

Abstract
Glucose regulation of pancreatic α-cell Ca2+ entry through voltage-dependent Ca2+ channels is essential for normal glucagon secretion and becomes defective during the pathogenesis of diabetes mellitus. The 2-pore domain K+ channel, TWIK-related acid-sensitive K+ channel 1 (TASK-1), is an important modulator of membrane voltage and Ca2+ entry. However, its role in α-cells has not been determined. Therefore, we addressed how TASK-1 channels regulate α-cell electrical activity, Ca2+ entry, and glucagon secretion. We find that TASK-1 channels expressed in human and rodent α-cells are blocked by the TASK-1 channel inhibitor A1899. Alpha-cell 2-pore domain K+ currents were also significantly reduced after ablation of mouse α-cell TASK-1 channels. Inhibition of TASK-1 channels with A1899 caused plasma membrane potential depolarization in both human and mouse α-cells, which resulted in increased electrical excitability. Moreover, ablation of α-cell TASK-1 channels increased α-cell electrical excitability under elevated glucose (11mM) conditions compared with control α-cells. This resulted in significantly elevated α-cell Ca2+ influx when TASK-1 channels were inhibited in the presence of high glucose (14mM). However, there was an insignificant change in α-cell Ca2+ influx after TASK-1 inhibition in low glucose (1mM). Glucagon secretion from mouse and human islets was also elevated specifically in high (11mM) glucose after acute TASK-1 inhibition. Interestingly, mice deficient for α-cell TASK-1 showed improvements in both glucose inhibition of glucagon secretion and glucose tolerance, which resulted from the chronic loss of α-cell TASK-1 currents. Therefore, these data suggest an important role for TASK-1 channels in limiting α-cell excitability and glucagon secretion during glucose stimulation.
Elevated blood glucagon levels contribute to dysglycemia in type 2 diabetes (T2DM) and early stage type 1 diabetes (T1DM) (1–3). Thus, it is important to determine the mechanisms that modulate glucagon secretion as these could potentially be used to reduce hyperglucagonemia and hyperglycemia in diabetic states (4). Ca2+ entry through voltage-dependent Ca2+ channels (VDCCs) is essential for α-cell glucagon secretion and is elevated under low-glucose conditions (3, 5, 6). The ATP-sensitive potassium (KATP) channels are also involved in regulating glucagon secretion from islet α-cells (5, 7). During high-glucose conditions, inhibition of mouse α-cell KATP channel activity depolarizes the membrane potential (Δψp), leading to voltage-dependent inactivation of the VDCCs. This reduces Ca2+ influx and glucagon secretion (5, 7, 8). Conversely, increased KATP activity during low-glucose conditions hyperpolarizes the mouse α-cell Δψp, reducing voltage-dependent inactivation of VDCCs and leading to increased Ca2+ entry through VDCCs and elevated glucagon secretion (5, 8). Although KATP is an important mediator of acute changes in α-cell Ca2+ in response to glucose, what is not understood is how α-cells eventually hyperpolarize during continued glucose stimulation (6, 9–11). Because KATP would be inhibited during glucose stimulation, hyperpolarization in α-cells during elevated glucose conditions must be mediated by a non-KATP channel (6, 9–11).
Pancreatic α-cells have non-KATP K+ channels that are active at all physiological voltages and have biophysical properties that are similar to 2-pore domain K+ (K2P) channels (12). Blocking α-cell KATP channels results in a significant decrease in membrane conductance (by ∼0.71 nS) when stepped from a holding potential of −80 to −70 mV (13). Although this clearly indicates that a majority of α-cell K+ currents are mediated via KATP, it also demonstrates that there are active non-KATP channels (12, 13). Furthermore, currents active between −80 and −60 mV are present in KATP null α-cells. These currents are predicted to play a role in regulating the α-cell Δψp when KATP is inhibited under high-glucose conditions (13). Although the identity of the channel(s) mediating these currents has not been determined, their biophysical properties resemble those of a K2P channel. K2P channels permit K+ efflux from the cell at the physiological membrane potentials attained by the α-cell (14, 15). Moreover, the remaining outward K+ currents of α-cells that are not KATP are small currents, resembling the “leak” conductance of K2P channels (16, 17). Because these currents resemble leak, many reports on α-cell K+ channels have potentially subtracted these currents from their α-cell recordings. Thus, the physiological importance of these small K+ currents may have been inadvertently overlooked. K2P currents may regulate α-cell glucagon secretion, potentially contributing to the dysglycemia of T1DM and T2DM. However, the specific function of K2P channels in regulating α-cell glucagon secretion is currently unknown. Ultimately, understanding the function of K2P channels in glucagon secretion may reveal novel therapeutic targets for the treatment of T1DM and T2DM (4).
The K2P channel subfamily contains fifteen different K+ channels of which 6 are expressed in the pancreatic islet (18, 19). Northern blot analysis has demonstrated that TASK-1, TALK-1, and TREK-2 are highly expressed in the pancreas (20–22). Transcriptome analysis has confirmed that these 3 K2P channels are expressed in islet-cells and also found that TALK-2, TASK-2, and TWIK-1 are expressed in the pancreatic islet (23, 24). Analysis of TWIK-1 immunofluorescence finds that this channel is expressed primarily in islet β-cells (25). In human α-cells, the most abundant islet K2P channel transcripts are TASK-1 and TALK-1 (19, 23). Although TASK-1 channels are highly expressed in pancreatic α-cells, their role in regulating glucagon secretion remains undetermined (19, 23, 26).
TASK-1 channels show outwardly rectifying small conductance currents. As input resistance increases, the influence of these small currents on the membrane potential becomes more pronounced. Indeed, the TASK-1 currents of neurons and pancreatic β-cells hyperpolarize the Δψp from where action potentials (APs) fire (26–28). Thus, inhibition of TASK-1 channels depolarizes the Δψp, leading to increased electrical excitability (28). The small conductance of TASK-1 channels balances the depolarizing conductance of excitable cells at a Δψp that permits AP firing. Often, these currents are too small to hyperpolarize the Δψp below the activation threshold of voltage-dependent sodium channels and VDCCs (28). However, when TASK-1 channels are fully activated (for example, by inhalation anesthetics), Δψp hyperpolarization leads to a reduction in neuronal AP firing by diminishing the activity of voltage-dependent sodium channels and VDCCs (28). Because α-cells also fire sodium-dependent APs, TASK-1 currents may play a role in regulating α-cell Δψp and electrical excitability.
Here, we investigated the role of α-cell TASK-1 channels in mouse and human islets. We find that inhibition of TASK-1 channels significantly depolarizes the α-cell Δψp and increased excitability under conditions of elevated glucose, which leads to increased Ca2+ influx and glucagon secretion. Importantly, the results indicate that TASK-1 regulates α-cell glucagon secretion in a glucose-dependent manner. Thus, these studies reveal for the first time that α-cell TASK-1 currents serve an important role in regulating glucose-dependent inhibition of glucagon secretion.
Materials and Methods
Chemicals
Glucose, Krebs-Ringer buffer (KRB) salts, tolbutamide, amphotericin B, and tetraethylammonium (TEA) were from Sigma. The TASK-1 channel inhibitor A1899 was produced by the Vanderbilt Chemical Synthesis Core as previously described (29).
Transgenic mice
Mice containing a floxed tandem-dimer red fluorescent protein (tdRFP) in the ROSA26 locus were crossed with mice crossed with glucagon promoter-Cre mice as previously described (30–32); these animals specifically express tdRFP in islet α-cells. Another transgenic line was also created via mating mice with floxed green calmodulin (GCaMP3) Ca2+ indicator in the Rosa26 locus (Jackson stock 014538) with the glucagon-Cre mice; these animals specifically express GCAMP3 in islet α-cells.
Mouse islet and α-cell isolation
Islets were isolated from pancreata of 2- to 4-month-old C57BL/6 mice, using collagenase digestion and Ficoll gradients as previously described (33). Human cadaveric islets from male and female nondiabetic donors were provided by multiple isolation centers organized by the Integrated Islet Distribution Program; the islets all had more than 85% viability. Islets were plated or dissociated in 0.005% trypsin, placed on glass coverslips, and cultured for 16 hours in RPMI 1640 medium supplemented with 10% fetal calf serum, concentrations of glucose specified, 100-IU mL−1 penicillin, and 100-mg mL−1 streptomycin. Cells and islets were maintained in a humidified incubator at 37°C under an atmosphere of 95% air–5% CO2.
Perforated-patch electrophysiology
Patch electrodes (2–4 MΩ) loaded with solution containing 140mM KCl, 1mM MgCl2·6H2O, 10mM EGTA, 10mM HEPES (pH 7.25 with KOH), and the pore-forming antibiotic amphotericin B were used to record islet attached α-cells (34). Islet-cell clusters were perfused with Krebs-Ringer-HEPES buffer containing 119 mmol L−1 NaCl, 2 mmol L−1 CaCl2, 4.7 mmol L−1 KCl, 10 mmol L−1 HEPES, 1.2 mmol L−1 MgSO4, and 1.2 mmol L−1 KH2 PO4 (adjusted to pH 7.35 with NaOH), with the indicated concentrations of glucose and compounds. Alpha-cells were identified via RFP expression and were sealed in voltage clamp at −80 mV, and good access was obtained over several minutes through perforations by amphotericin B (34). After being switched to current clamp the cells were recorded continuously at 10 kHz. To confirm that the human recordings were performed in α-cells, the cells were poststained for glucagon.
Whole-cell voltage clamp electrophysiological recordings
TASK-1 channel currents were recorded using whole-cell ruptured patch clamps with an Axopatch 200B amplifier and pCLAMP10 software (Molecular Devices). Patch electrodes (2–4 MΩ) were loaded with intracellular solution containing 140mM KCl, MgCl2·6H2O, 10mM EGTA, 10mM HEPES, and 5mM MgATP (pH 7.25 with KOH). Cells were perifused with a modified KRB containing 97.7mM N-methyl-d-glucamine, 26mM KCl, 25mM HEPES, 1.2mM MgSO4, 1.2mM KH2PO4, 14.4mM glucose, and 20mM TEA (adjusted to pH 7.35 with NaOH).
Measurement of cytoplasmic calcium
Fluorescent dye
Islets were dispersed into single cells and clusters of 5–10 cells and plated on MatTek plates (MatTek Corp) overnight. Islet-cells and clusters were incubated (20 min, 37°C) in KRB supplemented with 2 μmol/L of fura 2 acetoxymethyl ester (Molecular Probes). Fluorescence imaging was performed using a Nikon Eclipse TE2000-U microscope equipped with an epifluorescence illuminator (SUTTER, Inc) a CCD camera (HQ2; Photometrics, Inc) and Nikon Elements software (NIKON, Inc). Cells were perifused at 37°C at a flow of 2 mL/min with appropriate KRB-based solutions that contained glucose concentrations and compounds specified in the figures. The ratios of emitted fluorescence intensities at excitation wavelengths of 340 and 380 nm (F340/F380) were determined every 5 seconds.
Genetic Ca2+ indicator (GCaMP3)
Islets were isolated from mice expressing GCaMP3 in α-cells and plated on MatTek plates (MatTek Corp) and imaged for changes in green fluorescent protein (GFP) fluorescence every 2 seconds in response to the indicated conditions with a Zeiss 710 Laser Scanning Confocal Microscope (Carl Zeiss, Inc).
Western blotting and immunofluorescence analysis
Protein extracts were prepared from islets by extraction with dodium dodecyl sulfate loading buffer (1% sodium dodecyl sulfate, 30 mmol/L Tris-HCl [pH 6.8], 5% β-mercaptoethanol, 5% glycerol, and 0.1% bromophenol blue) with protease inhibitors at 80°C for 10 minutes. After electrophoresis through a 4%–12% denaturing polyacrylamide gel, proteins were prepared as a Western blotting on a nitrocellulose membrane (Bio-Rad). Anti-TASK-1 (NeuroMab; this antibody has been extensively validated for TASK-1 channel specificity with knockout tissues, see NeuroMab TASK-1 data sheet) or anti-Glyceraldehyde 3-phosphate dehydrogenase (Rockland Immunochemicals) was used to probe the membrane at 1:250 or 1:700 dilution, respectively, in PBS, 0.1% Tween, and 3% powdered dried milk followed by a horseradish peroxidase-coupled secondary antibody (Jackson ImmunoResearch) at 1:5000 in the same solution. The membranes were washed in PBS containing 0.1% Tween between and after antibody incubations; horseradish peroxidase was illuminated using Pico Signal (Pierce) and exposed on Kodak X-Omat Blue film.
Mouse pancreata were fixed in 4% paraformaldehyde and embedded with paraffin as previously described. Five-micrometer sections were deparaffinized, rehydrated, and stained overnight with primary antibodies against glucagon (1:500; Cell Signaling) and TASK-1 (1:500; NeuroMab). The sections were then washed and stained with secondary antibodies conjugated to Cyanine3 and dylight488 (1:500; Jackson ImmunoResearch). Images of the sections were obtained with a Zeiss LSM 710 confocal microscope.
Glucagon secretion measurements
Mouse islets were allowed to recover after isolation overnight in 5.6mM glucose. For glucagon measurements, 20 islets (n = 6 mice and n = 4 human islet donors) were incubated with DMEM supplemented with an additional 0.5mM CaCl2 containing 5.6mM glucose for 4 hours followed by DMEM supplemented with an additional 0.5mM CaCl2 containing the indicated glucose concentrations with and without 250nM A1899 for 45 minutes. Glucagon secretion was analyzed using an enzyme immunoassay detection kit (Ray Biotech), and data are presented as mean ± SEM.
Mouse glucose tolerance testing (GTT)
Mouse GTT was performed as previously described by injecting 2-mg/kg dextrose and monitoring blood glucose at the indicated time points after glucose injection (see reference 40 below). The mice were fed a normal chow diet, and GTT was performed at 10 weeks of age.
Results
TASK-1 channels are functionally expressed in mouse and human α-cells
TASK-1 RNA has been shown to be expressed in pancreatic islets of both human and mouse as well as α-cells via transcriptome analysis (23, 35). Therefore, we characterized TASK-1 protein in mouse and human α-cells. We stained dispersed islet-cells for TASK-1 and glucagon. Mouse and human α-cells stained positive for TASK-1 (Figure 1, A–E). To determine whether α-cell TASK-1 generates functional currents, whole-cell voltage clamp recordings were used to measure TASK-1 currents from dispersed α-cells. To ensure that α-cells were recorded we used a mouse model that expressed the tdRFP specifically in α-cells. This animal is a cross between a transgenic mouse with a glucagon promoter-Cre transgene and mice containing a ROSA26 promoter loxP-flanked stop codon cassette upstream of tdRFP (glucagon-Cre floxed tdRFP) (30). Islets from glucagon-Cre floxed tdRFP mice were dispersed into single cells and K2P currents were recorded from the tdRFP-expressing α-cells. Currents with biophysical characteristics that correspond to cloned TASK-1 channels were present in mouse α-cells (Figure 1, G and H) (27). The α-cell TASK-1-like currents were insensitive to TEA or tolbutamide and were also maintained when extracellular Ca2+ was removed (16.7 ± 1.4 pA/pF [normalized current at 60 mV]; n = 9) (Figure 1, G and H). Thus, the TASK-1-like currents were not mediated via KATP, voltage-gated or Ca2+ activated K+ channels. Furthermore, inhibition of TASK-1 channels with the specific and potent inhibitor of TASK-1, A1899 (250nM), revealed that TASK-1 is responsible for at least 45.3% of the K2P currents in mouse α-cells (9.18 ± 0.7 pA/pF [normalized current at 60 mV]; n = 9) (Figure 1, G and H) (29). This indicates that mouse α-cells contain a small K2P like current that resembles TASK-1.
Figure 1.
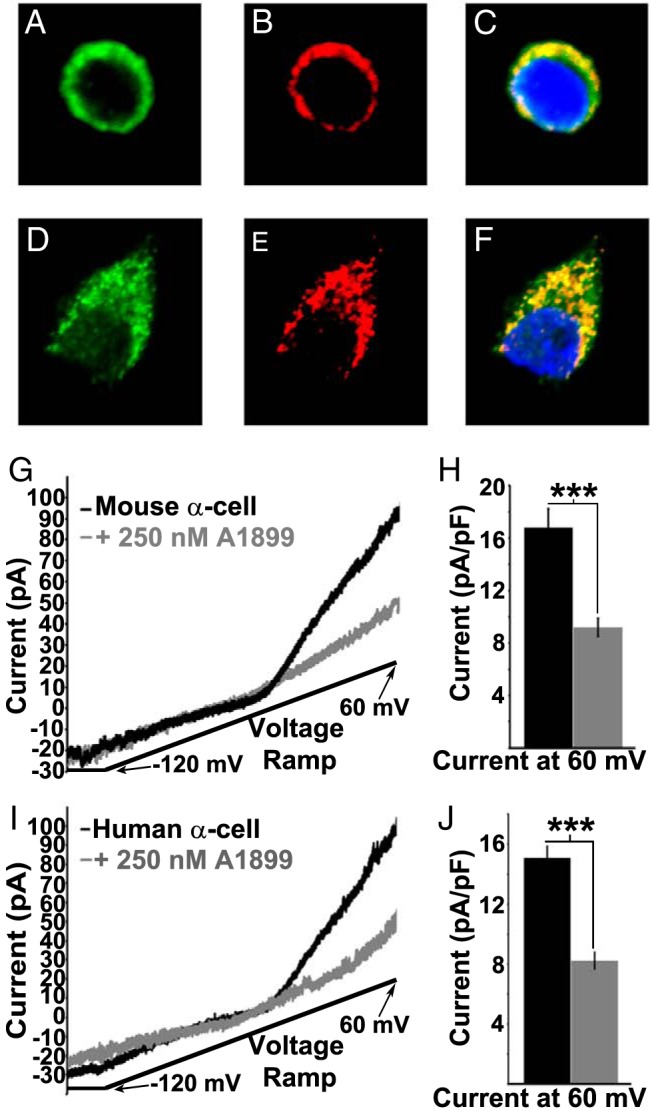
Mouse and human α-cells functionally express TASK-1 channels. A–C, Immunofluorescence of a mouse pancreatic α-cell for TASK-1 (green), glucagon (red), and nuclei (blue) with an overlay of all signals in C. D–F, Immunofluorescence of a human pancreatic α-cell for TASK-1 (green), glucagon (red), and nuclei (blue) with an overlay of all signals in F. G, Inhibition of mouse α-cell TASK-1 currents with 250nM A1899 (currents recorded in 14mM glucose, 100μM tolbutamide, and 10mM TEA; currents recorded in response to a voltage ramp from −120 to 60 mV over a 1-s interval). H, Average mouse α-cell TASK-1 current amplitude at +60 mV before (black bar) or 5 minutes after A1899 treatment (gray bar; currents are normalized to cell capacitance; ±SEM; ***, P < .001). I, Inhibition of human α-cell TASK-1 currents with 200nM A1899 (currents recorded in 14mM glucose, 100μM tolbutamide, and 10mM TEA). J, Average human α-cell TASK-1 current amplitude at +60 mV before or 5 minutes after A1899 treatment (currents are normalized to cell capacitance; ±SEM; ***, P < .001).
We went on to test whether human α-cells also contain a TASK-1 like current. Human islets were dispersed into single cells and recorded in response to a voltage ramp from −120 to 60 mV in the presence of TEA and tolbutamide without Na+ or Ca2+ (15.09 ± 0.7 pA/pF [normalized current at 60 mV]; n = 5) (Figure 1, I and J). After each recording, the cell type was determined via hormone staining and only α-cells that stained positive for glucagon were analyzed. The K2P currents recorded from human α-cells were significantly inhibited with the TASK-1 inhibitor A1899 by 45.5% (8.22 ± 0.5 pA/pF [normalized current at 60 mV]; n = 5) (Figure 1, I and J). Therefore, human α-cells also contain functional and active TASK-1 like channels at the plasma membrane.
To confirm that the A1899-sensitive K2P currents were mediated via TASK-1, we ablated α-cell TASK-1 channels and looked at the remaining K2P currents. This was accomplished by crossing mice containing “floxed” exon-2 of the TASK-1 gene KCNK3 (36) with mice containing a glucagon promoter-Cre transgene and mice containing a ROSA26 promoter loxP-flanked stop codon cassette upstream of the tdRFP protein (30). Currents from tdRFP-expressing cells from the glucagon-Cre floxed KCNK3 tdRFP mice show significantly diminished α-cell K2P currents (25.1 ± 4.3 pA/pF [normalized current at 60 mV]; n = 11) compared with control glucagon-Cre floxed tdRFP mice (13.6 ± 1.6 pA/pF [normalized current at 60 mV]; n = 12) (Figure 2, A and B). The loss in K2P currents in the TASK-1 ablated α-cells is similar to the reduction in α-cell K2P currents after A1899 treatments. Furthermore, the α-cell K2P currents recorded from glucagon-Cre floxed tdRFP mice were insensitive to 250nM A1899 (data not shown). Therefore, A1899 specifically inhibits the TASK-1 K2P channel in α-cells. Furthermore this data indicates that ablation of TASK-1 eliminates a significant amount of the α-cell K2P currents, which may play a role in regulating Δψp and Ca2+ entry.
Figure 2.
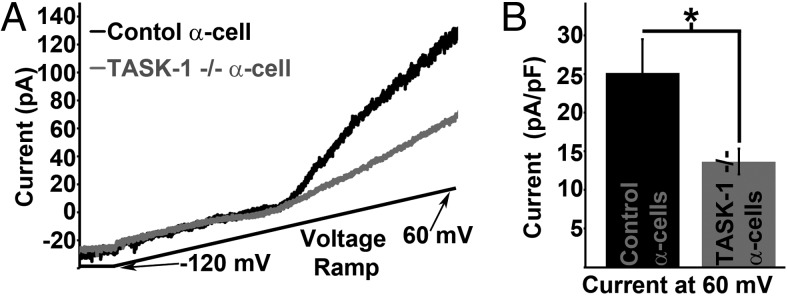
Ablation of α-cell TASK-1 channels reduces K2P current amplitude. A, Representative mouse α-cell K2P currents from control α-cells (black trace) and α-cells with TASK-1 ablation (gray trace, currents recorded in 14mM glucose, 100μM tolbutamide, and 10mM TEA; currents recorded in response to a voltage ramp from −120 to 60 mV over a 1-s interval). B, Average α-cell K2P current amplitude at +60 mV with (black bar) or without TASK-1 channels (gray bar; currents are normalized to cell capacitance; ±SEM; *, P < .05).
TASK-1 currents regulate pancreatic α-cell Δψp
The α-cell TASK-1 channels generate small amplitude K+ currents; however, modest changes in K+ flux in pancreatic α-cells are predicted to play an important role in modulating the Δψp (37, 38). Because α-cells in islet cell clusters respond physiologically to glucose stimulation, we dispersed islets into clusters of 5–10 cells and recorded the α-cells in these clusters. We then assessed α-cell TASK-1 channels for their influence on Δψp. Inhibition of α-cell TASK-1 channels with A1899 caused Δψp depolarization of both mouse (7.48 mV ± 1.32 mV; n = 13) and human α-cells in the presence of high glucose (14mM), which was maintained for the duration of A1899 treatment (Figure 3, A–C). To ensure that this was not a paracrine influence from other islet-cells in the cluster of cells that the α-cell was recorded in, we went on to assess the influence of A1899 on dispersed single α-cell Δψp. Although there is greater electrical activity in high glucose from dispersed α-cells, we still observed Δψp depolarization in response to A1899 treatment (n = 4) (Supplemental Figure 1). Thus, the Δψp depolarization in response to A1899 treatment is due to blockade of α-cell TASK-1 channels.
Figure 3.

TASK-1 inhibition increases α-cell electrical excitability. A and B, Representative mouse α-cell electrical activity in response to 14mM glucose alone (gray bar) and 14mM glucose with 250nM A1899 (black bar). C, Human α-cell electrical activity recorded in the presence of high glucose alone (14mM, gray bar) or with 250nM A1899 (black bar).
Although A1899 is a fairly specific inhibitor of TASK-1 channels, we went on to confirm that modulation of the α-cell Δψp by A1899 occurs via inhibition of TASK-1 by using the glucagon-Cre floxed KCNK3 tdRFP mouse model with α-cell TASK-1 ablation. Alpha-cells expressing tdRFP and lacking TASK-1 were recorded in islet cell clusters of approximately 5–10 cells and monitored for changes in Δψp in response to glucose (Figure 4). Switching control tdRFP-expressing α-cells (from glucagon-Cre floxed tdRFP mice) from 11mM to 1mM glucose resulted in a significant Δψp depolarization (5.84 ± 0.99 mV; n = 7), which increased AP firing frequency (Figure 4, A and B). However, tdRFP cells from glucagon-Cre floxed KCNK3 tdRFP mice showed only modest Δψp depolarization when switched from 11mM to 1mM glucose (2.1 ± 0.35 mV; n = 16) (Figure 4, C and D) compared with α-cells from glucagon-Cre floxed tdRFP mice (Figure 4, A and B). Furthermore, tdRFP cells from glucagon-Cre floxed KCNK3 tdRFP mice showed equally diminished Δψp polarization when switched from 1mM to 11mM glucose when compared with controls (data not shown). Thus, TASK-1 channels play an important role in limiting excitability in α-cells via hyperpolarization of the Δψp. The α-cell Δψp in low glucose (1mM) was also more depolarized in α-cells from the glucagon-Cre floxed KCNK3 tdRFP mice compared with α-cells from the glucagon-Cre floxed tdRFP mice. This resulted in a greater AP firing frequency in TASK-1 null α-cells (2.07 ± 0.15 Hz) in elevated glucose (14mM) conditions compared with control α-cells (0.86 ± 0.40 Hz). Moreover, the increase in AP firing frequency in TASK-1 null α-cells after a switch from high (11mM) to low (1mM) glucose was only 8.43 ± 5.22%, which is significantly less than the 68.55 ± 14.51% increase in control α-cell AP firing frequency after stimulation with low (1mM) glucose. However, α-cells with or without TASK-1 channels had no significant difference in AP firing frequency during stimulation with low (1mM) glucose. Thus, loss of α-cell TASK-1 channels caused significant Δψp depolarization and increased AP firing under elevated glucose conditions (11mM) when compared with control α-cells.
Figure 4.

Ablation of α-cell TASK-1 channels results in Δψp depolarization and increased electrical excitability. A and B, Representative electrical activity recorded from control mouse α-cells located in a cluster of other islet cells (5–10 cell clusters) in response to a switch from 11mM glucose (gray bar) to 1mM glucose (black bar) containing extracellular solution (n = 7). C and D, Representative electrical activity recorded from mouse α-cells without TASK-1 channels located in a cluster of other islet cells (5–10 cell clusters) in response to a switch from 11mM glucose (gray bar) to 1mM glucose (black bar) containing extracellular solution (n = 16).
TASK-1 limits α-cell calcium influx
Pancreatic α-cell Ca2+ entry is required for glucagon secretion and VDCC activity is in part regulated by Δψp depolarization that occurs during low glucose (1mM) stimulation (9, 10). Therefore, we next assessed α-cell Ca2+ influx in response to TASK-1 inhibition. We first assessed mouse α-cell Ca2+ under low glucose (1mM) conditions and interestingly TASK-1 inhibition caused no significant change in total Ca2+ influx (n = 109 cells) (Figure 5, A and B). However, inhibition of TASK-1 resulted in a significant increase of both human (n = 19 cells) and mouse (n = 34 cells) dispersed α-cell Ca2+ influx under high-glucose (14mM) conditions (Figure 5, C–F). Thus, these results indicate that TASK-1 channels limit Ca2+ entry into pancreatic α-cells primarily under high-glucose conditions.
Figure 5.

Inhibition of α-cell TASK-1 significantly increases Ca2+ influx under high-glucose (14mM) conditions. A, Dispersed mouse α-cell Ca2+ response after TASK-1 inhibition with 250nM A1899 (gray bar) in the presence of 1mM glucose (±SEM). B, Area under the Ca2+ influx curve of mouse α-cells in 1mM glucose (black bar, 2-min time course) and in 1mM glucose with A1899 (gray bar) from 3 to 5 minutes after A1899 treatment (±SEM). C, Representative dispersed mouse α-cell Ca2+ response to 14mM glucose (black bar) followed by 250nM A1899 treatment (gray bar). D, Area under the Ca2+ influx curve of mouse α-cells in 14mM glucose (black bar, 2-min time course) and in 14mM glucose with A1899 (gray bar) from 3 to 5 minutes after A1899 treatment (±SEM; ***, P < .001). E, Representative dispersed human α-cell Ca2+ response to 14mM glucose (black bar) followed by 250nM A1899 treatment (gray bar). F, Area under the Ca2+ influx curve of human α-cells in 14mM glucose (black bar, 2-min time course) and in 14mM glucose with A1899 (gray bar) from 3 to 5 minutes after A1899 treatment (±SEM; *, P < .05). G, Representative mouse intact islet α-cell Ca2+ responses to 14mM glucose (black bar) followed by 250nM A1899 treatment (gray bar). H, Area under the Ca2+ influx curve of intact mouse α-cells in 14mM glucose (black bar, 2-min time course) and in 14mM glucose with A1899 (gray bar) from 3 to 5 minutes after A1899 treatment (±SEM; ***, P < .001).
Because dispersed α-cells show different Ca2+ responses than intact islet α-cells, we went on to image Ca2+ levels in intact islet α-cells during TASK-1 inhibition (31, 39). Employing a mouse model with α-cell expression of the GCaMP3 Ca2+ indicator, we monitored changes in intact islet α-cell Ca2+ flux via confocal time-lapse microscopy of GFP fluorescent intensity. Similar to dispersed cells the intact mouse islet α-cells (n = 122 cells) in high glucose (11mM) showed a significant increase in intracellular Ca2+ (increased GFP fluorescence) after A1899 treatment (Figure 5, G and H). Therefore, TASK-1 inhibition results in α-cell Ca2+ influx. These results suggest that α-cell TASK-1 channels limit Ca2+ influx, which might impact glucagon secretion.
TASK-1 channels are important regulators of glucose inhibition of glucagon secretion
We next examined whether TASK-1 channel modulation of α-cell Ca2+ entry influences glucagon secretion. Inhibition of TASK-1 with 250nM A1899 significantly enhanced static glucagon secretion from both mouse and human islets in high glucose (14mM) (Figure 6, A and B). TASK-1 channel inhibition did not significantly influence glucagon secretion under low-glucose (1mM) conditions (Figure 6, A and B). This would be predicted to reduce glucose tolerance but interestingly we find that glucose tolerance was improved in mice with α-cell TASK-1 ablation compared with controls (Figure 6C). Interestingly, this was due to an increase in glucose inhibition of glucagon secretion from islets with α-cell TASK-1 ablation (Figure 6D). The differences in islet glucagon secretion in high glucose with A1899 vs α-cell TASK-1 ablation could be due to A1899 influences on other islet-cells, which will influence paracrine regulation of α-cell function. However, the differences are most likely due to the acute vs chronic inhibition of α-cell TASK-1 channels. Taken together the data suggests that TASK-1 channels play an important role in glucose-induced inhibition of α-cell electrical excitability, Ca2+ influx, and glucagon secretion.
Figure 6.

TASK-1 channels limit islet glucagon secretion during elevated glucose conditions. A, Static glucagon secretion from mouse islets in response to low-glucose (1mM) or high-glucose (14mM) stimulation with (gray bars) or without 250nM A1899 (black bars; n = 4, ±SEM; **, P < .01). B, Static glucagon secretion from human islets in response to low-glucose (1mM) or high-glucose (14mM) stimulation with (gray bars) or without 250nM A1899 (black bars; n = 3, ±SEM; *, P < .05). C, GTT on mice with (gray trace) or without (black trace) α-cell TASK-1 ablation (n = 5 mice per group, ±SEM; *, P < .05). D, Static glucagon secretion from mouse islets in response to low-glucose (1mM) or high-glucose (14mM) stimulation with (gray bars) or without (black bars) α-cell TASK-1 ablation (n = 4 per group, ±SEM; *, P < .05).
Discussion
The mechanism for glucose sensing in the α-cell is controversial but involves the activity of K+ channels (3, 40). Although it is widely accepted that glucose stimulation influences the α-cell membrane potential, glucose has been shown to both depolarize as well as hyperpolarize the α-cell membrane potential (3, 9, 10, 13). KATP channels are expressed in α-cells, and both activators as well as inhibitors of these channels can regulate the α-cell Δψp and glucagon secretion (11). One explanation for the glucose-dependent regulation of glucagon secretion is through KATP channel activity (7, 8). The KATP model postulates that a reduction in KATP activity during high energy/glucose conditions causes extended depolarization of the α-cell Δψp resulting in voltage-dependent inactivation of α-cell P/Q-type VDCCs, which diminishes Ca2+ influx near glucagon exocytotic machinery and limits glucagon secretion (7, 13). However, it has also been shown that the α-cell Δψp is hyperpolarized during conditions of hyperglycemia, below the activation threshold of P/Q-type VDCCs (9, 10). We also find that a majority of α-cells recorded show hyperpolarization after glucose stimulation. Because KATP channels would be inhibited under high-glucose conditions due to the elevated ATP/ADP ratio, the α-cell Δψp must be hyperpolarized by a non-KATP current during hyperglycemia. The exact K+ channel(s) responsible for α-cell Δψp hyperpolarization during glucose stimulation remains to be determined. The results presented here demonstrate that TASK-1 channels participate in hyperpolarizing the α-cell Δψp during glucose stimulation. The results suggest that TASK-1 channels serve an important role of modulating glucose induced inhibition of glucagon secretion through Δψp polarization, which reduces Ca2+ entry and glucagon secretion.
TASK-1 channels serve to regulate the Δψp from where APs fire in neurons and β-cells, and are highly expressed in the pancreatic α-cell (23, 41). Although α-cell TASK-1 channels are predicted to be active under both low and high-glucose conditions (27), TASK-1 channels only affect α-cell glucagon secretion at elevated glucose concentrations (≥11mM). Moreover, α-cell Ca2+ entry is only increased in α-cells under high-glucose conditions when TASK-1 is inhibited, whereas there is no significant change in α-cell Ca2+ influx in response to TASK-1 inhibition under low-glucose conditions (1mM). This may be due to differences in α-cell electrical excitability and Ca2+ influx under low and high-glucose conditions. When α-cells are exposed to low glucose, they maintain a depolarized Δψp and show increased AP firing that results in increased Ca2+ entry (9, 10). Thus, under these conditions the modest depolarization of the Δψp mediated via TASK-1 inhibition may not significantly influence AP firing frequency or Ca2+ influx, because the α-cells are already undergoing rapid AP firing. Furthermore, when α-cells fire APs, many repolarizing K+ channels are activated, such as voltage-gated and large calcium activated and voltage-gated and calcium-activated channels. This may reduce the ability of TASK-1 inhibition to depolarize the Δψp (42). However, when glucose concentrations are elevated, α-cell electrical activity is significantly decreased due to Δψp hyperpolarization. The significantly decreased rate of AP firing leads to reduced K+ channel activity and increased membrane resistance (11). Under these conditions small currents mediated via TASK-1 channels would be expected to play a significant role in regulating the Δψp, and when inhibited could lead to enough Δψp depolarization to significantly increase electrical excitability and Ca2+ influx. Future studies will determine whether these ionic mechanism(s) allow for the glucose dependence of TASK-1 channel modulation of α-cell Ca2+ influx and glucagon secretion.
Another possible mechanism for the glucose-dependent regulation of α-cell function by TASK-1 channels could be direct regulation of the TASK-1 channel in response to elevated metabolism and increased ATP/ADP levels. Previous reports have found that mitochondrial function and ATP are required for TASK-1 channel function. When TASK-1 is expressed as a heteromeric conformation with TASK-3, metabolic poisoning with azide results in a reduction of channel activity (43). Furthermore, elevations in the intracellular ATP concentration cause an increase in the open probability of TASK-1 channels in rat carotid body type1 cells (44). Thus, α-cell TASK-1 channels may be more active in elevated glucose conditions, which would hyperpolarize the Δψp. TASK-1 channels are not only regulated by metabolic signals by also by a number of signaling cascades that have been shown to influence α-cell function and glucagon secretion. For example, cAMP inhibits α-cell glucagon secretion at low levels and also inhibits TASK-1 channels via PKA-induced phosphorylation (45, 46). Thus, cAMP modulation of α-cell glucagon secretion could be mediated in part through inhibition of TASK-1 channels. Interestingly, certain conditions that lead to inhibition of K+ efflux through TASK-1 channels also result in Na+ entry through the channel (47). Therefore, α-cell TASK-1 channels may also serve to regulate Na+ entry, which would be predicted to influence Δψp depolarization, Ca2+ entry and glucagon secretion. Future studies will determine how α-cell TASK-1 channels are regulated, which may illuminate physiological mechanisms that influence glucagon secretion.
Although α-cell TASK-1 inhibition or ablation both result in Δψp depolarization under elevated glucose conditions, glucose inhibition of islet glucagon secretion is reduced in response to A1899 and increased in response to α-cell TASK-1 ablation. One possible explanation for this difference is that A1899 treatment of islets causes paracrine influences on α-cell glucagon secretion. Paracrine signals such as δ-cell somatostatin and/or β-cell insulin secretion might be influenced by A1899 and lead to reduced glucose inhibition of glucagon secretion (26). However, single α-cells undergo significant Δψp depolarization, in response to A1899, which results in an increase in Ca2+ influx that would presumably increase glucagon secretion. This indicates that paracrine signaling does not play a significant role in regulating α-cell Δψp in response to TASK-1 inhibition. Another possibility is that the increased glucose inhibition of glucagon secretion from TASK-1-deficient α-cells may be mediated by a mechanism that is induced to compensate for loss of these channels. Indeed, α-cell mass changes very rapidly in response to changes in blood glucagon levels via signals generated from the liver (48, 49), which may allow compensation for the loss of α-cell TASK-1 channels. Although compensation for α-cell TASK-1 ablation may occur, an increase in glucose inhibition of glucagon secretion from islets with TASK-1 α-cell ablation may also result from chronic Δψp depolarization. The prolonged Δψp depolarization observed in TASK-1-deficient α-cells under elevated glucose conditions could lead to voltage-dependent inactivation of P/Q-type VDCCs (9, 10); the activity of which are required for glucagon secretion. Thus, α-cell Δψp depolarization induced by ablation of TASK-1 channels in high glucose and by KATP inhibition in low glucose may both lead to voltage-dependent inactivation of P/Q-type VDCCs and inhibition of glucagon secretion. Future studies using a conditional knockout of α-cell TASK-1 channels will determine the mechanism for the differences in glucagon secretion with chronic and acute inhibition of TASK-1. Taken together the data from both chronic and acute inhibition of islet TASK-1 channels implicate an important role for these channels in modulating α-cell Δψp and glucagon secretion.
Although this is the first report to describe a role for α-cell K2P channels in modulating glucagon secretion, other K2P channels are also expressed in α-cells and have emerged as important regulators of islet function. Of the K2P channels expressed in human α-cells, TALK-1 is the second most abundant transcript after TASK-1 (23, 41). Furthermore, a nonsynonymous coding sequence polymorphism in the KCNK16 gene that codes for TALK-1 is associated with a predisposition for developing T2DM (50). This suggests the possibility that defects in α-cell TALK-1 channels may also influence glucagon secretion and the pathogenesis of T2DM (50). The importance of K2P channels during islet hormone secretion is becoming clear, thus, studies are required to determine whether these channels can be used as potential therapeutic targets for reducing hyperglucagonemia in T2DM patients (51).
In conclusion, this study suggests that TASK-1 plays an important role limiting glucagon secretion specifically under high energy/glucose conditions. TASK-1 hyperpolarizes the α-cell Δψp, decreasing excitability and Ca2+ influx during glucose stimulation, which reduces glucagon secretion. Thus, TASK-1 modulates glucose-dependent inhibition of α-cell glucagon secretion.
Acknowledgments
We thank Doug Bayliss for the floxed KCNK3 mouse line. We also thank Imju Jeong for electrophysiological assistance.
This work was supported by National Institutes of Health Grants DK096122 and DK081666 as well as a Pilot and Feasibility grant through the Vanderbilt University Diabetes Research Training Center P60 DK20593.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AP
- action potential
- GTT
- glucose tolerance testing
- GCaMP3
- green calmodulin Ca2+ indicator 3
- KATP
- ATP-sensitive potassium
- K2P
- 2-pore domain K+
- KRB
- Krebs-Ringer buffer
- SEM
- standard error of measurement
- Δψp
- membrane potential
- T2DM
- type 2 diabetes
- tdRFP
- tandem-dimer red fluorescent protein
- TEA
- tetraethylammonium
- VDCC
- voltage-dependent Ca2+ channel
- TASK-1
- TWIK-related acid-sensitive K+ channel 1
- TWIK-1
- Tandem of pore domains in a Weak Inward rectifying K+ channel.
References
- 1. Müller WA, Faloona GR, Unger RH. Hyperglucagonemia in diabetic ketoacidosis. Its prevalence and significance. Am J Med. 1973;54(1):52–57. [DOI] [PubMed] [Google Scholar]
- 2. Sherwin RS, Fisher M, Hendler R, Felig P. Hyperglucagonemia and blood glucose regulation in normal, obese and diabetic subjects. N Engl J Med. 1976;294(9):455–461. [DOI] [PubMed] [Google Scholar]
- 3. Quesada I, Tudurí E, Ripoll C, Nadal A. Physiology of the pancreatic α-cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol. 2008;199(1):5–19. [DOI] [PubMed] [Google Scholar]
- 4. Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122(1):4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. MacDonald PE, De Marinis YZ, Ramracheya R, et al. A K ATP channel-dependent pathway within α cells regulates glucagon release from both rodent and human islets of Langerhans. PLoS Biol. 2007;5(6):e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quoix N, Cheng-Xue R, Mattart L, et al. Glucose and pharmacological modulators of ATP-sensitive K+ channels control [Ca2+]c by different mechanisms in isolated mouse α-cells. Diabetes. 2009;58(2):412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang Q, Ramracheya R, Lahmann C, et al. Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 2013;18(6):871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rorsman P, Salehi SA, Abdulkader F, Braun M, MacDonald PE. K(ATP)-channels and glucose-regulated glucagon secretion. Trends Endocrinol Metab. 2008;19(8):277–284. [DOI] [PubMed] [Google Scholar]
- 9. Manning Fox JE, Gyulkhandanyan AV, Satin LS, Wheeler MB. Oscillatory membrane potential response to glucose in islet β-cells: a comparison of islet-cell electrical activity in mouse and rat. Endocrinology. 2006;147(10):4655–4663. [DOI] [PubMed] [Google Scholar]
- 10. Allister EM, Robson-Doucette CA, Prentice KJ, et al. UCP2 regulates the glucagon response to fasting and starvation. Diabetes. 2013;62(5):1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bokvist K, Olsen HL, Høy M, et al. Characterisation of sulphonylurea and ATP-regulated K+ channels in rat pancreatic A-cells. Pflugers Arch. 1999;438(4):428–436. [DOI] [PubMed] [Google Scholar]
- 12. Gromada J, Ma X, Hoy M, Bokvist K, Salehi A, Berggren PO, Rorsman P. ATP-sensitive K+ channel-dependent regulation of glucagon release and electrical activity by glucose in wild-type and SUR1−/− mouse α-cells. Diabetes. 2004;53(suppl 3):S181–S189. [DOI] [PubMed] [Google Scholar]
- 13. Gopel SO, Kanno T, Barg S, Weng XG, Gromada J, Rorsman P. Regulation of glucagon release in mouse -cells by KATP channels and inactivation of TTX-sensitive Na+ channels. J Physiol. 2000;528(Pt 3):509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kang D, Choe C, Kim D. Functional expression of TREK-2 in insulin-secreting MIN6 cells. Biochem Biophys Res Commun. 2004;323(1):323–331. [DOI] [PubMed] [Google Scholar]
- 15. Duprat F, Lauritzen I, Patel A, Honoré E. The TASK background K2P channels: chemo- and nutrient sensors. Trends Neurosci. 2007;30(11):573–580. [DOI] [PubMed] [Google Scholar]
- 16. Hatlapatka K, Willenborg M, Rustenbeck I. Plasma membrane depolarization as a determinant of the first phase of insulin secretion. Am J Physiol Endocrinol Metab. 2009;297(2):E315–E322. [DOI] [PubMed] [Google Scholar]
- 17. Ren J, Sherman A, Bertram R, et al. Slow oscillations of KATP conductance in mouse pancreatic islets provide support for electrical bursting driven by metabolic oscillations. Am J Physiol Endocrinol Metab. 2013;305(7):E805–E817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mathie A, Al-Moubarak E, Veale EL. Gating of two pore domain potassium channels. J Physiol. 2010;588(pt 17):3149–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benner C, van der Meulen T, Caceres E, Tigyi K, Donaldson CJ, Huising MO. The transcriptional landscape of mouse β cells compared to human β cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genom. 2014;15:620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bang H, Kim Y, Kim D. TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. J Biol Chem. 2000;275(23):17412–17419. [DOI] [PubMed] [Google Scholar]
- 21. Girard C, Duprat F, Terrenoire C, et al. Genomic and functional characteristics of novel human pancreatic 2P domain K(+) channels. Biochem Biophys Res Commun. 2001;282(1):249–256. [DOI] [PubMed] [Google Scholar]
- 22. Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M. TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J. 1997;16(17):5464–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bramswig NC, Everett LJ, Schug J, et al. Epigenomic plasticity enables human pancreatic α to β cell reprogramming. J Clin Invest. 2013;123(3):1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ku GM, Kim H, Vaughn IW, et al. Research resource: RNA-Seq reveals unique features of the pancreatic β-cell transcriptome. Mol Endocrinol. 2012;26(10):1783–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chatelain FC, Bichet D, Douguet D, et al. TWIK1, a unique background channel with variable ion selectivity. Proc Natl Acad Sci USA. 2012;109(14):5499–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dadi PK, Vierra NC, Jacobson DA. Pancreatic β-cell specific ablation of TASK-1 channels augments glucose-stimulated calcium entry and insulin secretion, improving glucose tolerance. Endocrinology. 2014;155(10):3757–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Talley EM, Lei Q, Sirois JE, Bayliss DA. TASK-1, a two-pore domain K+ channel, is modulated by multiple neurotransmitters in motoneurons. Neuron. 2000;25(2):399–410. [DOI] [PubMed] [Google Scholar]
- 28. Sirois JE, Lei Q, Talley EM, Lynch C, 3rd, Bayliss DA. The TASK-1 two-pore domain K+ channel is a molecular substrate for neuronal effects of inhalation anesthetics. J Neurosci. 2000;20(17):6347–6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Streit AK, Netter MF, Kempf F, et al. A specific two-pore domain potassium channel blocker defines the structure of the TASK-1 open pore. J Biol Chem. 2011;286(16):13977–13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur J Immunol. 2007;37(1):43–53. [DOI] [PubMed] [Google Scholar]
- 31. Le Marchand SJ, Piston DW. Glucose suppression of glucagon secretion: metabolic and calcium responses from α-cells in intact mouse pancreatic islets. J Biol Chem. 2010;285(19):14389–14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127(11):2317–2322. [DOI] [PubMed] [Google Scholar]
- 33. Philipson LH, Rosenberg MP, Kuznetsov A, et al. Delayed rectifier K+ channel overexpression in transgenic islets and β-cells associated with impaired glucose responsiveness. J Biol Chem. 1994;269(45):27787–27790. [PubMed] [Google Scholar]
- 34. Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37(1):15–26. [DOI] [PubMed] [Google Scholar]
- 35. Eizirik DL, Sammeth M, Bouckenooghe T, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012;8(3):e1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mulkey DK, Talley EM, Stornetta RL, et al. TASK channels determine pH sensitivity in select respiratory neurons but do not contribute to central respiratory chemosensitivity. J Neurosci. 2007;27(51):14049–14058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramracheya R, Ward C, Shigeto M, et al. Membrane potential-dependent inactivation of voltage-gated ion channels in α-cells inhibits glucagon secretion from human islets. Diabetes. 2010;59(9):2198–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang DM, Chai Y, Erickson JR, Heller Brown J, Bers DM, Lin YF. Modulation of sarcolemmal ATP-sensitive potassium channels by nitric oxide via sGC/PKG/ROS/ERK1/2/CaMKII signaling in ventricular cardiomyocytes. J Physiol. 2014;592(pt 5):971–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olsen HL, Theander S, Bokvist K, Buschard K, Wollheim CB, Gromada J. Glucose stimulates glucagon release in single rat α-cells by mechanisms that mirror the stimulus-secretion coupling in β-cells. Endocrinology. 2005;146(11):4861–4870. [DOI] [PubMed] [Google Scholar]
- 40. Jacobson DA, Wicksteed BL, Philipson LH. The α-cell conundrum: ATP-sensitive K+ channels and glucose sensing. Diabetes. 2009;58(2):304–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dorrell C, Schug J, Lin CF, et al. Transcriptomes of the major human pancreatic cell types. Diabetologia. 2011;54(11):2832–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Spigelman AF, Dai X, MacDonald PE. Voltage-dependent K(+) channels are positive regulators of α cell action potential generation and glucagon secretion in mice and humans. Diabetologia. 2010;53(9):1917–1926. [DOI] [PubMed] [Google Scholar]
- 43. Kreneisz O, Benoit JP, Bayliss DA, Mulkey DK. AMP-activated protein kinase inhibits TREK channels. J Physiol. 2009;587(pt 24):5819–5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Buckler KJ, Williams BA, Orozco RV, Wyatt CN. The role of TASK-like K+ channels in oxygen sensing in the carotid body. Novartis Found Symp. 2006;272:73–85; discussion 85–94, 131–140. [PubMed] [Google Scholar]
- 45. De Marinis YZ, Salehi A, Ward CE, et al. GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab. 2010;11(6):543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Olschewski A, Li Y, Tang B, et al. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ Res. 2006;98(8):1072–1080. [DOI] [PubMed] [Google Scholar]
- 47. Ma L, Zhang X, Zhou M, Chen H. Acid-sensitive TWIK and TASK two-pore domain potassium channels change ion selectivity and become permeable to sodium in extracellular acidification. J Biol Chem. 2012;287(44):37145–37153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gelling RW, Du XQ, Dichmann DS, et al. Lower blood glucose, hyperglucagonemia, and pancreatic α cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci USA. 2003;100(3):1438–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Longuet C, Robledo AM, Dean ED, et al. Liver-specific disruption of the murine glucagon receptor produces α-cell hyperplasia: evidence for a circulating α-cell growth factor. Diabetes. 2013;62(4):1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cho YS, Chen CH, Hu C, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet. 2012;44(1):67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mathie A, Veale EL. Therapeutic potential of neuronal two-pore domain potassium-channel modulators. Curr Opin Investig Drugs. 2007;8(7):555–562. [PubMed] [Google Scholar]