

Abstract
Recent studies have associated endocrine-disrupting chemical (EDC) exposure with the increased risk of cardiovascular disease in humans, but the underlying mechanisms responsible for these associations remain elusive. Many EDCs have been implicated in activation of the nuclear receptor pregnane X receptor (PXR), which acts as a xenobiotic sensor to regulate xenobiotic metabolism in the liver and intestine. Here we report an important role of intestinal PXR in linking xenobiotic exposure and hyperlipidemia. We identified tributyl citrate (TBC), one of a large group of Food and Drug Administration–approved plasticizers for pharmaceutical or food applications, as a potent and selective PXR agonist. TBC efficiently activated PXR and induced PXR target gene expression in vitro and in vivo. Interestingly, TBC activated intestinal PXR but did not affect hepatic PXR activity. Exposure to TBC increased plasma total cholesterol and atherogenic low-density lipoprotein cholesterol levels in wild-type mice, but not in PXR-deficient mice. TBC-mediated PXR activation stimulated the expression of an essential cholesterol transporter, Niemann-Pick C1-like 1 (NPC1L1), in the intestine. Promoter analysis revealed a DR-4 type of PXR response element in the human NPC1L1 promoter, and TBC promoted PXR recruitment onto the NPC1L1 promoter. Consistently, TBC treatment significantly increased lipid uptake by human and murine intestinal cells and deficiency of PXR inhibited TBC-elicited lipid uptake. These findings provide critical mechanistic insight for understanding the impact of EDC-mediated PXR activation on lipid homeostasis and demonstrate a potential role of PXR in mediating the adverse effects of EDCs on cardiovascular disease risk in humans.
Influences of the chemical environment on human health have recently become the subject of intense interest. Mounting evidence shows that endocrine-disrupting chemicals (EDCs) can interfere with complex endocrine signaling mechanisms and result in adverse consequences in humans and wildlife (1, 2). Recent findings have implicated exposure to EDCs in the etiology of cardiovascular disease (CVD) and metabolic disorders (1–6). For instance, higher bisphenol A (BPA) exposure has been consistently associated with CVD in multiple large-scale human population studies (4, 5, 7). Exposure to certain polychlorinated biphenyls (PCBs) induces hypercholesterolemia and promotes atherosclerosis in animals (8, 9). Circulating PCB levels have been associated with atherosclerotic plaques in elderly individuals (10). High circulating levels of phthalates are also associated with carotid atherosclerosis (11). However, the underlying mechanisms responsible for these associations remain largely unknown, which continues to hamper rational assessment of the health risks of EDC exposure.
Many EDCs such as phthalates, PCBs, and BPA and its analogs have been implicated in the activation of the pregnane X receptor (PXR) (also known as steroid and xenobiotic receptor) (12–15). PXR is a nuclear receptor activated by numerous endogenous hormones, dietary steroids, pharmaceutical agents, and xenobiotic chemicals (15–17). PXR functions as a xenobiotic sensor that induces expression of genes required for xenobiotic metabolism in the liver and intestine, including cytochromes P450 (CYPs), conjugating enzymes (eg, glutathione transferase), and ABC family transporters (eg, multidrug resistance 1 [MDR1]) (15, 18). In the past decade, the role of PXR as a xenobiotic sensor has been well established (15). However, the role of PXR in mediating the pathophysiological effects of EDCs in humans and animals remains elusive.
The identification of PXR as a xenobiotic sensor provided an important tool for the study of new mechanisms through which xenobiotic exposure affects diseases. Recent evidence indicates that PXR may also play an important role in the regulation of lipid homeostasis (19–24). For instance, it is well-known that many clinically relevant PXR ligands (eg, rifampicin and ritonavir) can elevate plasma lipid levels in patients and increase their CVD risk (25–28). A meta-analysis of 7 genome-wide association studies indicated that common genetic variants in PXR can affect plasma lipid levels in humans and 19 PXR single nucleotide polymorphisms were identified to significantly affect plasma low-density lipoprotein (LDL) cholesterol levels (29). We have recently demonstrated that chronic activation of PXR elicited by feeding mice the mouse PXR ligand pregnane 16α-carbonitrile (PCN) led to increased levels of plasma total cholesterol and the atherogenic lipoproteins LDL and very low-density lipoprotein (VLDL) in wild-type (WT) mice, but not in PXR-deficient (PXR−/−) mice (19). Activation of PXR also increased plasma total cholesterol and VLDL levels in apolipoprotein E *3-Leiden mice, which exhibit a human-like lipoprotein distribution on a cholesterol-rich diet (20). Very recently, we identified amprenavir, a widely used antiretroviral drug, as a potent PXR-selective agonist (24). Exposure to amprenavir significantly increased plasma total cholesterol and LDL cholesterol levels in WT mice, but not in PXR−/− mice (24).
Although emerging evidence is consistent with the hypothesis that modulation of PXR activity alters lipid homeostasis, the mechanisms underlying PXR ligand–elicited hyperlipidemia remain largely unknown. PXR is expressed at high levels in the liver and intestine, two organs that play a central role in whole-body lipid homeostasis. PXR has been reported to regulate several key hepatic lipogenic genes that promote dyslipidemia and hepatic steatosis, including CD36, SCD-1, lipin-1, Insig-1, and S14 (21, 30–32); however, the role of intestinal PXR in the regulation of lipid homeostasis remains elusive. Here we report that intestinal PXR plays an important role in linking EDC exposure and hypercholesterolemia. We identified several phthalate substitute plasticizers widely used in food packaging materials, medical devices, cosmetics, and pharmaceutical drugs as agonists of PXR. Tributyl citrate (TBC), one of a large group of Food and Drug Administration (FDA)–approved pharmaceutical plasticizers, is a potent and selective PXR agonist but does not activate other nuclear receptors. Interestingly, TBC activated intestinal PXR but did not affect hepatic PXR activity. Short-term TBC exposure increased plasma total cholesterol and atherogenic LDL cholesterol levels in WT mice, but not in PXR−/− mice. We found that TBC-mediated PXR activation stimulated the expression of the intestinal cholesterol transporter Niemann-Pick C1-like 1 (NPC1L1) and significantly increased lipid uptake by human and murine intestinal cells. We also identified a PXR-binding site in the human NPC1L1 promoter, indicating that NPC1L1 is a bona fide PXR target gene. These findings provide critical mechanistic insight for understanding the impact of EDC-mediated PXR activation on lipid homeostasis and demonstrate a potential role of PXR in mediating the adverse effects of EDCs on CVD risk in humans.
Materials and Methods
Reagents and plasmids
TBC, acetyl tributyl citrate (ATBC), di(2-ethylhexyl) phthalate (DEHP), acetyltriethyl citrate(ATEC), diisononyl phthalate (DiNP), triethyl citrate (TEC), di-n-butyl phthalate (DnBP), diisobutyl phthalate (DiBP), diethyl phthalate (DEP), PCN, and rifampicin (RIF) were purchased from Sigma-Aldrich. All of the chemicals were dissolved in dimethyl sulfoxide (DMSO). Human (h) and mouse (m) PXR expression vectors, GAL4 DNA-binding domain (DBD)-linked nuclear receptor ligand-binding domain vectors (GAL4-hPXR, GAL4-mPXR, GAL4-rat PXR, GAL4-retinoid acid receptor [RAR] α, GAL4-retinoid X receptor [RXR], GAL4-farnesoid X receptor [FXR], GAL4-liver X receptor [LXR], GAL4-peroxisome proliferator–activated receptor [PPAR] α, GAL4-PPARγ, and GAL4-vitamin D receptor [VDR]), and CMX-β-galactosidase expression vectors were described before (12, 33, 34). VP16-PXR, GAL4-nuclear receptor corepressor (NCoR), GAL4-silencing mediator of retinoid and thyroid hormone (SMRT), GAL4-steroid receptor coactivator-1 (SRC1), GAL4-PPAR binding protein (PBP), PXR-dependent CYP3A4 promoter reporter (CYP3A4XREM-luciferase), CYP3A2 promoter reporter [(CYP3A2)3-luciferase], and GAL4 reporter (MH100-luciferase) were described previously (12, 33, 35). For the human NPC1L1/DR-4 reporter, 4 copies of the DR-4 element (−9590 to −9574 bp) were synthesized and inserted into a pGL3 promoter vector (Promega) by annealing complementary oligonucleotides 5′-GCAGATCACTTGAGGTCAGG-3′ containing cohesive ends of the restriction enzyme sites, KpnI and MluI. The 2 mutant constructs were generated in a similar manner by using oligonucleotides 5′-GCAGAACACTTGAGAACAGG-3′ and 5′-GCAGATCTCTTGAGATCTGG-3′ for DR-4m1 and DR-4m2, respectively.
Cell culture and transfections
The human intestine epithelial cell line LS180 and hepatic cell line HepG2 were obtained from the American Type Culture Collection. The human hepatoma HepaRG cells were purchased from Life Technologies. Transfection assays were performed as described previously (12, 24). The cells were transfected with various expression plasmids as well as the corresponding luciferase reporter plasmids, together with CMX-β-galactosidase control plasmids using FuGENE 6 (Roche Diagnostics). The cells were then incubated with the corresponding ligands as indicated in the figure legends for 24 hours, and β-galactosidase and luciferase assays were performed as described previously (12, 33). Fold activation was calculated relative to solvent controls. Each data point represents the average of triplicate experiments ± SEM and was replicated in 3 to 5 independent experiments. For the mammalian 2-hybrid assays, LS180 cells were transfected with the GAL4 reporter, VP16-hPXR, and GAL-SRC1, GAL-PBP, GAL-NCoR, and GAL-SMRT (12, 33). The cells were then treated with compounds at the indicated concentrations.
Animals
C57BL/6 WT mice were purchased from The Jackson Laboratory. PXR-deficient mice (PXR−/−) on a C57BL/6 background were used as described previously (19). All of the animals were housed in the Division of Laboratory Animal Resources, University of Kentucky, as approved by the Institutional Animal Care and Use Committee in a specific pathogen–free environment with a light-dark cycle. Eight-week-old male WT or PXR−/− mice were fed a semisynthetic low-fat AIN76 diet containing 0.02% cholesterol (Research Diets) (19, 36, 37) and treated by oral gavage with vehicle (corn oil) or 10 mg/kg body weight (BW) TBC daily for 7 days. In addition, WT mice were also treated with vehicle control or 10 mg/kg/BW TBC daily by ip injection. On the day of euthanization, mice were fasted for 6 hours after the dark phase (feeding cycle) (19, 37). Mice were then anesthetized by ip injection with ketamine (Fort Dodge Animal Health). Mice were exsanguinated by left ventricular puncture, and blood was collected into EDTA-containing syringes. Plasma was prepared by spinning at 16 000 g for 10 minutes. The circulation was flushed with PBS, and intestinal and liver tissues were collected and stored in RNAlater solution (Life Technologies). Mouse primary hepatocytes and enterocytes were isolated as described previously (38, 39). Primary cells were cultured in multiwell plates and treated with the indicated compounds before being harvested for quantitative real-time PCR (qPCR) and Western blot analysis or cholesterol uptake assay.
Plasma analysis
Plasma total cholesterol and triglyceride concentrations were determined enzymatically by colorimetric methods as described previously (Roche) (24, 40). Lipoprotein fractions were isolated by spinning 60 μL of plasma in a TL-100 ultracentrifuge (Beckman Coulter) at its own density (1.006 g/mL) at 70 000 rpm for 3 hours to harvest the supernatant and then after adjustment of the infranatant with solid KBr to a density of 1.063 g/mL spinning it for 70 000 rpm for 18 hours to harvest the supernatant (40). The cholesterol content of each supernatant and the final infranatant were measured and taken to be VLDL (<1.006 g/mL), LDL (1.006 ≤ d ≤ 1.063 g/mL), and high-density lipoprotein (HDL) (d > 1.063 g/mL) cholesterol, respectively. Cholesterol concentrations in all 3 fractions were then determined enzymatically by a colorimetric method (Roche). Plasma cholesterol levels were determined enzymatically in the original plasma sample.
Cholesterol uptake assay
A cholesterol uptake assay was performed as described previously (41). In brief, micelles were prepared by mixing 9.7 mM taurocholate (Sigma-Aldrich), 6.47 mM egg yolk l-α-phosphatidylcholine (Sigma-Aldrich), and 1.5 mM cholesterol (Sigma-Aldrich), together with 1 μCi of [1,2-3H(N)]cholesterol (PerkinElmer)/μmol of cholesterol and evaporated under a mild stream of argon. The lipid film was hydrated in serum-free MEM containing 0.5% fatty acid–free BSA (Sigma-Aldrich) and incubated at 37°C in a rotating incubator. Solutions were filtered through a 0.45-μm surfactant-free cellulose acetate filter (Corning) and used to incubate the LS180 cells or primary enterocytes. After extensive PBS washing, the signals of the cells were counted by a β-counter.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed as described previously (38, 42). In brief, NPC1L1/DR-4 and CYP3A4/ER-6 probes were created by annealing the oligonucleotides 5′-GGGCAGATCACTTGAGGTCAGGAG-3′ (NPC1L1/DR-4), 5′-GGGCAGAACACTTGAGAACAGGAG-3′ (NPC1L1/DR-4m1), 5′-GGGCAGATCTCTTGAGaTCTGGAG-3′ (NPC1L1/DR-4m2), or 5′-ATATGAACTCAAAGGAGGTCAGTG-3′ (CYP3A4/ER6) to the complementary strand. Double-stranded oligonucleotides were end labeled using T4 polynucleotide kinase (New England Biosciences) and γ-[32P]ATP (PerkinElmer). Then 5 μL of in vitro–translated PXR or RXR protein was incubated with 2 μg of poly d(I-C) (Promega), 2 μL of bandshift buffer (50 mM MgCl2 and 340 mM KCl), and 6 μL of delta buffer (0.1 mM EDTA, 40 mM KCl, 25 mM HEPES [pH 7.6], 8% Ficoll 400, and 1 mM dithiothreitol) on ice for 10 minutes. 32P-labeled double-stranded oligonucleotide probe (100 000 cpm) was then added, and the reaction was incubated for another 30 minutes on ice. For the supershift assays, proteins were incubated with 2 μg of goat anti-PXR (sc-7739; Santa Cruz Biotechnology) or rabbit anti-PXR (sc-25381; Santa Cruz Biotechnology) antibodies for 1 hour before the addition of 32P-labeled probe. The binding complexes were subjected to electrophoresis in a 6% nondenaturing polyacrylamide gel containing 0.5× Tris-borate-EDTA (TBE). The gels were dried and visualized by exposure to X-ray film.
Chromatin immunoprecipitation (ChIP)
ChIP analysis was performed by using an antibody against PXR (sc-25381; Santa Cruz Biotechnology) and a SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling). The precipitated genomic DNA relative to inputs was analyzed by semiquantitative PCR using specific primers and DNA polymerase (Takara). The sequences of primer sets used for PCR are listed in Supplemental Table 1.
RNA isolation and qPCR analysis
Total RNA was isolated from mouse tissues and intestinal LS180, HepaRG, and primary cells using TRIzol reagent (Life Technologies) per the manufacturer-supplied protocol. qPCR was performed using gene-specific primers and the SYBR Green PCR kit (Life Technologies) as described previously (12, 24). The primer sets used in this study are listed in Supplemental Table 1.
Western blot analysis
Western blot analysis was performed as described previously (42). Proteins were isolated from cells or mouse tissues by homogenization in radioimmunoprecipitation assay buffer with complete mini protease inhibitor cocktail (Roche). Protein concentrations were determined by the Pierce BCA protein assay kit (Thermo Scientific). Anti-PXR antibodies were purchased from Santa Cruz Biotechnology (sc-7739), anti-NPC1L1 antibodies were purchased from Novus Biologicals (NB400–128), and anti-β-actin antibodies were purchased from Sigma-Aldrich (A2066).
Statistical analysis
All data are expressed as means ± SEM unless otherwise noted. Statistical analysis was performed using a two-sample, two-tailed Student t test unless otherwise noted, with P < .05 regarded as significant. One-way ANOVA was used when multiple comparisons were made, followed by the Dunnett t test for multiple comparisons to a control.
Results
FDA-approved phthalate substitute plasticizers can activate PXR
Based on previous findings that several plastic-associated chemicals including BPA and phthalates can activate PXR (12, 43), we examined several widely used phthalates and phthalate substitute plasticizers for PXR activation in transient transfection assays. Consistent with previous reports (44, 45), DEHP, DiNP, and ATBC can activate PXR and induce PXR-mediated CYP3A4-luciferase reporter activities (Figure 1A). TBC, an FDA-approved phthalate substitute plasticizer used in food contact substances and as a pharmaceutical excipient, was a more potent PXR agonist than any of the other tested plasticizers and induced reporter gene activity in a dose-dependent manner (Figure 1A). We then tested the ability of TBC to activate a panel of other nuclear receptors, including mouse PXR, human RARα, RXR, FXR, LXRα, PPARα, PPARγ, VDR, constitutive androstane receptor, and estrogen receptor α (Figure 1B). TBC can activate both human and mouse PXR but was unable to activate any of the other nuclear receptors. Thus, TBC is a PXR-selective agonist.
Figure 1.

Activation of PXR by phthalates and phthalate substitute plasticizers. A, LS180 cells were cotransfected with full-length human PXR and a CYP3A4-luciferase reporter. Cells were treated with DMSO vehicle, TBC, DEHP, ATBC, ATEC, DiNP, TEC, (DnBP, DiBP, and DEP at the indicated concentrations for 24 hours. B, LS180 cells were cotransfected with a GAL4 reporter and a series of GAL4 DBD-nuclear receptor ligand-binding domain constructs. Cells were treated with DMSO vehicle or 10 μM TBC for 24 hours. CAR, constitutive androstane receptor; ERα, estrogen receptor α. C and D, LS180 cells were cotransfected with a GAL4 reporter, VP16-hPXR, and expression vector for GAL4 DBD or GAL4 DBD linked to the receptor interaction domains of PXR coactivators (GAL-SRC1 or GAL-PBP) (C) or PXR corepressors (GAL-SMRT or GAL-NCoR) (D). Cells were treated with DMSO vehicle, TBC, or RIF at the indicated concentrations for 24 hours. Data are shown as fold induction of normalized luciferase activity compared with that for DMSO treatment and represent the means from triplicate experiments.
Nuclear receptor coregulators play critical roles in nuclear receptor signaling. We used a mammalian 2-hybrid assay to evaluate the effects TBC on PXR coregulator interaction. TBC promoted the specific interactions between PXR and the coactivators SRC-1 and PBP (Figure 1C). Consistent with our previous report (12, 33), unliganded PXR interacted with corepressors, NCoR and SMRT (Figure 1D), and TBC disrupted this interaction as did the potent human PXR ligand RIF (Figure 1D). Thus, binding of TBC to PXR inhibits PXR-corepressor interaction and promotes PXR-coactivator recruitment, thereby inducing PXR transcriptional activation in a concentration-dependent manner.
TBC activates intestinal PXR but does not affect hepatic PXR activity
We next used human hepatoma HepaRG cells (46) and intestinal LS180 cells (24, 33) to test the effects of TBC exposure on PXR activity and target gene expression. The known human PXR ligand RIF induced the expression of bona fide PXR target genes involved in phase I (CYP3A4), phase II (UGT1A1), and phase III (MDR1) metabolism in both cell lines. Interestingly, TBC stimulated PXR target gene expression in intestinal LS180 cells but not in HepaRG cells (Figure 2A). We then isolated mouse primary hepatocytes and enterocytes to confirm these findings. As expected, the mouse PXR ligand PCN induced PXR target gene expression in both hepatocytes and enterocytes (Figure 2B). However, TBC was only able to induce PXR target gene expression in enterocytes but not in hepatocytes (Figure 2B). We also performed transfection assays in LS180 and HepG2 cell lines. Consistently, TBC did not activate PXR in HepG2 cells but was able to induce PXR reporter activity in LS180 cells after being treated for as few as 3 hours (Supplemental Figure 1).
Figure 2.

TBC activates intestinal PXR but does not affect hepatic PXR activity. A, Human HepaRG hepatoma cells and LS180 intestinal cells were treated with control medium or medium containing 10 μM TBC or RIF. Total RNA was isolated, and gene expression levels of human PXR target genes were analyzed by qPCR (n = 3; **, P < .01; ***, P < .001). B, Primary hepatocytes and enterocytes were isolated from WT mice and were incubated with control medium or medium containing 10 μM TBC or PCN. Total RNA was isolated, and gene expression levels of mouse PXR target genes were analyzed by qPCR (n = 3; *P < .05; **P < .01; ***P < .001). C, Eight-week-old WT mice were treated with vehicle or 10 mg/kg BW TBC or PCN daily by oral gavage for 1 week. Total RNA was isolated from liver and small intestine. Expression levels of PXR target genes, CYP3A11, GSTA1, and MDR1a, were measured by qPCR (n = 4–5; *, P < .05; **, P < .01; and ***, P < .001 compared with the control group).
To further investigate the tissue-specific effects of TBC on PXR activity in vivo, WT mice were treated with vehicle (corn oil), 10 mg/kg BW TBC, or PCN daily by oral gavage for 1 week. Consistent with our previous report (38), PCN activated PXR and induced target gene expression in both liver and intestine (Figure 2C). However, TBC only stimulated expression of known PXR target genes in intestine but not in liver (Figure 2C). To examine whether lower TBC doses can activate intestinal PXR in vivo, we also treated mice with 2.5 or 5 mg/kg/d TBC by oral gavage for 1 week. Neither 5 nor 2.5 mg/kg/d TBC activated PXR or induced PXR target gene expression in intestine (Supplemental Figure 2). Next, to determine whether the tissue-specific effect of TBC on intestinal PXR is related to poor absorption into the circulation, WT mice were also treated with 10 mg/kg/d TBC by ip delivery. Interestingly, TBC still cannot induce PXR target gene expression in liver even by ip delivery (Supplemental Figure 3). Therefore, it is unlikely that the inability of TBC to activate hepatic PXR is due to poor absorption by the intestine. Taken together, these results suggest that exposure to TBC at 10 mg/kg/d via oral delivery activates intestinal PXR but does not affect hepatic PXR signaling.
TBC exposure elevates plasma lipid levels in WT mice, but not in PXR−/− mice
We previously reported that activation of PXR by feeding PCN at a high dose (200 mg/kg in diet) for 2 weeks significantly increased plasma total cholesterol levels and atherogenic LDL and VLDL levels in WT mice (19). Consistent with our previous report, WT mice treated with a relatively low dose of PCN (10 mg/kg BW/d via oral delivery) for 1 week also had significantly increased plasma total, LDL, and VLDL cholesterol levels (Figure 3A). Although TBC only activates intestinal PXR, TBC treatment significantly increased total cholesterol levels compared with those in control WT mice (Figure 3A). TBC treatment did not affect HDL or VLDL cholesterol levels but significantly increased LDL cholesterol levels in WT mice (Figure 3A). The relatively stronger effects of PCN on plasma lipid levels could be due to its effects on hepatic PXR signaling. In contrast, TBC did not affect any of the lipoprotein levels in PXR−/− mice (Figure 3B), indicating that TBC-elicited hypercholesterolemia effects are mediated by PXR signaling. Taken together, these results suggest that activation of intestinal PXR by TBC is sufficient to increase plasma cholesterol levels.
Figure 3.

Exposure to TBC induces hyperlipidemia in WT but not in PXR−/− mice. A, Eight-week-old male WT mice were treated with vehicle or 10 mg/kg BW TBC or PCN daily by oral gavage for 1 week. Plasma total cholesterol levels and lipoprotein levels (LDL, VLDL, and HDL) were measured (n = 6–10; *, P < .05 and ***, P < .001 compared with the control group). B, Eight-week-old male PXR−/− mice were treated with vehicle or 10 mg/kg BW TBC daily by oral gavage for 1 week. Plasma total cholesterol levels and lipoprotein levels (LDL, VLDL, and HDL) were measured (n = 4–5).
TBC stimulates the expression of intestinal cholesterol transporter NPC1L1 and promotes cholesterol uptake by intestinal cells
Small intestinal lipid absorption is the key step for lipid accumulation in the body. Interestingly, we found that TBC stimulated expression of the transporters responsible for intestinal lipid absorption including CD36 and NPC1L1 in intestines of WT mice, but not in intestines of PXR−/− mice (Figure 4A). The known PXR target gene, CD36 (21), encodes a fatty acid transporter that plays an important role in intestinal fatty acid absorption and chylomicron production (47, 48). NPC1L1 is an essential transporter mediating intestinal cholesterol absorption (49–51). Western blot analysis also confirmed that TBC induced intestinal NPC1L1 protein levels in WT mice but not in PXR−/− mice (Figure 4B). Consistent with a previous report (52), PXR activation did not affect expression levels of the cholesterol efflux transporters ABCG5 and ABCG8. In addition, the known PXR ligand PCN was also able to induce NPC1L1 expression in intestine (Supplemental Figure 4), indicating that NPC1L1 is a downstream target of PXR.
Figure 4.

Activation of PXR by TBC stimulates the expression of the intestinal cholesterol transporter NPC1L1 in mice and increases cholesterol uptake by murine intestinal cells. A, WT and PXR−/− mice were treated with vehicle or 10 mg/kg BW TBC daily for 1 week. Total RNA was isolated from small intestine, and the expression levels of indicated genes were measured by qPCR (n = 5–6 per group; *, P < .05). B, Western blot analysis of intestinal NPC1L1 protein levels in control or TBC-treated WT and PXR−/− mice. The top band indicates glycosylated NPC1L1. C and D, Primary enterocytes isolated from WT and PXR−/− mice were treated with vehicle control or 10 μM TBC for 3 hours. NPC1L1 mRNA levels were measured by qPCR (n = 3; ***, P < .001), and protein levels were analyzed by Western blot (D). E, Primary enterocytes isolated from WT and PXR−/− mice were treated with vehicle control or 10 μM TBC for 2 hours, followed by incubation with [3H]cholesterol and TBC for 1 hour. The cellular cholesterol uptake was then measured (n = 3; *, P < .05; and **, P < .01).
To further investigate the effects of TBC on PXR activity and NPC1L1 expression, we isolated primary enterocytes from WT and PXR−/− mice. Consistent with in vivo results, TBC treatment increased NPC1L1 mRNA and protein levels in WT enterocytes (Figure 4, C and D). TBC-mediated NPC1L1 induction was abolished in PXR-deficient enterocytes. Because NPC1L1 plays a key role in cholesterol uptake by intestinal cells, we then performed cholesterol uptake assays using primary enterocytes. As expected, TBC treatment increased [3H]cholesterol uptake by primary enterocytes of WT mice but did not affect cholesterol uptake by PXR-deficient enterocytes (Figure 4E). Taken together, these results indicate a previously unrecognized role of PXR in the regulation of intestinal cell cholesterol uptake.
Activation of PXR by TBC transcriptionally regulates NPC1L1 expression and increases cholesterol uptake by human intestinal cells
To determine the impact of TBC-mediated PXR activation on NPC1L1 regulation and cholesterol uptake in human intestinal cells, we used small interfering RNA (siRNA) to successfully reduce PXR expression in human LS180 cells (Figure 5A). As expected, TBC treatment induced NPC1L1 mRNA levels in control LS180 cells and siRNA-mediated PXR knockdown decreased TBC-induced NPC1L1 induction (Figure 5B). In addition, TBC was also able to significantly increase [3H]cholesterol uptake by LS180 cells (Figure 5C). LS180 cells transfected with siRNA against PXR can still take up [3H]cholesterol upon TBC treatment, which may be due to incomplete inhibition of endogenous PXR expression by siRNA. However, TBC-mediated cholesterol update was significantly reduced by siRNA-mediated PXR knockdown. These results confirm that TBC-mediated PXR activation can increase NPC1L1 expression and cholesterol uptake in human intestinal cells.
Figure 5.
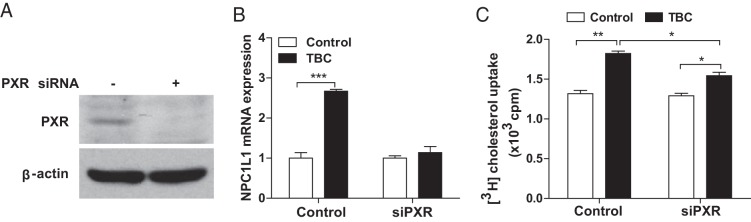
TBC promotes cholesterol uptake by human intestinal cells. A, Western blot analysis of PXR levels in human intestinal LS180 cells transfected with control siRNA or siRNA against PXR (siPXR). B, Control or siPXR LS180 cells were treated with 10 μM TBC for 3 hours, and NPC1L1 expression was analyzed by qPCR (n = 3; ***, P < .001). C, Control or siPXR LS180 cells were treated with 10 μM TBC for 2 hours followed by incubation with [3H]cholesterol and TBC for an additional 1 hour. The cellular cholesterol uptake was then measured (n = 3; *, P < .05; and **, P < .01).
We next analyzed the promoter of the human NPC1L1 gene and identified a DR-4-type (direct repeat spaced by 4 nucleotides) of nuclear receptor response element (AGATCACTTGAGGTCA), similar to DR-4 elements found in other PXR target genes (16). EMSA confirmed that the PXR and RXR heterodimer was able to bind to this DR-4 element as well as the positive control, the ER-6 (everted repeat spaced by 6 nucleotides) element in the CYP3A4 promoter (Figure 6A). The binding of NPC1L1/DR-4 or CYP3A4/ER-6 by PXR-RXR is specific as excess cold probe decreased PXR-RXR binding to those elements (Figure 6A). In addition, 2 different anti-PXR antibodies disrupted the protein-DNA complex (Figure 6B), suggesting that PXR is a component of the protein complex that binds to the NPC1L1/DR4 element. Further, mutations of the DR-4 elements were also able to abolish the binding of PXR-RXR dimers to both mutant DR-4 sites (Figure 6C).
Figure 6.

NPC1L1 is a direct transcriptional target of PXR. A, In vitro translated human PXR and RXR, as indicated, were incubated with 32P-labeled NPC1L1/DR-4 or CYP3A4/ER-6 probes and analyzed by EMSA. Ten- or 25-fold excess of unlabeled NPC1L1/DR-4 or CYP3A4/ER-6 probes were used for the competition experiments. B, PXR/RXR proteins were incubated with 2 different anti-PXR antibodies, goat anti-PXR (1) or rabbit anti-PXR antibodies (2) for 1 hour before the addition of the 32P-labeled NPC1L1/DR-4 probe. C, PXR/RXR proteins were incubated with 32P-labeled WT NPC1L1/DR-4 or mutated DR-4 (DR-4m1 and DR-4m2) probes and were analyzed by EMSA. D, LS180 cells were treated with 10 μM TBC for 3 hours, and ChIP analysis was performed to determine the recruitment of PXR onto the NPC1L1 or CYP3A4 promoter. E–G, LS180 cells were cotransfected with full-length hPXR and RXR expression plasmids along with a synthetic reporter containing 4 copies of NPC1L1/DR-4 element (E) or mutated DR-4 elements, DR-4m1 (F) and DR-4m2 (G). Cells were then treated with DMSO vehicle (control), TBC, or RIF at the indicated concentrations for 24 hours. Data are shown as fold induction of normalized luciferase activity compared with that for DMSO treatment and represent the means for triplicate experiments.
Next, ChIP analysis demonstrated that TBC can promote the recruitment of PXR onto the NPC1L1 promoter region containing the DR-4 element (Figure 6D). ChIP on the CYP3A4 promoter region containing the ER-6 element was included as a positive control. Last, 4 copies of the NPC1L1/DR-4 or mutated DR-4 elements were synthesized and inserted into a luciferase reporter vector. Transfection assays were performed to determine whether the DR-4 motif is necessary and sufficient for mediating PXR transactivation. Indeed, both TBC and RIF can activate PXR and increase the NPC1L1/DR-4 reporter gene activity in a dose-dependent manner (Figure 6E). In contrast, TBC and RIF had no effect on activity of 2 mutated DR-4 reporters (Figure 6, F and G). Collectively, activation of PXR by TBC transcriptionally regulates NPC1L1 expression and increases cholesterol uptake by human intestinal cells.
Discussion
Because of their variety and low costs, plastics are fundamental in modern life, and plastic production exceeded 300 million tons in 2010 (2). Plastic-associated chemicals are produced in high volume for use in the production of plastics, including the base chemical BPA and numerous plasticizers. The adverse effects of BPA and several phthalate plasticizers (eg, DEHP) on human health have attracted considerable attention and engendered controversy in the past few decades, partly due to their endocrine-disrupting properties. We and others have identified BPA, BPA analogs (eg, bisphenol B and bisphenol AF), and the widely used phthalate plasticizer DEHP as potent PXR agonists (12, 13, 43). In addition to these well-known EDCs, there are many phthalate substitute plasticizers that have not been tested for endocrine disruption. For example, citrate esters, including TBC, ATBC, and TEC, represent a large group of plasticizers that have been approved by the FDA to be used in food packaging materials, vinyl toys, medical devices, cosmetics, and pharmaceutical drugs (53–56). ATBC has recently been shown to activate PXR and induce PXR target gene expression (45). However, it was unclear whether ATBC or other phthalate substitute plasticizers have any adverse effects on the development of complex diseases. In the current study, we demonstrate that TBC is a more potent PXR agonist than ATBC in our assays. Similar to ATBC, TBC activated intestinal PXR and induced PXR target gene expression but did not affect hepatic PXR activity. Nevertheless, TBC-mediated intestinal PXR activation was sufficient to increase plasma cholesterol levels, especially atherogenic LDL levels in WT mice. We then identified a key intestinal cholesterol transporter, NPC1L1, as a direct transcriptional target of PXR. Activation of PXR stimulated NPC1L1 expression and promoted cholesterol uptake by intestinal cells, which may contribute to TBC-induced hyperlipidemia. These findings demonstrate a previously unrecognized role of intestinal PXR in the regulation of lipid homeostasis.
Whereas the role of PXR in xenobiotic metabolism has been well established, recent studies have revealed the role of PXR in dyslipidemia and atherosclerosis. We previously reported that chronic activation of PXR elicited by feeding mice the potent mouse PXR agonist PCN led to increased levels of plasma total cholesterol and VLDL and LDL in WT mice but not in PXR−/− mice (19). PCN-mediated PXR activation affected several genes involved in hepatic lipid homeostasis. Because TBC does not activate PXR in the liver, it is unlikely that hepatic PXR signaling contributes to TBC-elicited hypercholesterolemia. Cholesterol uptake from the intestinal lumen by the enterocytes is the rate-limiting step in cholesterol absorption (47). NPC1L1, a multitransmembrane protein containing a conserved N-terminal NPC1 domain and a putative sterol-sensing domain, has been established as an essential transporter in mediating intestinal cholesterol uptake (47, 49, 50). NPC1L1 expression is enriched in the small intestine and is in the brush border membrane of enterocytes (50). NPC1L1 takes up free cholesterol into cells through vesicular endocytosis and is required for intestinal cholesterol absorption (47, 49, 50). Indeed, NPC1L1-deficient mice had substantially reduced intestinal cholesterol absorption and plasma cholesterol (particularly LDL) levels and were completely resistant to diet-induced hypercholesterolemia (49, 50). NPC1L1 is also the molecular target of the clinically used drug ezetimibe, a potent cholesterol absorption inhibitor widely used to treat hypercholesterolemia (50). Very recently, inactivating mutations in NPC1L1 has been associated with reduced plasma LDL cholesterol levels and a reduced risk of CVD in a large-scale human study (57). Despite the established function of NPC1L1 in intestinal cholesterol absorption, the transcriptional regulation of NPC1L1 has not been fully understood. Our results suggest that PXR is an important regulator of NPC1L1 transcription. PXR can directly bind to a DR-4 motif in human NPC1L1 promoter and stimulate NPC1L1 expression upon ligand activation. Thus, PXR-mediated NPC1L1 up-regulation may contribute to TBC and other ligand-induced hypercholesterolemia.
Whereas NPC1L1 plays an essential role in intestinal cholesterol absorption, another PXR-regulated transporter, CD36, mediates enterocyte uptake of fatty acids, which are then converted to triglycerides for transport into chylomicrons (47, 48). We and others previously reported that activation of PXR induces CD36 expression and increases lipid accumulation in the liver (21), intestine (23), and macrophage (19). In addition, activation of intestinal PXR can also induce the expression levels of several enzymes involved in intestinal lipid transportation and chylomicron secretion including diacylglycerol acyltransferase 1 and 2 (23). Therefore, the functions of PXR in intestinal lipid homeostasis are complex, and further studies are needed to define the precise mechanisms through which intestinal PXR modulates lipid homeostasis in animal models as well as in humans.
It is intriguing that TBC can only activate intestinal PXR but not hepatic PXR. Several tissue-specific PXR ligands have been identified (35, 58, 59). For example, rifaximin, a nonsynthetic antibiotic, has been shown to be a potent intestine-specific PXR ligand (58). The tissue-specific effect of rifaximin on intestinal PXR was probably related to its poor absorption as rifaximin accumulated in the intestine after oral treatment. However, the reason that TBC cannot activate hepatic PXR is unlikely due to poor absorption by the intestine as TBC was unable to active hepatic PXR even after ip delivery. Further, TBC treatment did not affect PXR activity in HepaRG and HepG2 cells in vitro. It is possible that hepatic cells can rapidly catalyze TBC. Further studies, including development of appropriate HPLC-tandem mass spectrometry assays and examination of TBC metabolites in vivo and in vitro, will be required to elucidate the detailed mechanisms underlying the tissue-specific effects of TBC and similar chemicals. In addition, we previously demonstrated that tocotrienols, members of the vitamin E family, show tissue-specific induction of PXR target genes, particularly CYP3A4 (35). Tocotrienols can up-regulate expression of CYP3A4 in human primary hepatocytes but not in LS180 cells. Nuclear receptor coregulators play critical roles in nuclear receptor activation and are also involved in the mechanisms underlying the divergent activities of selective estrogen receptor modulators (60, 61). We found that NCoR is expressed at different levels in intestinal LS180 cells and primary hepatocytes and that tocotrienols can only partially disrupt the interaction of unliganded PXR and NCoR in LS180 cells (35). Therefore, it is plausible that the coregulator difference between liver and intestine may contribute to the tissue-specific effects of TBC.
A fundamental question about all EDC studies is whether low-dose exposure to EDCs can influence human endocrine functions and cause adverse effects. Currently, there is little information about human exposure to TBC. The dose of 10 mg/kg/d TBC used in the present study to treat animals was based on human exposure to other plasticizers such as DEHP, DnBP, and ATBC. A retrospective human biomonitoring study of German adults aged 20 to 29 years showed median daily intakes for DnBP and DEHP of 7 and 4 mg/kg BW/d, respectively. Fourteen percent of subjects showed DnBP intakes above the tolerable daily intake value of 10 mg/kg BW/d set by the European Food Safety Authority (62, 63). Patients taking drugs containing another citrate ester plasticizer, ATBC, may be exposed to as much as 20 mg of ATBC daily (45). The 10 mg/kg BW/d TBC dose used in our study was also significantly lower than concentrations experimentally used for DEHP (1000 mg/kg BW/d) and DnBP (2000 mg/kg BW/d) to treat mice in some studies (2, 64). We have also examined whether lower TBC concentrations can activate PXR in vivo by testing 2 lower doses, 2.5 and 5 mg/kg/d, but neither dose activated PXR. Therefore, the 10 mg/kg/d dose used in this study is close to the lower limit of the TBC concentration that activates PXR in vivo. Considering the wide use of TBC in consumer products (eg, plastic wrap, toys, and drugs), we believe that the dose, which was sufficient to activate PXR activity and cause adverse effects in vivo, was reasonable. It would be interesting to measure the internal TBC concentrations in human samples and correlate internal TBC concentrations to plasma lipid levels in the future. Such information would provide important insights about the relationship of EDC exposure and disease outcomes in humans.
In addition to TBC, we and others have identified many other environmentally significant chemicals (eg, BPA, bisphenol B, and DEHP) as PXR agonists. Further, we recently demonstrated that certain EDCs can synergistically activate human PXR (12). The synergism between different EDCs supports the need to include mixtures for future in vitro and in vivo studies, which may have important implications for environmental chemical risk assessment. In addition to CVD, activation of PXR has been shown to induce tumor aggressiveness in humans and mice (65). Further studies are needed to investigate whether TBC-mediated intestinal PXR activation can induce tumorigenesis in animal models.
In summary, we have demonstrated that the phthalate substitute plasticizer TBC is a potent agonist of PXR. Of particular interest is the finding that TBC activated intestinal PXR but did not affect hepatic PXR activity. Nevertheless, activation of intestinal PXR by TBC was sufficient to increase plasma cholesterol levels in mice. We then identified the intestinal cholesterol transporter NPC1L1 as a direct transcriptional target of PXR and found that activation of PXR increased cholesterol uptake by intestinal cells. These findings demonstrate a previously unrecognized role of PXR in the regulation of intestinal lipid homeostasis. Findings from this study will hopefully stimulate further investigations of phthalate substitute plasticizers, in particular, the mechanisms by which TBC and other chemicals activate PXR, the tissue-specific TBC activation of PXR, and PXR regulation of intestinal lipid homeostasis. Activation of PXR should be taken into consideration for future risk assessment of phthalate substitute plasticizers and related environmental chemicals.
Acknowledgments
This work was supported in part by the National Institutes of Health (Grants R01ES023470, R21ES022745, P20GM103527, and T32DK007778) and the American Heart Association (Grant 14POST18740064).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ATBC
- acetyl tributyl citrate
- ATEC
- acetyl triethyl citrate
- BPA
- bisphenol A
- BW
- body weight
- ChIP
- chromatin immunoprecipitation
- CVD
- cardiovascular disease
- CYP
- cytochrome P450
- DEHP
- di(2-ethylhexyl) phthalate
- DEP
- diethyl phthalate
- DiBP
- di-isobutyl phthalate
- DiNP
- diisononyl phthalate
- DMSO
- dimethyl sulfoxide
- DBD
- DNA-binding domain
- DBP
- dibutyl phthalate
- EDC
- endocrine-disrupting chemicals
- EMSA
- electrophoretic mobility shift assay
- FDA
- Food and Drug Administration
- FXR
- farnesoid X receptor
- HDL
- high-density lipoprotein
- LDL
- low-density lipoprotein
- m
- mouse
- LXR
- liver X receptor
- MDR1
- multidrug resistance 1
- NCoR
- nuclear receptor corepressor
- NPC1L1
- Niemann-Pick C1-like 1
- PBP
- peroxisome proliferator–activated receptor binding protein
- PCB
- polychlorinated biphenyl
- PCN
- pregnane 16α-carbonitrile
- PPAR
- peroxisome proliferator–activated receptor
- PXR
- pregnane X receptor
- qPCR
- quantitative real-time PCR
- RAR
- retinoid acid receptor
- RIF
- rifampin
- RXR
- retinoid X receptor
- siRNA
- small interfering RNA
- SMRT
- silencing mediator of retinoid and thyroid hormone
- TBC
- tributyl citrate
- TEC
- triethyl citrate
- VLDL
- very low-density lipoprotein
- VDR
- vitamin D receptor
- WT
- wild type.
References
- 1. Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Casals-Casas C, Desvergne B. Endocrine disruptors: from endocrine to metabolic disruption. Annu Rev Physiol. 2011;73:135–162. [DOI] [PubMed] [Google Scholar]
- 3. vom Saal FS, Myers JP. Bisphenol a and risk of metabolic disorders. JAMA. 2008;300:1353–1355. [DOI] [PubMed] [Google Scholar]
- 4. Lang IA, Galloway TS, Scarlett A, et al. Association of urinary bisphenol a concentration with medical disorders and laboratory abnormalities in adults. JAMA. 2008;300:1303–1310. [DOI] [PubMed] [Google Scholar]
- 5. Melzer D, Osborne NJ, Henley WE, et al. Urinary bisphenol a concentration and risk of future coronary artery disease in apparently healthy men and women. Circulation. 2012;125:1482–1490. [DOI] [PubMed] [Google Scholar]
- 6. Lind L, Lind PM. Can persistent organic pollutants and plastic-associated chemicals cause cardiovascular disease? J Intern Med. 2012;271:537–553. [DOI] [PubMed] [Google Scholar]
- 7. Melzer D, Rice NE, Lewis C, Henley WE, Galloway TS. Association of urinary bisphenol a concentration with heart disease: evidence from NHANES 2003/06. PLoS One. 2010;5:e8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nagaoka S, Miyazaki H, Aoyama Y, Yoshida A. Effects of dietary polychlorinated biphenyls on cholesterol catabolism in rats. Br J Nutr. 1990;64:161–169. [DOI] [PubMed] [Google Scholar]
- 9. Arsenescu V, Arsenescu RI, King V, Swanson H, Cassis LA. Polychlorinated biphenyl-77 induces adipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ Health Perspect. 2008;116:761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lind PM, van Bavel B, Salihovic S, Lind L. Circulating levels of persistent organic pollutants (POPs) and carotid atherosclerosis in the elderly. Environ Health Perspect. 2012;120:38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lind PM, Lind L. Circulating levels of bisphenol A and phthalates are related to carotid atherosclerosis in the elderly. Atherosclerosis. 2011;218:207–213. [DOI] [PubMed] [Google Scholar]
- 12. Sui Y, Ai N, Park SH, et al. Bisphenol A and its analogues activate human pregnane X receptor. Environ Health Perspect. 2012;120:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tabb MM, Kholodovych V, Grün F, Zhou C, Welsh WJ, Blumberg B. Highly chlorinated PCBs inhibit the human xenobiotic response mediated by the steroid and xenobiotic receptor (SXR). Environ Health Perspect. 2004;112:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Masuyama H, Hiramatsu Y, Kunitomi M, Kudo T, MacDonald PN. Endocrine disrupting chemicals, phthalic acid and nonylphenol, activate pregnane X receptor-mediated transcription. Mol Endocrinol. 2000;14:421–428. [DOI] [PubMed] [Google Scholar]
- 15. Zhou C, Verma S, Blumberg B. The steroid and xenobiotic receptor (SXR), beyond xenobiotic metabolism. Nucl Recept Signal. 2009;7:e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blumberg B, Sabbagh W, Jr, Juguilon H, et al. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 1998;12:3195–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kliewer SA, Moore JT, Wade L, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. [DOI] [PubMed] [Google Scholar]
- 18. Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23:687–702. [DOI] [PubMed] [Google Scholar]
- 19. Zhou C, King N, Chen KY, Breslow JL. Activation of PXR induces hypercholesterolemia in wild-type and accelerates atherosclerosis in apoE deficient mice. J Lipid Res. 2009;50:2004–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Haan W, de Vries-van der Weij J, Mol IM, et al. PXR agonism decreases plasma HDL levels in ApoE3-Leiden.CETP mice. Biochim Biophys Acta. 2009;1791:191–197. [DOI] [PubMed] [Google Scholar]
- 21. Zhou J, Zhai Y, Mu Y, et al. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem. 2006;281:15013–15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ricketts ML, Boekschoten MV, Kreeft AJ, et al. The cholesterol-raising factor from coffee beans, cafestol, as an agonist ligand for the farnesoid and pregnane X receptors. Mol Endocrinol. 2007;21:1603–1616. [DOI] [PubMed] [Google Scholar]
- 23. Cheng J, Krausz KW, Tanaka N, Gonzalez FJ. Chronic exposure to rifaximin causes hepatic steatosis in pregnane X receptor-humanized mice. Toxicol Sci. 2012;129:456–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Helsley RN, Sui Y, Ai N, Park SH, Welsh WJ, Zhou C. Pregnane X receptor mediates dyslipidemia induced by the HIV protease inhibitor amprenavir in mice. Mol Pharmacol. 2013;83:1190–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khogali AM, Chazan BI, Metcalf VJ, Ramsay JH. Hyperlipidaemia as a complication of rifampicin treatment. Tubercle. 1974;55:231–233. [DOI] [PubMed] [Google Scholar]
- 26. Carr A, Samaras K, Chisholm DJ, Cooper DA. Pathogenesis of HIV-1-protease inhibitor-associated peripheral lipodystrophy, hyperlipidaemia, and insulin resistance. Lancet. 1998;351:1881–1883. [DOI] [PubMed] [Google Scholar]
- 27. Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med. 2005;6:421–425. [DOI] [PubMed] [Google Scholar]
- 28. Eirís JM, Lojo S, Del Río MC, et al. Effects of long-term treatment with antiepileptic drugs on serum lipid levels in children with epilepsy. Neurology. 1995;45:1155–1157. [DOI] [PubMed] [Google Scholar]
- 29. Lu Y, Feskens EJ, Boer JM, Müller M. The potential influence of genetic variants in genes along bile acid and bile metabolic pathway on blood cholesterol levels in the population. Atherosclerosis. 2010;210:14–27. [DOI] [PubMed] [Google Scholar]
- 30. He J, Gao J, Xu M, et al. PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes. 2013;62:1876–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moreau A, Téruel C, Beylot M, et al. A novel pregnane X receptor and S14-mediated lipogenic pathway in human hepatocyte. Hepatology. 2009;49:2068–2079. [DOI] [PubMed] [Google Scholar]
- 32. Roth A, Looser R, Kaufmann M, et al. Regulatory cross-talk between drug metabolism and lipid homeostasis: constitutive androstane receptor and pregnane X receptor increase Insig-1 expression. Mol Pharmacol. 2008;73:1282–1289. [DOI] [PubMed] [Google Scholar]
- 33. Zhou C, Poulton EJ, Grün F, et al. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol Pharmacol. 2007;71:220–229. [DOI] [PubMed] [Google Scholar]
- 34. Zhou C, Tabb MM, Nelson EL, et al. Mutual repression between steroid and xenobiotic receptor and NF-κB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest. 2006;116:2280–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou C, Tabb MM, Sadatrafiei A, Grün F, Blumberg B. Tocotrienols activate the steroid and xenobiotic receptor, SXR, and selectively regulate expression of its target genes. Drug Metab Dispos. 2004;32:1075–1082. [DOI] [PubMed] [Google Scholar]
- 36. Teupser D, Persky AD, Breslow JL. Induction of atherosclerosis by low-fat, semisynthetic diets in LDL receptor-deficient C57BL/6J and FVB/NJ mice: comparison of lesions of the aortic root, brachiocephalic artery, and whole aorta (en face measurement). Arterioscler Thromb Vasc Biol. 2003;23:1907–1913. [DOI] [PubMed] [Google Scholar]
- 37. Sui Y, Xu J, Rios-Pilier J, Zhou C. Deficiency of PXR decreases atherosclerosis in apoE-deficient mice. J Lipid Res. 2011;52:1652–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou C, Assem M, Tay JC, et al. Steroid and xenobiotic receptor and vitamin D receptor crosstalk mediates CYP24 expression and drug-induced osteomalacia. J Clin Invest. 2006;116:1703–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nassir F, Wilson B, Han X, Gross RW, Abumrad NA. CD36 is important for fatty acid and cholesterol uptake by the proximal but not distal intestine. J Biol Chem. 2007;282:19493–19501. [DOI] [PubMed] [Google Scholar]
- 40. Zhou C, Pridgen B, King N, Xu J, Breslow JL. Hyperglycemic Ins2AkitaLdlr−/− mice show severely elevated lipid levels and increased atherosclerosis: a model of type 1 diabetic macrovascular disease. J Lipid Res. 2011;52:1483–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cai L, Eckhardt ER, Shi W, et al. Scavenger receptor class B type I reduces cholesterol absorption in cultured enterocyte CaCo-2 cells. J Lipid Res. 2004;45:253–262. [DOI] [PubMed] [Google Scholar]
- 42. Sui Y, Park SH, Xu J, et al. IKKβ links vascular inflammation to obesity and atherosclerosis. J Exp Med. 2014;211:869–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takeshita A, Koibuchi N, Oka J, Taguchi M, Shishiba Y, Ozawa Y. Bisphenol-A, an environmental estrogen, activates the human orphan nuclear receptor, steroid and xenobiotic receptor-mediated transcription. Eur J Endocrinol. 2001;145:513–517. [DOI] [PubMed] [Google Scholar]
- 44. DeKeyser JG, Laurenzana EM, Peterson EC, Chen T, Omiecinski CJ. Selective phthalate activation of naturally occurring human constitutive androstane receptor splice variants and the pregnane X receptor. Toxicol Sci. 2011;120:381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takeshita A, Igarashi-Migitaka J, Nishiyama K, Takahashi H, Takeuchi Y, Koibuchi N. Acetyl tributyl citrate, the most widely used phthalate substitute plasticizer, induces cytochrome p450 3a through steroid and xenobiotic receptor. Toxicol Sci. 2011;123:460–470. [DOI] [PubMed] [Google Scholar]
- 46. Kanebratt KP, Andersson TB. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab Dispos. 2008;36:1444–1452. [DOI] [PubMed] [Google Scholar]
- 47. Abumrad NA, Davidson NO. Role of the gut in lipid homeostasis. Physiol Rev. 2012;92:1061–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nauli AM, Nassir F, Zheng S, et al. CD36 is important for chylomicron formation and secretion and may mediate cholesterol uptake in the proximal intestine. Gastroenterology. 2006;131:1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Davis HR, Jr, Zhu LJ, Hoos LM, et al. Niemann-Pick C1 like 1 (NPC1L1) is the intestinal phytosterol and cholesterol transporter and a key modulator of whole-body cholesterol homeostasis. J Biol Chem. 2004;279:33586–33592. [DOI] [PubMed] [Google Scholar]
- 50. Altmann SW, Davis HR, Jr, Zhu LJ, et al. Niemann-Pick C1 like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303:1201–1204. [DOI] [PubMed] [Google Scholar]
- 51. Jia L, Betters JL, Yu L. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu Rev Physiol. 2011;73:239–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yu L, Gupta S, Xu F, et al. Expression of ABCG5 and ABCG8 is required for regulation of biliary cholesterol secretion. J Biol Chem. 2005;280:8742–8747. [DOI] [PubMed] [Google Scholar]
- 53. Finkelstein M, Gold H. Toxicology of the citric acid esters: tributyl citrate, acetyl tributyl citrate, triethyl citrate, and acetyl triethyl citrate. Toxicology. 1959;1:283–298. [DOI] [PubMed] [Google Scholar]
- 54. Meyers DB, Autian J, Guess WL. Toxicity of plastics used in medical practice. II. Toxicity of citric acid esters used as plasticizers. J Pharm Sci. 1964;53:774–777. [DOI] [PubMed] [Google Scholar]
- 55. El-Gendy NA. Pharmaceutical plasticizers for drug delivery systems. Curr Drug Deliv. 2012;9:148–163. [DOI] [PubMed] [Google Scholar]
- 56. Hirata-Koizumi M, Takahashi M, Matsumoto M, Kawamura T, Ono A, Hirose A. Toxicity effects of phthalate substitute plasticizers used in toys (in Japanese). Kokuritsu Iyakuhin Shokuhin Eisei Kenkyusho Hokoku. 2012;130:31–42. [PubMed] [Google Scholar]
- 57. Myocardial Infarction Genetics Consortium I, Stitziel NO, Won HH, et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371:2072–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ma X, Shah YM, Guo GL, et al. Rifaximin is a gut-specific human pregnane X receptor activator. J Pharmacol Exp Ther. 2007;322:391–398. [DOI] [PubMed] [Google Scholar]
- 59. Cheng J, Shah YM, Ma X, et al. Therapeutic role of rifaximin in inflammatory bowel disease: clinical implication of human pregnane X receptor activation. J Pharmacol Exp Ther. 2010;335:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 61. McDonnell DP, Connor CE, Wijayaratne A, Chang CY, Norris JD. Definition of the molecular and cellular mechanisms underlying the tissue-selective agonist/antagonist activities of selective estrogen receptor modulators. Recent Prog Horm Res. 2002;57:295–316. [DOI] [PubMed] [Google Scholar]
- 62. Halden RU. Plastics and health risks. Annu Rev Public Health. 2010;31:179–194. [DOI] [PubMed] [Google Scholar]
- 63. Wittassek M, Wiesmüller GA, Koch HM, et al. Internal phthalate exposure over the last two decades—a retrospective human biomonitoring study. Int J Hyg Environ Health. 2007;210:319–333. [DOI] [PubMed] [Google Scholar]
- 64. Lapinskas PJ, Brown S, Leesnitzer LM, et al. Role of PPARα in mediating the effects of phthalates and metabolites in the liver. Toxicology. 2005;207:149–163. [DOI] [PubMed] [Google Scholar]
- 65. Wang H, Venkatesh M, Li H, et al. Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice. J Clin Invest. 2011;121:3220–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]