] [PubMed] [Google Scholar]
- 46.Weil BR, Herrmann JL, Abarbanell AM, Manukyan MC, Poynter JA, Meldrum DR. Intravenous infusion of mesenchymal stem cells is associated with improved myocardial function during endotoxemia. Shock. 2011;36:235–241. doi: 10.1097/SHK.0b013e318225f6ae. [DOI] [PubMed] [Google Scholar]
- 47.Krasnodembskaya A, Samarani G, Song Y, Zhuo H, Su X, Lee JW, Gupta N, Petrini M, Matthay MA. Human mesenchymal stem cells reduce mortality and bacteremia in gram-negative sepsis in mice in part by enhancing the phagocytic activity of blood monocytes. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1003–L1013. doi: 10.1152/ajplung.00180.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang CL, Leu S, Sung HC, Zhen YY, Cho CL, Chen A, Tsai TH, Chung SY, Chai HT, Sun CK, Yen CH, Yip HK. Impact of apoptotic adipose-derived mesenchymal stem cells on attenuating organ damage and reducing mortality in rat sepsis syndrome induced by cecal puncture and ligation. J Transl Med. 2012;10:244. doi: 10.1186/1479-5876-10-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shin S, Kim Y, Jeong S, Hong S, Kim I, Lee W, Choi S. The therapeutic effect of human adult stem cells derived from adipose tissue in endotoxemic rat model. Int J Med Sci. 2013;10:8–18. doi: 10.7150/ijms.5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mei SH, Haitsma JJ, Dos Santos CC, Deng Y, Lai PF, Slutsky AS, Liles WC, Stewart DJ. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. Am J Respir Crit Care Med. 2010;182:1047–1057. doi: 10.1164/rccm.201001-0010OC. [DOI] [PubMed] [Google Scholar]
- 51.Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, Cyster JG, Luther SA. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. 2007;8:1255–1265. doi: 10.1038/ni1513. [DOI] [PubMed] [Google Scholar]
- 52.Lee JW, Epardaud M, Sun J, Becker JE, Cheng AC, Yonekura AR, Heath JK, Turley SJ. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat Immunol. 2007;8:181–190. doi: 10.1038/ni1427. [DOI] [PubMed] [Google Scholar]
- 53.Cohen JN, Guidi CJ, Tewalt EF, Qiao H, Rouhani SJ, Ruddell A, Farr AG, Tung KS, Engelhard VH. Lymph node–resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J Exp Med. 2010;207:681–688. doi: 10.1084/jem.20092465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li J, Zhang Y, Lou J, Zhu J, He M, Deng X, Cai Z. Neutralisation of peritoneal IL-17A markedly improves the prognosis of severe septic mice by decreasing neutrophil infiltration and proinflammatory cytokines. PLOS One. 2012;7:e46506. doi: 10.1371/journal.pone.0046506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bouffi C, Bony C, Courties G, Jorgensen C, Noël D. IL-6-dependent PGE2 secretion by mesenchymal stem cells inhibits local inflammation in experimental arthritis. PLOS One. 2010;5:e14247. doi: 10.1371/journal.pone.0014247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen B, Hu J, Liao L, Sun Z, Han Q, Song Z, Zhao RC. Flk-1+ mesenchymal stem cells aggravate collagen-induced arthritis by up-regulating interleukin-6. Clin Exp Immunol. 2010;159:292–302. doi: 10.1111/j.1365-2249.2009.04069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malhotra D, Fletcher AL, Turley SJ. Stromal and hematopoietic cells in secondary lymphoid organs: Partners in immunity. Immunol Rev. 2013;251:160–176. doi: 10.1111/imr.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Almdahl SM, Bøgwald J, Hoffman J, Sjunneskog C, Seljelid R. The effect of splenectomy on Escherichia coli sepsis and its treatment with semisoluble aminated glucan. Scand J Gastroenterol. 1987;22:261–267. doi: 10.3109/00365528709078589. [DOI] [PubMed] [Google Scholar]
- 59.Mannick JB. Immunoregulatory and antimicrobial effects of nitrogen oxides. Proc Am Thorac Soc. 2006;3:161–165. doi: 10.1513/pats.200505-048BG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 61.Benz D, Cadet P, Mantione K, Zhu W, Stefano G. Tonal nitric oxide and health: Antibacterial and -viral actions and implications for HIV. Med Sci Monit. 2002;8:RA27–RA31. [PubMed] [Google Scholar]
- 62.Fang FC. Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies. Nat Rev Microbiol. 2004;2:820–832. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 63.Olekhnovitch R, Ryffel B, Müller AJ, Bousso P. Collective nitric oxide production provides tissue-wide immunity during Leishmania infection. J Clin Invest. 2014;124:1711–1722. doi: 10.1172/JCI72058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gao Y. The multiple actions of NO. Pflugers Arch. 2010;459:829–839. doi: 10.1007/s00424-009-0773-9. [DOI] [PubMed] [Google Scholar]
- 65.Cauwels A, Buys ES, Thoonen R, Geary L, Delanghe J, Shiva S, Brouckaert P. Nitrite protects against morbidity and mortality associated with TNF- or LPS-induced shock in a soluble guanylate cyclase–dependent manner. J Exp Med. 2009;206:2915–2924. doi: 10.1084/jem.20091236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lupp C, Baasner S, Ince C, Nocken F, Stover JF, Westphal M. Differentiated control of deranged nitric oxide metabolism: A therapeutic option in sepsis? Crit Care. 2013;17:311. doi: 10.1186/cc12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spronk PE, Ince C, Gardien MJ, Mathura KR, Oudemans-van Straaten HM, Zandstra DF. Nitroglycerin in septic shock after intravascular volume resuscitation. Lancet. 2002;360:1395–1396. doi: 10.1016/s0140-6736(02)11393-6. [DOI] [PubMed] [Google Scholar]
- 68.López A, Lorente JA, Steingrub J, Bakker J, McLuckie A, Willatts S, Brockway M, Anzueto A, Holzapfel L, Breen D, Silverman MS, Takala J, Donaldson J, Arneson C, Grove G, Grossman S, Grover R. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: Effect on survival in patients with septic shock. Crit Care Med. 2004;32:21–30. doi: 10.1097/01.CCM.0000105581.01815.C6. [DOI] [PubMed] [Google Scholar]
- 69.Link A, Hardie DL, Favre S, Britschgi MR, Adams DH, Sixt M, Cyster JG, Buckley CD, Luther SA. Association of T-zone reticular networks and conduits with ectopic lymphoid tissues in mice and humans. Am J Pathol. 2011;178:1662–1675. doi: 10.1016/j.ajpath.2010.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hähnlein J, Ramwadhdoebe TH, Semmelink JF, Maijer KI, Choi IY, Smits NAM, Berger FH, Maas M, Gerlag DM, Geitenbeek TB, Tak PP, van Baarsen LGM. Expression of the autoimmune regulator Aire in human lymph node stromal cells. Ann Rheum Dis. 2014;73:A82–A83. [Google Scholar]
- 71.Zeng M, Smith AJ, Wietgrefe SW, Southern PJ, Schacker TW, Reilly CS, Estes JD, Burton GF, Silvestri G, Lifson JD, Carlis JV, Haase AT. Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J Clin Invest. 2011;121:998–1008. doi: 10.1172/JCI45157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ren G, Su J, Zhang L, Zhao X, Ling W, L’huillie A, Zhang J, Lu Y, Roberts AI, Ji W, Zhang H, Rabson AB, Shi Y. Species variation in the mechanisms of mesenchymal stem cell-mediated immunosuppression. Stem Cells. 2009;27:1954–1962. doi: 10.1002/stem.118. [DOI] [PubMed] [Google Scholar]
- 73.Najar M, Raicevic G, Boufker HI, Fayyad Kazan H, De Bruyn C, Meuleman N, Bron D, Toungouz M, Lagneaux L. Mesenchymal stromal cells use PGE2 to modulate activation and proliferation of lymphocyte subsets: Combined comparison of adipose tissue, Wharton’s Jelly and bone marrow sources. Cell Immunol. 2010;264:171–179. doi: 10.1016/j.cellimm.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 74.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Osuchowski MF, Remick DG, Lederer JA, Lang CH, Aasen AO, Aibiki M, Azevedo LC, Bahrami S, Boros M, Cooney R, Cuzzocrea S, Jiang Y, Junger WG, Hirasawa H, Hotchkiss RS, Li XA, Radermacher P, Redl H, Salomao R, Soebandrio A, Thiemermann C, Vincent JL, Ward P, Yao YM, Yu HP, Zingarelli B, Chaudry IH. Abandon the mouse research ship? Not just yet! Shock. 2014;41:463–475. doi: 10.1097/SHK.0000000000000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Girard TD, Opal SM, Ely EW. Insights into severe sepsis in older patients: From epidemiology to evidence-based management. Clin Infect Dis. 2005;40:719–727. doi: 10.1086/427876. [DOI] [PubMed] [Google Scholar]
- 77.Ayala-Ramírez OH, Domínguez-Berjón MF, Esteban-Vasallo MD. Trends in hospitalizations of patients with sepsis and factors associated with inpatient mortality in the Region of Madrid, 2003–2011. Eur J Clin Microbiol Infect Dis. 2014;33:411–421. doi: 10.1007/s10096-013-1971-0. [DOI] [PubMed] [Google Scholar]
- 78.van der Poll T, Opal SM. Host–pathogen interactions in sepsis. Lancet Infect Dis. 2008;8:32–43. doi: 10.1016/S1473-3099(07)70265-7. [DOI] [PubMed] [Google Scholar]
- 79.Parekkadan B, Fletcher AL, Li M, Tjota MY, Bellemare-Pelletier A, Milwid JM, Lee JW, Yarmush ML, Turley SJ. Aire controls mesenchymal stem cell-mediated suppression in chronic colitis. Mol Ther. 2012;20:178–186. doi: 10.1038/mt.2011.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Howard M, Muchamuel T, Andrade S, Menon S. Interleukin 10 protects mice from lethal endotoxemia. J Exp Med. 1993;177:1205–1208. doi: 10.1084/jem.177.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.