

Abstract
The life-threatening, emotional, and economic burdens of premature birth have been greatly alleviated by antenatal glucocorticoid (GC) treatment. Antenatal GCs accelerate tissue development reducing respiratory distress syndrome and intraventricular hemorrhage in premature infants. However, they can also alter developmental processes in the brain and trigger adverse behavioral and metabolic outcomes later in life. This review summarizes animal model and clinical studies that examined the impact of antenatal GCs on the developing brain. In addition, we describe studies that assess glucocorticoid receptor (GR) action in neural stem/progenitor cells (NSPCs) in vivo and in vitro. We highlight recent work from our group on two GR pathways that impact NSPC proliferation, ie, a nongenomic GR pathway that regulates gap junction intercellular communication between coupled NSPCs through site-specific phosphorylation of connexin 43 and a genomic pathway driven by differential promoter recruitment of a specific GR phosphoisoform.
Glucocorticoids (GCs) are essential for mobilizing biological processes not required for fetal viability during the transition of the mammalian fetus from intrauterine to extrauterine life. The overall action of endogenous fetal GCs is to trigger organ maturation, enabling the lungs, liver, gastrointestinal tract, thyroid gland, adrenals, and kidneys to function and sustain life outside the uterine environment (1). Various tissues in the developing fetus express the glucocorticoid receptor (GR), and it is these primary organs that undergo a maturational shift to prepare the infant for parturition and ex utero survival. For example, in the lungs GCs trigger thinning of the alveolar septae and rapid maturation of alveoli, production of collagen and elastin, and production and release of surfactant proteins and phospholipids (2–5). GCs also improve the ability of the lungs to resorb fluids by increasing ion channels in the pulmonary epithelium and up-regulating β-adrenergic receptors (4, 6, 7). In the liver, GCs increase protein and glycogen synthesis as well as alter the expression of gluconeogenic enzymes, fatty acid synthase, aminotransferases, and thyroid hormone metabolism (1, 8–12). GC responses in the gut lead to increases in the number and height of villi and migration of enterocytes. As a result, digestive activity and hormone release are augmented (1, 13–18). The increase in the fetal kidney's resorptive ability and decrease in the fraction of excreted sodium are due in part to GC up-regulation of the Na+/H+ exchanger and Na+/K+ ATPase (19–23). Erythropoietin production decreases as do renin levels and angiotensin II receptor expression, but the renin-angiotensin system becomes more responsive to hypovolemia (24–28). GCs also hasten thyroid maturation, increasing thyroid hormones that are critical to neurodevelopment (1). Finally, in the fetal adrenals GCs impact cytoarchitecture of the zona fasciculata and induce cytochrome P450s, phenylethanolamine N-methyltransferases, and ACTH receptors (1). These specific developmental requirements for GCs are reflected in the ontogeny of circulating GC levels in the fetus. Specifically, human fetal serum cortisol levels as measured in the umbilical cord demonstrate a fall in midgestation and a rapid rise in late gestation (Table 1) (29).
Table 1.
Serum Cortisol Levels in the Human Fetus (29)
| Weeks of Gestation | Average Fetal Serum Cortisol, ng/mL |
|---|---|
| 15–17 | 8.4 |
| 17.5–20 | 4 |
| 35–36 | 20 |
| 37.5–40 | 45.1 |
Therapeutic use of synthetic GCs in pregnant women to reduce complications of premature birth
Synthetic GCs are prescribed to women at risk for preterm labor to decrease the morbidity and mortality associated with prematurity. This therapeutic modality was originally tested in fetal sheep in the late 1960s (30) and became a part of clinical practice after a 1972 landmark study. In that report, Liggins and Howie (30, 31) found that a 2-day course of synthetic antenatal GCs decreased respiratory distress syndrome (RDS) and intraventricular hemorrhage (IVH), specifically in infants under 32 weeks' gestation whose mother had at least 24 hours between steroid administration and delivery. Subsequent large cohort studies determined that antenatal GC therapy also reduced necrotizing enterocolitis and infant mortality (32, 33). Two types of synthetic GCs have historically been used for antenatal administration, dexamethasone (Dex) and betamethasone (Beta). Although Dex and Beta are very similar structurally, varying only in the position of one methyl group, they differ in their recommended dosing schedules, placental metabolism, affinity for GR in some fetal tissue, and potency of nongenomic effects (34–36). Recent studies suggest that the preparation of Dex or Beta may also contribute to their differing clinical effects as well (37, 38).
Currently used regimens for antenatal GCs were determined empirically and later established by a 1994 National Institutes of Health Consensus Conference and a meta-analysis of 18 trials by Crowley (32) and have remained unchanged since the study by Liggins and Howie in 1972 (31). The plasma unbound GC level achieved by this regimen is physiological; Ballard et al (39) found that it is comparable with stress-level plasma GCs as a result of endogenous cortisol production in premature infants with RDS. However, decreasing GCs to one single dose of Beta has long been considered, and a study using sheep has found that this permits lung maturation and improved cardiac and renal function (40, 41). Historically, multiple courses of GCs have been administered if the delivery does not occur within 7 days, although the recently published Multiple Courses of Antenatal Corticosteroids for Preterm Birth study suggests that multiple courses of antenatal GCs lead to a decreased length, weight, and head circumference at birth, compared with a single course (42). However, in multiple placebo controlled trials using weekly administration of GCs, the incidence of RDS and mortality was similar to one single course (43).
Complicating the issue is a recent Cochrane review suggesting there is benefit with regard to RDS in multiple courses of antenatal GCs when given to women who remain at risk of preterm delivery after 7 days since the initial course (44). Currently the same dosing regimen is used for singletons and multiple gestations (45), including whether multiple gestations are composed of one or more placentas. A recent study suggests that, at least for Beta, umbilical cord concentrations of Beta are similar for singleton or multigestational pregnancies and also suggests that there is no significant difference in cord concentrations if the mother is obese or of optimal habitus (46). The route of administration has traditionally been intramuscular because oral antenatal GCs have been linked to an increased risk of early-onset neonatal sepsis and IVH (47). Finally, neither gender nor race is taken into account in dosing regimens, despite a clear difference in some outcomes of Caucasian males and females compared with those of African descent (48, 49).
Recent studies have examined whether one particular synthetic GC preparation is substantially more beneficial than another. Although Dex may generate a more pronounced decrease in IVH and possibly time under neonatal intensive care (50), Beta may be more effective at preventing respiratory complications in very low-birth weight infants (51) and in mice (37) and may decrease incidence of periventricular leukomalacia in human studies (52). However, conflicting results have precluded the designation of an optimal GC dosing regimen (50, 51). In the United States, Beta is preferred over Dex for antenatal use in women at risk for preterm delivery.
A more controversial historical use of antenatal GCs is for the treatment of congenital adrenal hyperplasia (CAH). CAH is an autosomal recessive disorder primarily affecting the adrenal cortex. Ninety-five percent of CAH cases are caused by deficiency of 21-hydroxylase, the enzyme that converts 17-hydroxyprogesterone to 11-deoxycortisol. This results in deficiencies of cortisol and aldosterone and excess of androgens (53). Although classical CAH may result in childhood virilization in males, in utero virilization and the development of ambiguous genitalia uniquely affect females, and for this indication, GCs have been given early in gestation and continued throughout pregnancy for any fetus at risk for CAH based on parental genetics (53, 54). However, seven eighths of infants treated, all males and unaffected females, receive no benefit due to the need to treat before the earliest possible time for prenatal genetic testing (54). Additionally, in both rodent and primate models, GC treatment for CAH has been associated with hypertension, hyperinsulinemia, hyperglycemia, fatty liver on a high-fat diet, hypothalamus-pituitary-adrenal (HPA) hyperreactivity, and poorer learning and memory (55–57). Children who received treatment have experienced low birth weight (LBW), failure to thrive, developmental delay, mood disturbance, poor school performance, and social anxiety (58–61). Given this, antenatal GCs for prevention of female virilization in CAH are currently reserved for clinical trials (62).
Unlike endogenous GCs, Dex and Beta are not inactivated by enzymes found in the placenta (63). Furthermore, GCs easily cross the blood-brain barrier via simple diffusion (64) and therefore have access in the fetal brain to GR. In contrast to their beneficial effects on various fetal organ systems, synthetic GCs may negatively impact development of the fetal brain. In nonhuman primates treated with antenatal GCs and either delivered prematurely or at term, treatment was associated with lower brain and cerebellum weight and reductions in the dentate gyrus and cornu Ammon (CA) region of the hippocampus. These cellular abnormalities are associated with lower levels of the presynaptic protein synaptophysin and microtubule associated proteins in the frontal region (65). At 20 months of age, young monkeys who received antenatal GCs still had a reduction in hippocampal volume, which may be due to a chronically elevated cortisol level causing neurotoxicity (66). In nonhuman primates, antenatal GCs were associated with poorer concentration, learning and memory, and hyperactivity (67, 68). Similarly in rats and mice, antenatal GCs were associated with decreased learning and memory along with increased anxiety (69, 70). Antenatal GC administration was associated with changes in basal and stress-induced HPA axis function in a variety of mammals, but these effects are not generalizable across age and species. In species with young born at an advanced stage of development, antenatal synthetic GCs suppress HPA axis function early in life and in adulthood and briefly increase it in the juvenile period. In species with young born in an underdeveloped state, the HPA axis function is generally elevated by antenatal GCs throughout the life span (71).
In humans, antenatal GCs have been associated with structural changes in the brain. For example, antenatal GCs trigger cortical thinning specifically in the rostral anterior cingulate cortex in children 6–10 years of age who were born at term (63) as well as decreased cortical surface area and complexity of cortical folding in infants born at term (72) and periventricular leukomalacia in premature infants assessed at 2 years adjusted age (73). In clinical studies, antenatal GCs have been linked to neuropsychiatric changes in children, including attention deficits (74), decreased scores in cognitive tests (75), distractibility, and aggressive behavior (76). Thinning of the left rostral anterior cingulate cortex, as reported above, is associated with increased incidence of affective disorders (63). Although the mechanism(s) responsible for these alterations are not fully established, they may involve changes in neural stem cell proliferation and/or differentiation, thereby disrupting the development of neuronal circuits essential for higher order cognitive or behavioral function. GC effects in isolated neurons and glia are widespread, triggering a decrease in glucose uptake, inhibition of proliferation, decreased neuronal excitability, and increased dendritic atrophy (64). In a rodent model of antenatal GC administration, a single course of Beta produced significant anatomical differences in interneurons of the hippocampus (77). Antenatal GCs leads to potentially long-lasting alterations of the HPA axis in humans as well (78). For example, antenatal GCs are associated with increased stress reactivity (greater elevation in cortisol level when faced with stress) in female children, even when these children were born at term and maternal stress during pregnancy was controlled for (79).
Maternal stress and programming of the fetal brain
In addition to synthetic GCs, maternal stress has been shown to detrimentally affect fetal programming and brain development through elevated levels of endogenous GCs. In several animal studies, prenatal stress has been associated with reduced volume in several brain regions, including the amygdala, cerebral cortex, hippocampus, and the corpus callosum (80). Human studies have also shown that the timing of maternal stress has differential effects on fetal development. Maternal stress during late gestation leads to a decreased stress response in childhood and adolescence as measured by cortisol levels. By contrast, maternal stress throughout the second half of gestation increased stress responses. Girls born to mothers who experienced anxiety during the first trimester were found to have an increased susceptibility to affective disorders, whereas children whose mothers experienced stress during late gestation were more prone to developing attention deficit hyperactivity disorder (71).
A possible mechanism by which maternal stress alters fetal development is by changing gene expression patterns in the placenta. Male placentas of mice exposed to prenatal stress early in gestation (early prenatal stress) were found to have significantly increased expression of genes regulating growth and development. These changes were associated with maladaptive stress behaviors in adult early prenatal stress male mice (81). In humans, endogenous GCs have also been found to affect placental function by stimulating the placenta to synthesize and release CRH, which could subsequently stimulate the fetal HPA axis (82). Importantly, maternal stress affects pathways distinct from synthetic GCs. For instance, endogenous GCs bind to both GR and mineralocorticoid receptor with equal affinity, whereas synthetic GCs bind only to GR. Synthetic GCs can also bind other orphan nuclear receptors in the brain. Furthermore, placental 11β-hydroxysteroid dehydrogenase (HSD) preferentially inactivates endogenous GCs to cortisone during early gestation; however, levels of 11β-HSD decrease during late gestation, a period in which maternal stress may potentiate the actions of synthetic GCs (82). Finally, endogenous GCs are secreted in a pulse-like fashion, resulting in transient, acute changes in transcription unlike synthetic GCs, which continue to affect transcription after they have been withdrawn. This biological rhythmic secretion of GCs may serve a physiological role by programming cells to respond rapidly to stress, a response that may be impaired due to prolonged exposure to synthetic GCs (83).
Studying the impact of GCs on neural development using primary stem cell cultures
To provide mechanistic details of the neurodevelopmental consequences of antenatal GC exposure, our group (84, 85) and others have studied the effects of GCs on primary murine fetal neural stem/progenitor cell (NSPC) cultures. GCs exert an antiproliferative effect on NSPCs via multiple mechanisms. In rat NSPCs cultured as neurospheres, Dex treatment triggers cyclin D1 degradation via the ubiquitin-proteasome system, thereby inhibiting cell-cycle progression and proliferation (86). In a follow-up study, Dex mediated a decrease in NSPC proliferation through an independent mechanism, the down-regulation of BRUCE/Apollon, a member of the inhibitors of apoptosis protein family found in neuroblasts (87). Dex regulates BIR repeat-containing ubiquitin-conjugating enzyme (BRUCE) at the mRNA and protein level, partially by up-regulating the deubiquitinating enzyme Usp8/Ubpy, which then stabilizes the ubiquitin ligase Nrdp1 and enables it to target BRUCE for degradation (87). Similar studies using gene expression profiling in NSPCs derived from whole brains, found that Dex up-regulated ferritin heavy chain 1 and IGF binding protein 3, which exerted inhibitory effects on cyclin D1 and nestin, a marker of NSPCs (88). These studies also demonstrated that Dex down-regulated the endothelin receptor type B, a change that has been associated with apoptosis in the dentate gyrus and cerebellum and decreased proliferation in the cerebellum (88).
The antiproliferative effects of GCs on NSPCs have also been demonstrated in vivo. Embryonic rats treated with Dex at embryonic day (E) 14 or E15.5 for 3 days exhibited decreased levels of proliferating NSPCs in the striatum and hippocampus (86) as well as dentate gyrus (70). Administering Dex to neonatal rats from postnatal days 1–7 led to apoptosis and depletion of the NSPC pool in the subgranular zone of the dentate gyrus (89, 90). Endogenous GCs may in fact underlie the reduction in the NSPC pool with increasing age (89). Although not within the scope of this review, GCs have also been found to have similar antiproliferative effect on adult NSPCs in vitro and in vivo (91–93).
Early prenatal exposure to GCs is associated with behavioral changes later in life. Evidence that early exposure to GCs leads to long-lasting changes in the molecular profile of NSPCs comes from several sources. In NSPCs isolated from E15 rat cerebral cortices, the antiproliferative effects of Dex are associated with acute and chronic up-regulation of the cell cycle inhibitors p16 and p21. These alterations are accompanied by changes in target genes involved in senescence, such as Bmi1 and Hmga1 (94). Because senescence is related to mitochondrial dysfunction and susceptibility to oxidative stress, Dex down-regulated the mitochondrial proteins nicotinamide adenine dinucleotide hydroxide dehydrogenase 3 and cytochrome b and increased the production of reactive oxygen species and apoptosis when challenged with an oxidative stress inducer (94). The Dex-induced changes in mitochondrial and senescence genes and Dex-induced changes in DNA methylation after several passages suggest an epigenetic reprogramming of NSPCs (94).
Evidence for cell type-specific effects of Dex has come from several studies. Whereas prior studies used 10−6 M Dex in neural stem cells (94), Yu et al (89) found that 10−5 M Dex induced apoptosis in a rat hippocampal culture affecting mitotic and resting cell populations and both neurons and NSPCs but not astrocytes. In human NSPC cultures derived from gestation weeks 16–19, Dex similarly decreased proliferation but also decreased the percentage of neurons in differentiating cultures while increasing the proportion of glia (95). These Dex effects were mediated by GR binding to the promoter of Dickkopf1, leading to up-regulation of its expression and subsequent repression of canonical Wnt signaling (95). Several lines of evidence also support a role for GC-induced changes in oligodendrocytes (96, 97). In utero exposure to GC is associated with hypomyelination, although these effects are not due to direct changes in oligodendrocyte progenitor cell proliferation, maturation, or survival and are thought to be secondary to deficits in the primary cellular targets, microglia, and astrocytes (98). Dose- and drug-specific responses determine cellular outcome with high doses of Dex, reducing myelination by altering genomic responses (such as Olig 1 in microglia), whereas astrocyte production relies on the GR nongenomic or rapid pathway (99).
Although many studies demonstrate antiproliferative and proapoptotic effects of GCs, one study reported a dose-dependent proproliferative effect of GCs, including Dex, Beta, and hydrocortisone, on human induced pluripotent stem cell-derived NSPCs, leading to an increase in the number of microtubule-associated protein 2-positive neurons (100). In particular, hydrocortisone stimulated NSPC and neuronal proliferation, even under conditions of oxidative stress, which was hypothesized to be due to the effects of mineralocorticoid receptor activation by hydrocortisone or selective 11β-HSD2 inactivation of hydrocortisone (100).
Similar to Li et al, our studies use primary NSPCs isolated from E14.5 mouse cerebral cortex and cultured as three-dimensional neurospheres (84). To maintain their undifferentiated state, neurospheres are cultured on ultralow adherence plates in the presence of epidermal growth factor (EGF) and/or fibroblast growth factor-1 (FGF-1) (Figure 1). EGF and FGF are crucial for promoting renewal and expansion of the NSPCs. NSPCs derived from E14.5 brain are advantageous for analysis of antenatal GC effects because GR is expressed in NSPC domains in the developing cerebral cortex in vivo at this age (101). Notably, this is a time when embryonic exposure to endogenous GCs is minimal. The major advantage of this model lies in the ability of neurospheres to properly recapitulate the in vivo differentiation order of neurons and glia under in vitro conditions (102, 103) as well as in the maintenance of cell-cell coupling even in the in vitro setting (85), allowing the study of gap junction intracellular communication. Finally, alterations in the development of the cerebral cortex, such as those caused by a fetal exposure to GCs can contribute to neurodevelopmental diseases such as schizophrenia, anxiety, depression, autism spectrum disorders, and attention deficit hyperactivity disorder (104).
Figure 1.
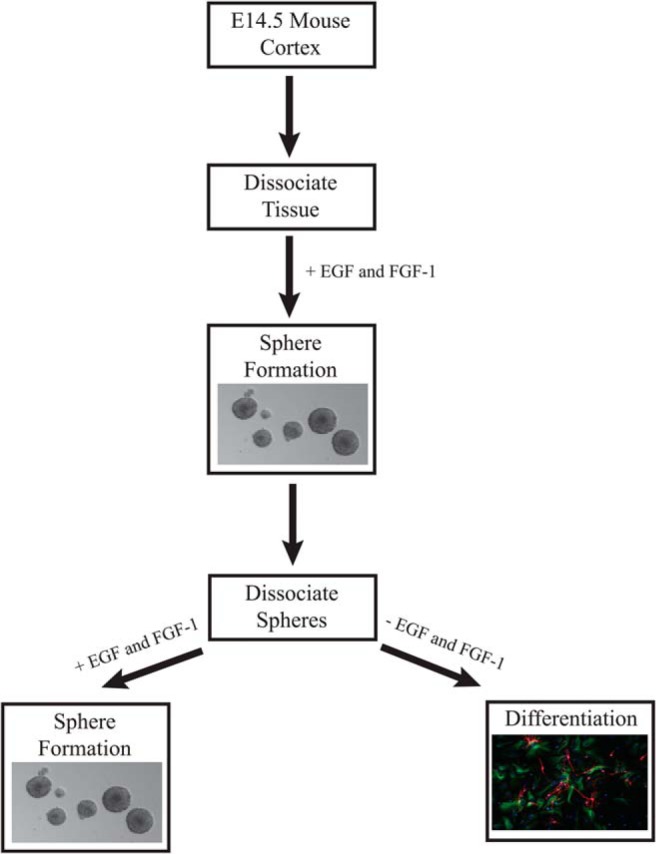
Outline of NSPC culture system. Cerebral cortices from E14.5 mice are dissociated into single cells and cultured in the presence of EGF and FGF-1 to form neurospheres (85). After the dissociation of neurospheres, continued growth in EGF and FGF-1 will produce secondary neurospheres, whereas plating in the absence of EGF and FGF-1 can lead to differentiation.
Using NSPCs isolated from the cerebral cortex, recent work from our laboratory has identified a novel rapid signaling pathway that impacts synchronized calcium waves in coupled cells and proliferation (85). Specifically, plasma membrane-associated GR regulates gap junction intercellular communication in coupled NSPCs through a rapid c-Src- and MAPK-dependent phosphorylation of the gap junction protein connexin 43 (Cx43) (85). Furthermore, the mobilization of this rapid GR signaling pathway occurs in lipid rafts and requires caveolin-1 (Cav-1) (85). The impact of Cx43-dependent propagation of synchronized calcium waves on NSPC proliferation observed in our primary cultures (85) has also been observed in the developing cerebral cortex in vivo and may therefore be a general feature of coordinated NSPC proliferation responsible for the generation of appropriately connected neural circuits (105).
As observed with other steroid receptors (106), the rapid signaling pathway we identified in NPSCs is linked to classical genomic GR signaling leading to gene-specific effects on GR targets (84). Specifically, analysis of Dex treated (ie, 4 h) NSPCs derived from mice null for Cav-1 by microarray revealed approximately 100 genes with altered GR transcriptional regulation (84). The mechanistic basis for Cav-1 effects on GR transcription targets is due in part to its impact on site-specific GR phosphorylation. For example, in the absence of Cav-1, GC-induced GR phosphorylation at serine 224 (mouse equivalent of human GR serine 211) is dramatically reduced, whereas phosphorylation at serine 234 (mouse equivalent of human GR serine 226) is unaffected (84). Furthermore, some GR target genes with reduced transcriptional responses to GC in Cav-1 null NSPCs (ie, Fkbp-5 and Sgk-1) exhibit diminished promoter recruitment of GR phosphorylated at serine 224 as revealed by chromatin immunoprecipitation assays (84). Analogous to the unique cistromes of individual GR phosphoisoforms (107), selective effects of the Cav-1 on GR phosphorylation could impact GR target gene selection in NSPCs and thereby influence various responses of these cells to GCs (Figure 2). For example, the lack of an antiproliferative response to GCs in Cav-1 null NSPCs could be due to the loss of hormone induction of Sgk-1, a gene previously established to mediate antiproliferative responses of GCs in cultured human hippocampal progenitor cells (108).
Figure 2.
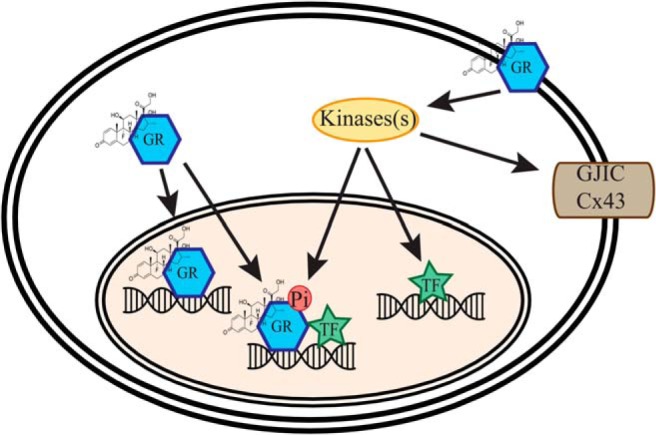
Model of integration of genomic and nongenomic GR signaling in NSPCs. Ligand binding (Dex shown in figure) to plasma membrane-associated GR can trigger nongenomic activation of specific protein kinases that can either directly regulate gap junction intercellular communication (GJIC) through phosphorylation of Cx43 or genomic actions through direct phosphorylation of GR or other transcription factors.
Future perspectives: reaching an improved therapy: how can the study of NSPCs inform antenatal synthetic GC treatment?
As stated above, both Dex and Beta are administered antenatally to reduce the complications of prematurity. Although upon initial glance the two GCs appear functionally equivalent, the action of these two hormones varies significantly and current studies reveal no consistently better GC for antenatal use (37). Strikingly, although Beta and Dex have similar affinities for the GR and similar genomic potencies, Dex has a 5-fold greater potency for the rapid action of GR than Beta in rat thymocytes (36). Different GR signaling modalities (ie, genomic vs rapid) may exert discrete influences on the developing brain and peripheral organ systems. Thus, the analysis of GR signaling pathways and functional responses of NSPC to novel therapeutic alternatives to synthetic GC administration could provide insights into treatment modalities that maintain beneficial effects in vulnerable organ systems in the premature infant (eg, lung) but limit their adverse neurodevelopmental effects. In addition, to improve antenatal GC therapies, we need a better understanding of the genetic or uterine environmental factors that could modulate GC responses. In term-born children treated during the fetal period with GCs, there is no difference in cortisol reactivity if the child was treated with Dex or Beta (79). However, despite reporting sexually dimorphic changes in cortisol secretion, this study did not separate the Dex/Beta-treated groups by sex prior to analysis. Gender-specific responses of NSPCs to GCs may exist because there are significant gender differences in outcome and infant mortality in response to antenatal GCs in both humans and animal models (79, 109, 110) (reviewed in reference 111). For example, LBW males have a higher risk of IVH and greater mortality than LBW girls (109). Fetal gender is not taken into account in current treatment guidelines, so there is a clear need to develop antenatal GC regimens that would be selectively robust in preterm male vs preterm female infants.
In summary, as additional molecular details of GR action in NSPCs are uncovered, antenatal synthetic GC regimens established more than 40 years ago may be updated to continue to provide life-saving benefits of GCs to premature infants while limiting adverse neurological outcomes that may take many years to be revealed.
Acknowledgments
This work was supported by National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases Grant U24 DK097746.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Beta
- betamethasone
- CAH
- congenital adrenal hyperplasia
- Cav-1
- caveolin-1
- Cx43
- connexin 43
- Dex
- dexamethasone
- E
- embryonic day
- EGF
- epidermal growth factor
- FGF-1
- fibroblast growth factor-1
- GC
- glucocorticoid
- GR
- glucocorticoid receptor
- HPA
- hypothalamus-pituitary-adrenal
- HSD
- hydroxysteroid dehydrogenase
- IVH
- intraventricular hemorrhage
- LBW
- low birth weight
- NSPC
- neural stem/progenitor cell
- RDS
- respiratory distress syndrome.
References
- 1. Fowden AL, Li J, Forhead AJ. Glucocorticoids and the preparation for life after birth: are there long-term consequences of the life insurance? Proc Nutr Soc. 1998;57:113–122. [DOI] [PubMed] [Google Scholar]
- 2. Crone RK, Davies P, Liggins GC, Reid L. The effects of hypophysectomy, thyroidectomy, and postoperative infusion of cortisol or adrenocorticotrophin on the structure of the ovine fetal lung. J Dev Physiol. 1983;5:281–288. [PubMed] [Google Scholar]
- 3. Schellenberg JC, Liggins GC. Elastin and collagen in the fetal sheep lung. I. Ontogenesis. Pediatr Res. 1987;22:335–338. [DOI] [PubMed] [Google Scholar]
- 4. Warburton D, Parton L, Buckley S, Cosico L, Enns G, Saluna T. Combined effects of corticosteroid, thyroid hormones, and β-agonist on surfactant, pulmonary mechanics, and β-receptor binding in fetal lamb lung. Pediatr Res. 1988;24:166–170. [DOI] [PubMed] [Google Scholar]
- 5. Kitterman JA, Liggins GC, Campos GA, et al. Prepartum maturation of the lung in fetal sheep: relation to cortisol. J Appl Physiol Respir Environ Exerc Physiol. 1981;51:384–390. [DOI] [PubMed] [Google Scholar]
- 6. Ingbar DH, Duvick S, Savick SK, et al. Developmental changes of fetal rat lung Na-K-ATPase after maternal treatment with dexamethasone. Am J Physiol. 1997;272:L665–L672. [DOI] [PubMed] [Google Scholar]
- 7. Wallace MJ, Hooper SB, Harding R. Effects of elevated fetal cortisol concentrations on the volume, secretion, and reabsorption of lung liquid. Am J Physiol. 1995;269:R881–R887. [DOI] [PubMed] [Google Scholar]
- 8. Liggins GC. The role of cortisol in preparing the fetus for birth. Reprod Fertil Dev. 1994;6:141–150. [DOI] [PubMed] [Google Scholar]
- 9. Jones CT, Rolph TP. Metabolism during fetal life: a functional assessment of metabolic development. Physiol Rev. 1985;65:357–430. [DOI] [PubMed] [Google Scholar]
- 10. Fowden AL, Mijovic J, Silver M. The effects of cortisol on hepatic and renal gluconeogenic enzyme activities in the sheep fetus during late gestation. J Endocrinol. 1993;137:213–222. [DOI] [PubMed] [Google Scholar]
- 11. Wu SY, Klein AH, Chopra IJ, Fisher DA. Alterations in tissue thyroxine-5′-monodeiodinating activity in perinatal period. Endocrinology. 1978;103:235–239. [DOI] [PubMed] [Google Scholar]
- 12. Jeffray TM, Berdusco ET, Wallace M, Fowden A, Challis JR. Effects of incremental cortisol and adrenalectomy on plasma corticosteroid binding capacity in fetal sheep. Can J Physiol Pharmacol. 1995;73:1568–1573. [DOI] [PubMed] [Google Scholar]
- 13. Trahair JF, Perry RA, Silver M, Robinson PM. Studies on the maturation of the small intestine in the fetal sheep. II. The effects of exogenous cortisol. Q J Exp Physiol. 1987;72:71–79. [DOI] [PubMed] [Google Scholar]
- 14. Sangild PT, Hilsted L, Nexo E, Fowden AL, Silver M. Secretion of acid, gastrin, and cobalamin-binding proteins by the fetal pig stomach: developmental regulation by cortisol. Exp Physiol. 1994;79:135–146. [DOI] [PubMed] [Google Scholar]
- 15. Sangild PT, Sjostrom H, Noren O, Fowden AL, Silver M. The prenatal development and glucocorticoid control of brush-border hydrolases in the pig small intestine. Pediatr Res. 1995;37:207–212. [DOI] [PubMed] [Google Scholar]
- 16. Sangild PT, Westrom BR, Fowden AL, Silver M. Developmental regulation of the porcine exocrine pancreas by glucocorticoids. J Pediatr Gastroenterol Nutr. 1994;19:204–212. [DOI] [PubMed] [Google Scholar]
- 17. Sangild PT, Westrom BR, Silver M, Fowden AL. Maturational effects of cortisol on the exocrine abomasum and pancreas in fetal sheep. Reprod Fertil Dev. 1995;7:655–658. [DOI] [PubMed] [Google Scholar]
- 18. Sangild T, Silver M, Fowden AL, Turvey A, Foltmann B. Adrenocortical stimulation of stomach development in the prenatal pig. Biol Neonate. 1994;65:378–389. [DOI] [PubMed] [Google Scholar]
- 19. Stonestreet BS, Hansen NB, Laptook AR, Oh W. Glucocorticoid accelerates renal functional maturation in fetal lambs. Early Hum Dev. 1983;8:331–341. [DOI] [PubMed] [Google Scholar]
- 20. Slotkin TA, Seidler FJ, Kavlock RJ, Gray JA. Fetal dexamethasone exposure accelerates development of renal function: relationship to dose, cell differentiation and growth inhibition. J Dev Physiol. 1992;17:55–61. [PubMed] [Google Scholar]
- 21. Towstoless MK, McDougall JG, Wintour EM. Gestational changes in renal responsiveness to cortisol in the ovine fetus. Pediatr Res. 1989;26:6–10. [DOI] [PubMed] [Google Scholar]
- 22. Celsi G, Wang ZM, Akusjarvi G, Aperia A. Sensitive periods for glucocorticoids' regulation of Na+,K(+)-ATPase mRNA in the developing lung and kidney. Pediatr Res. 1993;33:5–9. [DOI] [PubMed] [Google Scholar]
- 23. Guillery EN, Karniski LP, Mathews MS, et al. Role of glucocorticoids in the maturation of renal cortical Na+/H+ exchanger activity during fetal life in sheep. Am J Physiol. 1995;268:F710–F717. [DOI] [PubMed] [Google Scholar]
- 24. Lim GB, Dodic M, Earnest L, Jeyaseelan K, Wintour EM. Regulation of erythropoietin gene expression in fetal sheep by glucocorticoids. Endocrinology. 1996;137:1658–1663. [DOI] [PubMed] [Google Scholar]
- 25. Wood CE, Cheung CY, Brace RA. Fetal heart rate, arterial pressure, and blood volume responses to cortisol infusion. Am J Physiol. 1987;253:R904–R909. [DOI] [PubMed] [Google Scholar]
- 26. Wood CE, Keil LC, Rudolph AM. Physiological inhibition of ovine fetal plasma renin activity by cortisol. Endocrinology. 1984;115:1792–1796. [DOI] [PubMed] [Google Scholar]
- 27. Carbone GM, Sheikh AU, Zehnder T, Rose JC. Effect of chronic infusion of cortisol on renin gene expression and renin response to hemorrhage in fetal lambs. Pediatr Res. 1995;37:316–320. [DOI] [PubMed] [Google Scholar]
- 28. Segar JL, Bedell K, Page WV, Mazursky JE, Nuyt AM, Robillard JE. Effect of cortisol on gene expression of the renin-angiotensin system in fetal sheep. Pediatr Res. 1995;37:741–746. [DOI] [PubMed] [Google Scholar]
- 29. Murphy BE. Human fetal serum cortisol levels related to gestational age: evidence of a midgestational fall and a steep late gestational rise, independent of sex or mode of delivery. Am J Obstet Gynecol. 1982;144:276–282. [DOI] [PubMed] [Google Scholar]
- 30. Liggins GC. Premature delivery of foetal lambs infused with glucocorticoids. J Endocrinol. 1969;45:515–523. [DOI] [PubMed] [Google Scholar]
- 31. Liggins GC, Howie RN. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics. 1972;50:515–525. [PubMed] [Google Scholar]
- 32. Crowley PA. Antenatal corticosteroid therapy: a meta-analysis of the randomized trials, 1972 to 1994. Am J Obstet Gynecol. 1995;173:322–335. [DOI] [PubMed] [Google Scholar]
- 33. Roberts D, Dalziel S. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev. 2006:CD004454. [DOI] [PubMed] [Google Scholar]
- 34. Blanford AT, Murphy BE. In vitro metabolism of prednisolone, dexamethasone, betamethasone, and cortisol by the human placenta. Am J Obstet Gynecol. 1977;127:264–267. [DOI] [PubMed] [Google Scholar]
- 35. Levitz M, Jansen V, Dancis J. The transfer and metabolism of corticosteroids in the perfused human placenta. Am J Obstet Gynecol. 1978;132:363–366. [DOI] [PubMed] [Google Scholar]
- 36. Buttgereit F, Brand MD, Burmester GR. Equivalent doses and relative drug potencies for non-genomic glucocorticoid effects: a novel glucocorticoid hierarchy. Biochem Pharmacol. 1999;58:363–368. [DOI] [PubMed] [Google Scholar]
- 37. Wapner R, Jobe AH. Controversy: antenatal steroids. Clin Perinatol. 2011;38:529–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Merrill JD, Ballard RA. Clinical use of antenatal corticosteroids: benefits and risks. Pediatr Rev. 2000;1:E91–E98. [DOI] [PubMed] [Google Scholar]
- 39. Ballard PL, Granberg P, Ballard RA. Glucocorticoid levels in maternal and cord serum after prenatal betamethasone therapy to prevent respiratory distress syndrome. J Clin Invest. 1975;56:1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ervin MG, Padbury JF, Polk DH, Ikegami M, Berry LM, Jobe AH. Antenatal glucocorticoids alter premature newborn lamb neuroendocrine and endocrine responses to hypoxia. Am J Physiol Regul Integr Comp Physiol. 2000;279:R830–R838. [DOI] [PubMed] [Google Scholar]
- 41. Smith LM, Ervin MG, Wada N, Ikegami M, Polk DH, Jobe AH. Antenatal glucocorticoids alter postnatal preterm lamb renal and cardiovascular responses to intravascular volume expansion. Pediatr Res. 2000;47:622–627. [DOI] [PubMed] [Google Scholar]
- 42. Wapner RJ, Sorokin Y, Mele L, et al. Long-term outcomes after repeat doses of antenatal corticosteroids. N Engl J Med. 2007;357:1190–1198. [DOI] [PubMed] [Google Scholar]
- 43. Murphy KE, Hannah ME, Willan AR, et al. Multiple courses of antenatal corticosteroids for preterm birth (MACS): a randomised controlled trial. Lancet. 2008;372:2143–2151. [DOI] [PubMed] [Google Scholar]
- 44. Crowther CA, McKinlay CJ, Middleton P, Harding JE. Repeat doses of prenatal corticosteroids for women at risk of preterm birth for improving neonatal health outcomes. Cochrane Database Syst Rev. 2011;CD003935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hashimoto LN, Hornung RW, Lindsell CJ, Brewer DE, Donovan EF. Effects of antenatal glucocorticoids on outcomes of very low birth weight multifetal gestations. Am J Obstet Gynecol. 2002;187:804–810. [DOI] [PubMed] [Google Scholar]
- 46. Gyamfi C, Mele L, Wapner RJ, et al. The effect of plurality and obesity on betamethasone concentrations in women at risk for preterm delivery. Am J Obstet Gynecol. 2010;203:219.e1–219.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Della Torre M, Hibbard JU, Jeong H, Fischer JH. Betamethasone in pregnancy: influence of maternal body weight and multiple gestation on pharmacokinetics. Am J Obstet Gynecol. 2010;203:254.e1–254.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shankaran S, Lin A, Maller-Kesselman J, et al. Maternal race, demography, and health care disparities impact risk for intraventricular hemorrhage in preterm neonates. J Pediatr. 2014;164:1005–1011.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cuestas E, Bas J, Pautasso J. Sex differences in intraventricular hemorrhage rates among very low birth weight newborns. Gend Med. 2009;6:376–382. [DOI] [PubMed] [Google Scholar]
- 50. Brownfoot FC, Crowther CA, Middleton P. Different corticosteroids and regimens for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev. 2008;CD006764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Feldman DM, Carbone J, Belden L, Borgida AF, Herson V. Betamethasone vs dexamethasone for the prevention of morbidity in very-low-birthweight neonates. Am J Obstet Gynecol. 2007;197:284.e1–284.e4. [DOI] [PubMed] [Google Scholar]
- 52. Jobe AH, Soll RF. Choice and dose of corticosteroid for antenatal treatments. Am J Obstet Gynecol. 2004;190:878–881. [DOI] [PubMed] [Google Scholar]
- 53. Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet. 2005;365:2125–2136. [DOI] [PubMed] [Google Scholar]
- 54. Hirvikoski T, Nordenstrom A, Wedell A, Ritzen M, Lajic S. Prenatal dexamethasone treatment of children at risk for congenital adrenal hyperplasia: the Swedish experience and standpoint. J Clin Endocrinol Metab. 2012;97:1881–1883. [DOI] [PubMed] [Google Scholar]
- 55. Miller WL. Dexamethasone treatment of congenital adrenal hyperplasia in utero: an experimental therapy of unproven safety. J Urol. 1999;162:537–240. [PubMed] [Google Scholar]
- 56. Seckl JR. Prenatal glucocorticoids and long-term programming. Eur J Endocrinol. 2004;151(suppl 3):U49–U62. [DOI] [PubMed] [Google Scholar]
- 57. de Vries A, Holmes MC, Heijnis A, et al. Prenatal dexamethasone exposure induces changes in nonhuman primate offspring cardiometabolic and hypothalamic-pituitary-adrenal axis function. J Clin Invest. 2007;117:1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hirvikoski T, Lindholm T, Lajic S, Nordenstrom A. Gender role behaviour in prenatally dexamethasone-treated children at risk for congenital adrenal hyperplasia—a pilot study. Acta Paediatr. 2011;100:e112–e119. [DOI] [PubMed] [Google Scholar]
- 59. Hirvikoski T, Nordenstrom A, Lindholm T, Lindblad F, Ritzen EM, Lajic S. Long-term follow-up of prenatally treated children at risk for congenital adrenal hyperplasia: does dexamethasone cause behavioural problems? Eur J Endocrinol. 2008;159:309–316. [DOI] [PubMed] [Google Scholar]
- 60. Hirvikoski T, Nordenstrom A, Lindholm T, et al. Cognitive functions in children at risk for congenital adrenal hyperplasia treated prenatally with dexamethasone. J Clin Endocrinol Metab. 2007;92:542–548. [DOI] [PubMed] [Google Scholar]
- 61. Lajic S, Wedell A, Bui TH, Ritzen EM, Holst M. Long-term somatic follow-up of prenatally treated children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1998;83:3872–3880. [DOI] [PubMed] [Google Scholar]
- 62. Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:4133–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Davis EP, Sandman CA, Buss C, Wing DA, Head K. Fetal glucocorticoid exposure is associated with preadolescent brain development. Biol Psychiatry. 2013;74:647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Damsted SK, Born AP, Paulson OB, Uldall P. Exogenous glucocorticoids and adverse cerebral effects in children. Eur J Paediatr Neurol. 2011;15:465–477. [DOI] [PubMed] [Google Scholar]
- 65. Coe CL, Lubach GR. Developmental consequences of antenatal dexamethasone treatment in nonhuman primates. Neurosci Biobehav Rev. 2005;29:227–235. [DOI] [PubMed] [Google Scholar]
- 66. Uno H, Lohmiller L, Thieme C, et al. Brain damage induced by prenatal exposure to dexamethasone in fetal rhesus macaques. I. Hippocampus. Brain Res Dev Brain Res. 1990;53:157–167. [DOI] [PubMed] [Google Scholar]
- 67. Hauser J, Dettling-Artho A, Pilloud S, et al. Effects of prenatal dexamethasone treatment on postnatal physical, endocrine, and social development in the common marmoset monkey. Endocrinology. 2007;148:1813–1822. [DOI] [PubMed] [Google Scholar]
- 68. Rodriguez JS, Zurcher NR, Keenan KE, Bartlett TQ, Nathanielsz PW, Nijland MJ. Prenatal betamethasone exposure has sex specific effects in reversal learning and attention in juvenile baboons. Am J Obstet Gynecol. 2011;204:545.e1–545.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hauser J, Feldon J, Pryce CR. Direct and dam-mediated effects of prenatal dexamethasone on emotionality, cognition and HPA axis in adult Wistar rats. Horm Behav. 2009;56:364–375. [DOI] [PubMed] [Google Scholar]
- 70. Noorlander CW, Visser GH, Ramakers GM, Nikkels PG, de Graan PN. Prenatal corticosteroid exposure affects hippocampal plasticity and reduces lifespan. Dev Neurobiol. 2008;68:237–246. [DOI] [PubMed] [Google Scholar]
- 71. Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming, part 1: outcomes. Nat Rev Endocrinol. 2014;10:391–402. [DOI] [PubMed] [Google Scholar]
- 72. Modi N, Lewis H, Al-Naqeeb N, Ajayi-Obe M, Dore CJ, Rutherford M. The effects of repeated antenatal glucocorticoid therapy on the developing brain. Pediatr Res. 2001;50:581–585. [DOI] [PubMed] [Google Scholar]
- 73. Spinillo A, Viazzo F, Colleoni R, Chiara A, Maria Cerbo R, Fazzi E. Two-year infant neurodevelopmental outcome after single or multiple antenatal courses of corticosteroids to prevent complications of prematurity. Am J Obstet Gynecol. 2004;191:217–224. [DOI] [PubMed] [Google Scholar]
- 74. Crowther CA, Doyle LW, Haslam RR, et al. Outcomes at 2 years of age after repeat doses of antenatal corticosteroids. N Engl J Med. 2007;357:1179–1189. [DOI] [PubMed] [Google Scholar]
- 75. MacArthur BA, Howie RN, Dezoete JA, Elkins J. School progress and cognitive development of 6-year-old children whose mothers were treated antenatally with betamethasone. Pediatrics. 1982;70:99–105. [PubMed] [Google Scholar]
- 76. French NP, Hagan R, Evans SF, Mullan A, Newnham JP. Repeated antenatal corticosteroids: effects on cerebral palsy and childhood behavior. Am J Obstet Gynecol. 2004;190:588–595. [DOI] [PubMed] [Google Scholar]
- 77. Bustamante C, Valencia M, Torres C, et al. Effects of a single course of prenatal betamethasone on dendritic development in dentate gyrus granular neurons and on spatial memory in rat offspring. Neuropediatrics. 2014;45:354–361. [DOI] [PubMed] [Google Scholar]
- 78. Waffarn, Davis EP. Effects of antenatal corticosteroids on the hypothalamic-pituitary-adrenocortical axis of the fetus and newborn: experimental findings and clinical considerations. Am J Obstet Gynecol. 2012;207:446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Alexander N, Rosenlocher F, Stalder T, et al. Impact of antenatal synthetic glucocorticoid exposure on endocrine stress reactivity in term-born children. J Clin Endocrinol Metab. 2012;97:3538–3544. [DOI] [PubMed] [Google Scholar]
- 80. Charil A, Laplante DP, Vaillancourt C, King S. Prenatal stress and brain development. Brain Res Rev. 2010;65:56–79. [DOI] [PubMed] [Google Scholar]
- 81. Mueller BR, Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci. 2008;28:9055–9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 2: Mechanisms. Nat Rev Endocrinol. 2014;10:403–411. [DOI] [PubMed] [Google Scholar]
- 83. Desvergne B, Heligon C. Steroid hormone pulsing drives cyclic gene expression. Nat Cell Biol. 2009;11:1051–1053. [DOI] [PubMed] [Google Scholar]
- 84. Peffer ME, Chandran UR, Luthra S, et al. Caveolin-1 regulates genomic action of the glucocorticoid receptor in neural stem cells. Mol Cell Biol. 2014;34(14):2611–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Samarasinghe RA, Di Maio R, Volonte D, et al. Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc Natl Acad Sci USA. 2011;108:16657–16662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sundberg M, Savola S, Hienola A, Korhonen L, Lindholm D. Glucocorticoid hormones decrease proliferation of embryonic neural stem cells through ubiquitin-mediated degradation of cyclin D1. J Neurosci. 2006;26:5402–5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sippel M, Rajala R, Korhonen L, et al. Dexamethasone regulates expression of BRUCE/Apollon and the proliferation of neural progenitor cells. FEBS Lett. 2009;583:2213–2217. [DOI] [PubMed] [Google Scholar]
- 88. Li SY, Wang P, Tang Y, Huang L, Wu YF, Shen HY. Analysis of methylprednisolone-induced inhibition on the proliferation of neural progenitor cells in vitro by gene expression profiling. Neurosci Lett. 2012;526:154–159. [DOI] [PubMed] [Google Scholar]
- 89. Yu S, Patchev AV, Wu Y, Lu J, et al. Depletion of the neural precursor cell pool by glucocorticoids. Ann Neurol. 2010;67:21–30. [DOI] [PubMed] [Google Scholar]
- 90. Ichinohashi Y, Sato Y, Saito A, et al. Dexamethasone administration to the neonatal rat results in neurological dysfunction at the juvenile stage even at low doses. Early Hum Dev. 2013;89:283–288. [DOI] [PubMed] [Google Scholar]
- 91. Alonso G. Prolonged corticosterone treatment of adult rats inhibits the proliferation of oligodendrocyte progenitors present throughout white and gray matter regions of the brain. Glia. 2000;31:219–231. [DOI] [PubMed] [Google Scholar]
- 92. Kim JB, Ju JY, Kim JH, et al. Dexamethasone inhibits proliferation of adult hippocampal neurogenesis in vivo and in vitro. Brain Res 2004;1027:1–10. [DOI] [PubMed] [Google Scholar]
- 93. Schroter A, Lustenberger RM, Obermair FJ, Thallmair M. High-dose corticosteroids after spinal cord injury reduce neural progenitor cell proliferation. Neuroscience. 2009;161:753–763. [DOI] [PubMed] [Google Scholar]
- 94. Bose R, Moors M, Tofighi R, Cascante A, Hermanson O, Ceccatelli S. Glucocorticoids induce long-lasting effects in neural stem cells resulting in senescence-related alterations. Cell Death Dis. 2010;1:e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Moors M, Bose R, Johansson-Haque K, Edoff K, Okret S, Ceccatelli S. Dickkopf 1 mediates glucocorticoid-induced changes in human neural progenitor cell proliferation and differentiation. Toxicol Sci. 2012;125:488–495. [DOI] [PubMed] [Google Scholar]
- 96. Huang WL, Harper CG, Evans SF, Newnham JP, Dunlop SA. Repeated prenatal corticosteroid administration delays myelination of the corpus callosum in fetal sheep. Int J Dev Neurosci. 2001;19:415–425. [DOI] [PubMed] [Google Scholar]
- 97. Kim JW, Kim YJ, Chang YP. Administration of dexamethasone to neonatal rats induces hypomyelination and changes in the morphology of oligodendrocyte precursors. Comp Med. 2013;63:48–54. [PMC free article] [PubMed] [Google Scholar]
- 98. Jenkins SI, Pickard MR, Khong M, et al. Identifying the cellular targets of drug action in the central nervous system following corticosteroid therapy. ACS Chem Neurosci. 2014;5:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zia MT, Vinukonda G, Vose LR, et al. Postnatal glucocorticoid-induced hypomyelination, gliosis, and neurologic deficits are dose-dependent, preparation-specific, and reversible. Exp Neurol. 2015;263:200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ninomiya E, Hattori T, Toyoda M, Umezawa A, Hamazaki T, Shintaku H. Glucocorticoids promote neural progenitor cell proliferation derived from human induced pluripotent stem cells. Springerplus. 2014;3:527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tsiarli MA, Paula Monaghan A, Defranco DB. Differential subcellular localization of the glucocorticoid receptor in distinct neural stem and progenitor populations of the mouse telencephalon in vivo. Brain Res. 2013;1523:10–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Qian X, Shen Q, Goderie SK, et al. Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron. 2000;28:69–80. [DOI] [PubMed] [Google Scholar]
- 103. Shen Q, Wang Y, Dimos JT, Fasano CA, et al. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat Neurosci. 2006;9:743–751. [DOI] [PubMed] [Google Scholar]
- 104. Schmitt A, Malchow B, Hasan A, Falkai P. The impact of environmental factors in severe psychiatric disorders. Front Neurosci. 2014;8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Malmersjo S, Rebellato P, Smedler E, et al. Neural progenitors organize in small-world networks to promote cell proliferation. Proc Natl Acad Sci USA. 2013;110:E1524–E1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hammes SR, Levin ER. Minireview: recent advances in extranuclear steroid receptor actions. Endocrinology. 2011;152:4489–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chen W, Dang T, Blind RD, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22:1754–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Anacker C, Cattaneo A, Musaelyan K, et al. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc Natl Acad Sci USA. 2013;110:8708–8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mohamed MA, Aly H. Male gender is associated with intraventricular hemorrhage. Pediatrics. 2010;125:e333–e339. [DOI] [PubMed] [Google Scholar]
- 110. Carbone DL, Zuloaga DG, Hiroi R, Foradori CD, Legare ME, Handa RJ. Prenatal dexamethasone exposure potentiates diet-induced hepatosteatosis and decreases plasma IGF-I in a sex-specific fashion. Endocrinology. 2012;153:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ballabh P. Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatr Res. 2010;67:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]