

Abstract
Background
The spectrum of RB1gene mutations in Retinoblastoma (RB) patients and the necessity of multiple traditional methods for complete variant analysis make the molecular diagnosis a cumbersome, labor-intensive and time-consuming process. Here, we have used targeted next generation sequencing (NGS) approach with in-house analysis pipeline to explore its potential for the molecular diagnosis of RB.
Methods
Thirty-three patients with RB and their family members were selected randomly. DNA from patient blood and/or tumor was used for RB1 gene targeted sequencing. The raw reads were obtained from Illumina Miseq. An in-house bioinformatics pipeline was developed to detect both single nucleotide variants (SNVs) and small insertions/deletions (InDels) and to distinguish between somatic and germline mutations. In addition, ExomeCNV and Cn. MOPS were used to detect copy number variations (CNVs). The pathogenic variants were identified with stringent criteria, and were further confirmed by conventional methods and cosegregation in families.
Results
Using our approach, an array of pathogenic variants including SNVs, InDels and CNVs were detected in 85% of patients. Among the variants detected, 63% were germline and 37% were somatic. Interestingly, nine novel pathogenic variants (33%) were also detected in our study.
Conclusions
We demonstrated for the first time that targeted NGS is an efficient approach for the identification of wide spectrum of pathogenic variants in RB patients. This study is helpful for the molecular diagnosis of RB in a comprehensive and time-efficient manner.
Keywords: Retinoblastoma, Targeted next generation sequencing, Molecular diagnosis
Background
Retinoblastoma (RB, OMIM#180200), the most common pediatric eye tumor in the retina is initiated by inactivating biallelic variants of RB1 gene [1]. Retinoblastoma occurs in hereditary and non-hereditary forms, with most bilateral and some unilateral RB cases being hereditary. The non-heritable form predominantly leads to unilateral tumors where in both variants have occurred in somatic cells and are not transmitted [2]. It is essential to identify and distinguish the germline and somatic variations in RB1 for predicting the accurate risk of RB in future siblings and offsprings. The retinoblastoma susceptibility gene, RB1 (Genbank accession number L11910.1; NCBI RefSeq NM_000321.2) is located on chromosome 13q14.2 and is composed of 27 exons distributed along 183 kb of genomic sequence. A wide spectrum of heterogeneous RB1 gene variants that includes – single nucleotide variations (SNVs), small insertions/deletions (InDels) and structural variations (SVs) had been reported in RB patients [3]. Some of the variants such as nonsense and frameshift are associated with bilateral RB, while other types have unilateral RB or milder phenotypic expression [4].
Predictive genetic testing of RB can help to save the vision and avoid unnecessary (and invasive) eye examinations for patients and their close relatives in a cost effective manner. Currently, the routine procedure for genetic testing of RB1 involves multiple methods of mutation detection in the coding regions and intron-exon boundaries using Sanger sequencing, and deletion/duplication analysis by genotyping methods such as multiplex ligation-dependent probe amplification (MLPA), quantitative multiplex PCR (QMPCR) [5]. The major limitations of Sanger sequencing are the extended time taken for screening all 27 exons individually and limited data (2X) generated from the sequencing runs. Thus, identifying the spectrum of heterogeneous variants in RB1 gene makes the molecular diagnosis of RB challenging and time-consuming.
Accurate identification of RB1 pathogenic variants in a reduced time is very important for diagnosis, confirmation, genetic counseling, risk assessment, and carrier screening of RB patients and their family members. Next Generation sequencing (NGS) has been found to be a time-efficient and accurate approach for the molecular diagnosis of simple to complex diseases including cancer [6-8]. Due to this improved efficiency, NGS has been widely used as diagnostic tool for retinal dystrophies [9-12]. In the present study, we have used targeted next generation sequencing approach with in-house bioinformatics pipeline for the molecular diagnosis of RB for the first time.
Methods
Clinical diagnosis and patient samples
A total of 21 families with bilateral RB and 12 families with unilateral RB were selected for this study (Table 1). The clinical diagnosis of RB was made by thorough clinical examination and radiological investigations (CT/MRI and USG B scan) along with Retcam imaging in Aravind Eye Hospital Madurai, India. Retinal examination was performed in family members to detect small scars/pigmentary changes, which are suggestive of regressed RB. The blood samples were collected from patients and family members. In addition, fresh tumor samples were collected from enucleated patient eyes. The present study was approved by the Institutional Ethics Committee of Aravind Medical Research Foundation, Madurai, India (Registration Number: ECR/182/Arvind/Inst/TN/2013). All the patient samples were collected after getting the informed consent from the families.
Table 1.
Clinical & family history of RB patients and samples selected for NGS
| Patient no | Age (months)/Sex | Laterality | Family history | Samples analysed by NGS |
|---|---|---|---|---|
| RB1 | 4/F | Bilateral | Father with regressed RB | Patient’s blood |
| RB2 | 1/F | Bilateral | Father with regressed RB | Patient’s blood |
| RB3 | 33/F | Bilateral | Nil | Patient’s blood |
| RB4 | 4/M | Bilateral | Mother and sibling affected with RB | Patient’s blood |
| RB5 | 0/F | Bilateral | Nil | Patient’s blood |
| RB6 | 31/F | Bilateral | Three siblings affected with RB | Patient’s blood |
| RB7 | 39/F | Bilateral | Father affected with RB | Patient’s and Father’s blood |
| RB8 | 44/M | Unilateral | Nil | Patient’s blood and tumor |
| RB9 | 7/F | Unilateral | Nil | Patient’s tumor |
| RB10 | 0/M | Unilateral | Nil | Patient’s tumor |
| RB11 | 5/F | Unilateral | One sibling affected with RB | Patient’s and Sibling’s blood |
| RB12 | 14/M | Unilateral | Nil | Patient’s blood and tumor |
| RB13 | 12/M | Bilateral | Nil | Patient’s blood |
| RB14 | 44/F | Bilateral | Father and sibling affected with RB | Patient’s blood |
| RB15 | 11/M | Bilateral | Father affected with RB | Patient’s blood |
| RB16 | 0/M | Bilateral | Nil | Patient’s blood |
| RB17 | 0/M | Bilateral | Father and sibling affected with RB | Patient’s blood |
| RB18 | 26/F | Bilateral | Nil | Patient’s blood |
| RB19 | 36/F | Bilateral | Nil | Patient’s blood |
| RB20 | 31/M | Bilateral | Nil | Patient’s blood |
| RB21 | 12/M | Unilateral | Nil | Patient’s tumor |
| RB22 | 18/F | Unilateral | Nil | Patient’s tumor |
| RB23 | 19/M | Bilateral | Father and sibling affected with RB | Patient’s blood |
| RB24 | 26/F | Bilateral | Nil | Patient’s blood |
| RB25 | 8/F | Bilateral | Nil | Patient’s blood and tumor |
| RB26 | 12/F | Bilateral | Nil | Patient’s blood |
| RB27 | 66/F | Bilateral | Nil | Patient’s blood |
| RB28 | 58/F | Unilateral | Nil | Patient’s tumor |
| RB29 | 28/M | Unilateral | Nil | Patient’s tumor |
| RB30 | 18/M | Unilateral | Nil | Patient’s tumor |
| RB31 | 88/M | Unilateral | Nil | Patient’s tumor |
| RB32 | 27/M | Unilateral | Nil | Patient’s tumor |
| RB33 | 3/M | Bilateral | Nil | Patient’s blood |
The age at which first sign was detected is given in months. Laterality was confirmed by the clinical investigations and imaging (CT Scan/MRI, Ultrasound, Retcam). Family history was ascertained by three or four generation pedigree. NGS was performed on patient’s tumor wherever available. In three patients (RB8, RB12 and RB25), both blood and tumor samples were analyzed. In two families, affected members were also included for NGS along with patients (RB7 and RB11).
DNA isolation
Genomic DNA was isolated from blood samples (2 ml for patients and 5 ml for parents) by salting out method [13] and tumor by QIAamp® DNA Mini Kit (Qiagen, Germantown, MD) following the manufacturer’s protocol. The quality and quantity of the DNA was checked by Nanodrop 1000 spectrophotometer (Thermo Scientific, Waltham, USA).
Library preparation and targeted next generation sequencing
Targeted NGS was performed in total of 33 patients. Of those, 12 were tumor and 21 were blood samples. In three patients, tumor/blood matched pairs were included. In two families, the affected family members along with the patient were also analysed (Table 1). A Primer library was custom-designed to amplify 27 exons, exon/intron boundaries and promoter region of RB1 gene using the Illumina Truseq custom Amplicon and Agilent SureSelect in-solution hybridization capture kits by the service provider (Scigenom, Kochi, India). Briefly, 2 μg of each genomic DNA was sheared into 100-500 bp fragments. Regions of interest were enriched using the above methods and libraries were prepared. The high sensitivity DNA chips were used in Agilent Bioanalyzer, to validate the enrichment process. Quantitative PCR was used to measure the quantity of the library before sequencing. Captured libraries were sequenced in a multiplexed fashion on Miseq with paired end run to obtain 2×150 bp reads with at least 100X depth of coverage. The coding region with <20X depth of coverage were covered by Sanger sequencing.
SNV and InDel detection
Obtained raw sequence reads from Miseq were analysed using bioinformatics pipeline as shown in Figure 1. Data was quality filtered using fastQC tool [14]. The filtered reads were mapped to Hg19 reference sequence using Burrow-Wheeler Aligner (BWA version 0.7.5a-r405) [15]. Resulting BAM files were locally realigned using GenomeAnalysisTK-3.1-1 (GATK) Indel-realigner [16] tool to minimize the mismatches across the reads. GATK haplotype caller was performed to retrieve germline single nucleotide variants (SNVs) and small insertions/deletions (InDels) with phred score 20 and minimum depth 5 from all the samples. MuTect-1.1.4 [17] and GATK Indelocator tools were used to identify somatic SNVs and InDels from the tumor samples with blood matched control respectively. Wherever matched blood sample is not available, the blood sample with similar coverage was used. All the SNVs and InDels were subjected to identify rare and potential variants. The rare variants were identified using ANNOVAR [18] by filtering common variants with alternative allelic frequency higher than 1% based on 1000 Genomes project (http://www.1000genomes.org/data), dbSNP135 (http://www.ncbi.nlm.nih.gov//SNP) and ESP (http://evs.gs.washington.edu/EVS). Of those, non-synonymous/synonymous SNVs, coding InDels, and intronic variants that were less than 10 bp beyond the canonical splice site junction were selected. The potential variants were identified using ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), COSMIC (http://cancer.sanger.ac.uk/cosmic) and In-house (reported pathogenic variants) databases. The detection of germline SNVs and Indels was fully automated. Detected variants were further manually assessed with the help of IGV-2.3.25 viewer to avoid mapping errors.
Figure 1.
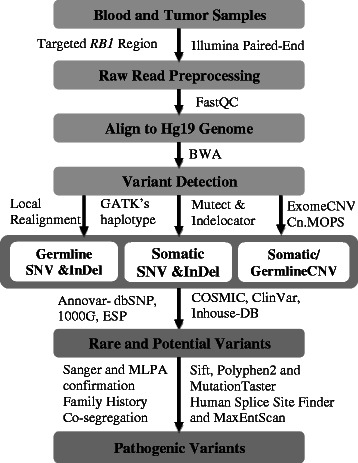
Analysis pipeline to identify pathogenic variants in tumor and blood samples from retinoblastoma patients.
Copy number variation (CNV) detection
The locally realigned BAM files were used to detect CNV. ExomeCNV [19], a statistical tool that uses coverage and alternative allele frequencies to estimate CNV, was used to detect somatic CNVs from the tumor/blood pairs as described above. Whereas, Cn. MOPS [20], a read count based CNV caller was used to detect the germline CNVs in the blood samples. More than five blood samples with similar exon coverage were used to improve the sensitivity of Cn. MOPS. Log Ratios (LogR) ≥ ±1 were set for Deletion/Duplication analysis in both the tools; median LogR score were used for Cn. MOPS.
Identification of pathogenic variants
In order to identify pathogenic variants, we used the following criteria. i) known pathogenic variants; ii) if not, variants that could give rise to premature protein termination, frameshift, canonical splice site alterations and large exonic deletions; iii) nonsynonymous SNVs if Sift [21], Polyphen2 [22] and MutationTaster [23] all suggested pathogenic, and iv) splice variants selected from both Human splice site finder [24] and MaxEntScan [25].
Confirmation of variants by Sanger sequencing and MLPA
All the pathogenic variants were confirmed by Sanger sequencing, MLPA and cosegregation analysis in blood and tumor samples of patients, and blood samples of family members. PCR amplification of the corresponding exon around the variant site of the RB1 gene was performed. Each 25 ul reaction contained 20 ng of genomic DNA, 10XPCR Buffer, 100 mM dNTPS, 10uM of each forward and reverse primer and 1U of Taq DNA polymerase (Sigma Aldrich, Missouri, USA) with cycling conditions and PCR primers described previously [26]. Cycle sequencing was performed using the BigDye Terminator kit version 3.1 and purified products were analyzed on a 3130 Genetic Analyzer (Life Technologies, USA). MLPA was performed with SALSA MLPA kit P047-RB1 kit (MRC-Holland, Amsterdam, The Netherlands) according to manufacturer’s instructions. Fragment analysis was performed with Gene Mapper software (Life Technologies, USA) and data was analyzed using Coffalyser software (MRC, Holland) where DNA copy number ratios of tumor samples were computed using the matched blood sample. For genes targeted by multiple probes, copy number ratios were averaged. A threshold ratio of >1.3 denotes duplication and a ratio of <0.7 denotes deletion. Size fractionation was carried out by agarose gel electrophoresis to confirm deletions ranging from 10 to 30 bp.
Targeted sequencing using Ion-Torrent Personal Genome machine (PGM)
In order to compare the data obtained from Miseq, eight patients were selected randomly for the cross platform comparison. Of those, six were blood from patients RB2, RB4, RB7, RB13, RB24, RB25 and two were tumor/blood matched pairs of samples from RB8 and RB12. Sequencing was performed with Ion-Torrent PGM at University of Pennsylvania, Philadelphia using the protocol as described [27]. Briefly, multiplex PCR was performed to generate the PCR fragments of all 27 exons of RB1 using Qiagen multiplex PCR kit. Ion Xpress Plus gDNA Fragment Library Preparation kit was used for shearing, adapter ligation and nick repair. Emulsion PCR was performed with One Touch2 system and enrichment with One Touch ES using 200 bp chemistry. Purification was performed at each step with Ampure beads (Agencourt) and quality was checked using the Bioanalyzer. The final enriched libraries were sequenced on Ion PGM with 318 chip. The sequence reads were aligned against the human RB1 genomic sequence [GenBank Accession L11910.1] and variant calling were made using Ion Torrent Suite (Life Technologies) as described [27]. The reads were automatically barcode-sorted followed by removal of the reads with low quality. The BAM and BAI files of Ion PGM runs were checked visually on IGV-2.3.25 viewer to avoid sequencing errors associated with homopolymer regions.
Results
Targeted sequencing characteristics
Thirty three patients with RB as shown in Table 1 from unrelated Indian families were selected for this study of targeted RB1 sequencing. Illumina Truseq Custom Amplicon was used for target amplification in 23 samples (RB1-RB23) and Agilent SureSelect enrichment method was used for other 10 samples (RB24-RB33). Paired end sequencing in Miseq covered nearly 3000 bases of RB1 encompassing 27 exons along with their flanking intron and promoter regions. The mean depth of coverage was found to be ~ 200X with Truseq and ~150X with SureSelect. The average sequencing coverage of the targeted regions was 98.0% in Truseq compared to 99.8% in SureSelect as exons 14 and 20 were not covered sufficiently (<20X) in Truseq. The missed regions were covered by Sanger sequencing. Therefore, complete coverage of all the target bases was ensured to provide high quality bases for sensitive and efficient variant detection. An automatic in-house variant calling pipeline was developed using freely available tools to detect germline SNVs and InDels for all the samples, wherein the tumor samples were checked for somatic SNVs and InDels. With stringent criteria, pathogenic SNVs and InDels were identified and patients with no pathogenic variants were further analysed for copy number variations (CNVs). ExomeCNV and Cn. MOPS were used to detect somatic and germline CNVs in tumor and blood samples respectively. All the pathogenic variants were further confirmed by conventional methods and cosegregation. Somatic events were re-confirmed by their absence in same patient blood sample.
Identification of germline SNVs and InDels in RB patients
Blood samples of 21 bilateral (familial and sporadic) patients and one familial unilateral patient (Table 1), were analyzed to detect SNVs and InDels. Pathogenic variants were identified in 15 patients, of which eight were novel and seven were previously reported (Table 2). Surprisingly, all the reported pathogenic variants were found to be nonsense variants, resulting in premature protein termination. Five of them were shown to be de novo as only the patient had the mutation and not the family members, and remaining two were inherited from one of their parents (Table 1 and 2).
Table 2.
RB1 variants identified by targeted NGS in blood samples of RB patients
| Patient no | cDNA change | Amino acid change | Functional consequence | Cosegregation in family |
|---|---|---|---|---|
| RB1 | c.-212_-195del | Promoter Deletion | Heterozygous Father | |
| RB2 | c.1399C > T | p.R467X | Premature Protein Termination | Heterozygous Father |
| RB4 | c.265-9 T > A | Altered Splicing | Heterozygous Mother and Sibling | |
| RB11 | c.46_74del | p.A16AfsX14 | Frameshift | Heterozygous Mother and three Siblings |
| RB13 | c.751C > T | p.R251X | Premature Protein Termination | De novo |
| RB14 | c.2520 + 4 A > G | Altered Splicing | Heterozygous Father | |
| RB15 | c.2115_2118del | p.M705IfsX8 | Frameshift | Heterozygous Father |
| RB16 | c.1363C > T | p.R455X | Premature Protein Termination | De novo |
| RB17 | c.1960 + 2 T > A | Altered Splicing | Heterozygous Father and Sibling | |
| RB18 | c.38_66del | p.A13AfsX17 | Frameshift | De novo |
| RB19 | c.1399 C > T | p.R467X | Premature Protein Termination | Heterozygous Mother |
| RB24 | c.1961_1963del | p.654_655del | Altered Splicing | De novo |
| RB25 | c.1072C > T | p.R358X | Premature Protein Termination | De novo |
| RB26 | c.521 T > A | p.L174X | Premature Protein Termination | De novo |
| RB27 | c.160G > T | p.E54X | Premature Protein Termination | De novo |
Novel variants are marked in bold. Cosegregation of the variants was confirmed by Sanger sequencing analysis of the variants in family members.
The novel pathogenic variants either caused aberrant splicing or frameshift due to deletions. Four variants identified in patients RB4, RB14, RB17 and RB24 were found to affect splicing based on the HSF and MaxEntScan tools (Table 2). Patient RB4 was found to have a heterozygous c.265-9 T > A intronic variant at the upstream of acceptor splice site, which was predicted to activate a cryptic splice site (9 bases prior to exon 3) that may result in frameshift. In case of patient RB14 and RB17, heterozygous intronic variants near canonical splice sites might result in altered splicing. Three patients RB4, RB14 and RB17 had affected members in the family showing the same variant as that of the patient. As an example, cosegregation of variant in the family of Patient RB4 was shown in Figure 2A. Interestingly, a de novo heterozygous in-frame deletion of three bases identified at the start site of exon 20 in patient RB24 was considered to be deleterious, which might result in splicing defect (Figure 2B).
Figure 2.
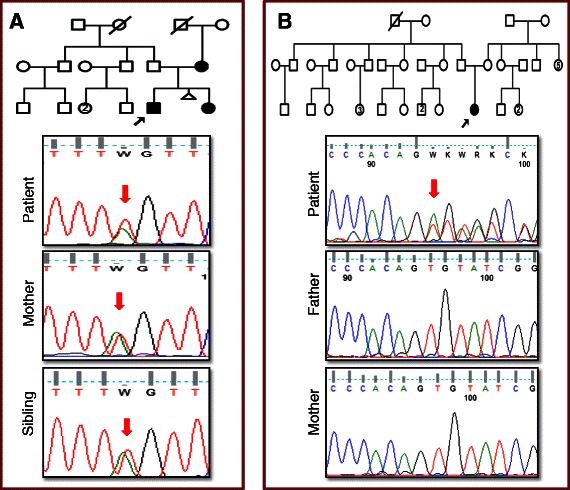
Confirmation of novel pathogenic splice variants. (A) Cosegregation of variants in the family was confirmed by Sanger sequencing of blood samples of Patient RB4, his mother and sibling, who had heterozygous c.265-9 T > A variant that created a new splice site acceptor. (B) Patient RB24 had a de novo heterozygous in-frame deletion of three bases identified at the start site of exon 20. Red arrows denote the variant.
A heterozygous deletion of 17 bases at the upstream of ORF in the promoter region was detected in patient RB1, which was confirmed by agarose gel electrophoresis. The deletion was also detected in father, who was diagnosed as regressed RB (Figure 3A). In addition, frameshift deletions were detected in three patients. One patient RB15 had deletion of four bases which was also detected in his father. A deletion of 29 bases in exon 1 was identified in patient RB18. In family of patient RB11, a deletion of 29 bases at another locus of exon 1 was observed in all members except father (Figure 3B), where one sibling was affected with RB (Table 1).
Figure 3.
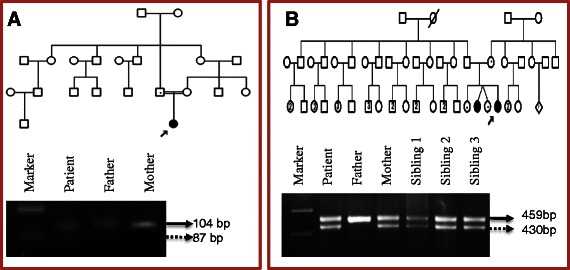
Agarose gel electrophoresis for the confirmation of small deletions. (A) 17 bp deletion in the Promoter region was observed in blood samples of patient RB1 and his father. (B) 29 bp deletion in Exon 1 was observed in blood samples of patient RB11, her mother and siblings. The size of actual and deleted product is indicated by straight and dotted arrows respectively in both gels.
Identification of somatic SNVs and InDels in RB tumor samples
Somatic variants were detected in tumor samples of 7 out of 11 patients with sporadic unilateral RB (Table 1 and Table 3). Homozygous variants were identified in 4 patients (RB10, RB12, RB22 and RB29) and two heterozygous variants were identified in other 3 patients (RB8, RB9 and RB31). Of the homozygous variants, three were nonsense variants in patient RB10, RB12 and RB29, while one novel frameshift variant was identified in Patient RB22. One nonsense and another splice site variant were identified in both Patients RB8 and RB9, and two nonsense variants were identified in patient RB31. In addition, a somatic loss of heterozygosity (LOH) was detected in tumor sample of Patient RB25, where the germline heterozygous nonsense variant (c.1072C > T) was converted to homozygous (Table 2). All the somatic variants and zygosity were confirmed by Sanger sequencing in patient tumor and blood samples. Our results are consistent with the Knudson’s two hit hypothesis [28] in all the patients as we have identified either homozygous or two heterozygous variants.
Table 3.
RB1 variants identified by targeted NGS in tumor samples of RB patient
| Patient no | cDNA change | Amino acid change | Functional consequence |
|---|---|---|---|
| RB9 | c.380G > A/c.1363C > T | p.S127N/p.R455X | Missense-Altered splicing/Premature Protein Termination |
| RB10 | c.763C > T* | p.R255X* | Premature Protein Termination |
| RB12 | c. 1072C > T* | p.R358X* | Premature Protein Termination |
| RB22 | c.1732_1733delGinsTT* | p.D578LfsX6 * | Frameshift |
| RB29 | c.1654C > T* | p.R552X* | Premature Protein Termination |
| RB31 | c.409 G > T/c.751 C > T | p.E137X/p.R251X | Premature Protein Termination Premature Protein Termination |
Novel variant is marked in bold. In patients RB10, RB12, RB22 and RB29, homozygous variants (marked with *) were identified. All the variants given in the table were somatic variants as they were detected only in patient’s tumor but not in blood samples of patient and family members.
Detection of copy number variations (CNVs)
Eleven samples with no pathogenic SNVs and InDels were subjected to the analysis of CNVs. For blood samples, we utilized the tool Cn. MOPS, which detected five heterozygous germline CNVs in four samples. Deletion found in each patient sample RB3, RB5, RB6, RB7 was confirmed by MLPA (Table 4). Of those, deletion of exon 10-12 in patient RB7 cosegregated with phenotype (Figure 4A). Another deletion (exon 22) in patient RB6 detected by Cn. MOPS, was not found by MLPA. Somatic deletions including a homozygous deletion of Exon10 in patient RB21 and a heterozygous deletion of Exons 7-27 in patient RB32 (Figure 4B) were observed using ExomeCNV, which were further confirmed by MLPA (Table 4). Overall, 80% and 100% sensitivity were observed in detecting germline and somatic CNVs respectively.
Table 4.
Copy number variations (CNVs) identified in tumor/blood samples of Retinoblastoma patients
| Patient no | CNV | logR | Method used | Cosegregation in family | MLPA confirmation |
|---|---|---|---|---|---|
| RB3 | Deletion of whole RB1 | -1.0 | Cn. MOPS | De novo | Yes |
| RB5 | Deletion of exons 4-6 | -1.0 | Cn. MOPS | De novo | Yes |
| RB6 | Deletion of exon 22 | -1.0 | Cn. MOPS | - | No |
| RB6 | Deletion of exons 24-25 | -5.5 | Cn. MOPS | Heterozygous Father | Yes |
| RB7 | Deletion of exons 10-12 | -1.2 | Cn. MOPS | Heterozygous Father | Yes |
| RB21 | Deletion of exon 10 | -5.4 | ExomeCNV | - | Yes |
| RB32 | Deletion of exons 7-27 | -1.1 | ExomeCNV | - | Yes |
Two programs, Cn. MOPS and ExomeCNV were used to identify germline and somatic CNVs from blood and tumor samples respectively. CNVs identified were confirmed and cosegregation was observed by MLPA. The exon 22 deletion identified in patient RB6 was not detected by MLPA. Except the exon 10 homozygous deletion identified in patient RB21, all other CNVs were heterozygous.
Figure 4.
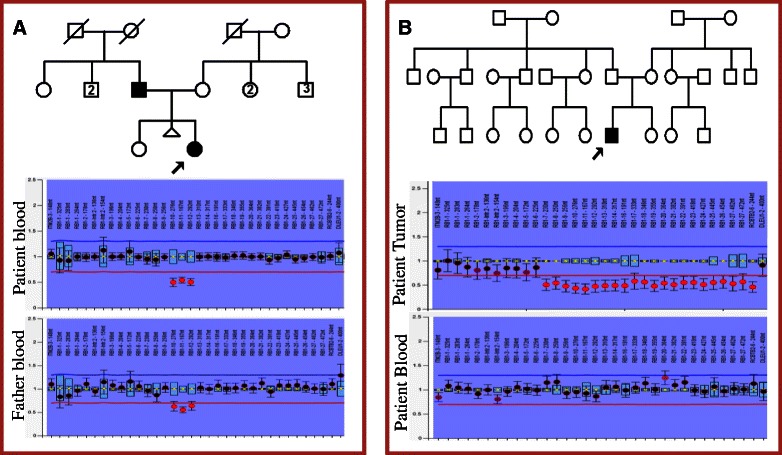
Confirmation of copy number variations (CNVs) by MLPA. (A) Patient RB7 had an affected father and both of them showed deletion of exons 10-12. (B) Patient RB32 had a somatic deletion of exons 7-27 which was not detected in blood. The deletions were denoted by the red spots below the deletion cut-off line (red) in the ratio chart.
Cross platform comparison of Illumina Miseq and ion-torrent PGM results
The five pathogenic variants detected in patients RB2, RB4, RB13, RB24 and RB25 (Table 2) were concordant with Ion PGM results. Both platforms detected somatic variants in tumor samples (Table 3) and their absence in blood samples of same patients (RB8 and RB12). However, deletion found in patient RB7 (Table 4) was not detected by Ion Torrent Suite. Further, analysis by Cn. Mops could not be carried out because of small sample size.
Unsolved cases
No pathogenic variants were detected in five patients (RB20, RB23, RB28, RB30 and RB33) with our approach. Rare variants not following our criteria for pathogenicity and deep intronic variant were excluded. For example, in two unsolved cases (RB20 and RB23) one missense variant (Exon19, c.A1846G, p.K616E) was detected. Although it was reported in Human Gene Mutation Database (HGMD) [29], it was not predicted as pathogenic with SIFT, Polyphen2 and MutationTaster tools. It was also observed in more than 12 patients with low coverage and not detected with Sanger sequencing. In another example, one deep intronic variant (exon 3, c.380 + 150 T > A), reported in COSMIC database (ID = COSM164493) and detected in patient RB30, RB23 and RB33 was excluded from the analysis. Moreover it was found to be polymorphism in dbSNP and observed in more than 80% of our patient samples.
Discussion
Retinoblastoma, the most common childhood intraocular tumor has complex genetic basis of cancer development, initiated by biallelic inactivation of RB1 gene [28]. Genetic testing of RB1 will be beneficial to provide counselling for families. However, genetic analysis of heterogeneous spectrum of variants in RB1 gene is no trivial task [4] and essentially requires comprehensive approach. Here, we have used NGS approach for the molecular analysis of Indian patients with RB, based on RB1 gene target enrichment, multiplexing and bioinformatics pipeline. We used in-house pipeline to successfully detect both pathogenic germline and somatic variants in RB patients. With our approach, we were able to identify heterogeneous spectrum of RB1 gene variants including SNVs, InDels and CNVs. All the variants detected were validated using Sanger sequencing, MLPA and size fractionation methods. Thus, our approach, achieving a diagnostic rate of 85%, proved to be efficient for the molecular diagnosis of RB. Moreover, the cross platform comparison with Ion-Torrent PGM results further confirmed the efficiency of NGS.
An important consideration about NGS for diagnosis is identifying the pathogenic variants among the large number of variants detected. In order to identify pathogenic variants, we used stringent criteria after several modifications during the pipeline development. The filtering process has been set to include synonymous and polymorphic variants as potential variants if they are present in cancer and disease databases. While those not present in any databases were classified as rare variants. By applying stringent criteria, we could detect known and novel pathogenic variants with no false positives. For example, a novel intronic variant (c.265-9 T > A) in patient RB4 (Figure 2A) creates a cryptic splice site and is most likely a pathogenic variant. We further confirmed its pathogenicity by cosegregation with phenotype. However, another splice variant (c.1961_1963del) in patient RB24 (Figure 2B) as predicted as most likely pathogenic did not co-segregate with phenotype. Ultimately, functional studies are necessary for assigning pathogenicity to these novel variants.
The limitation of the targeted NGS is the uneven capture efficiency that reduced the sensitivity of detection of CNVs. The capture efficiency was highly variable with the library prepared with Illumina-Truseq and also there were no coverage of exon 14 and few regions of exon 20. This drawback was overcome with the Agilent Sureselect method. However, variable depth of coverage was noted in exons 1 and 27 (10-200X). Hence uniform capture efficiency with a higher depth of RB1 sequencing will resolve the issues.
In addition to the technical limitation of the targeted NGS, complete RB1 sequencing is needed to detect the missed variants in the deep intronic and untranslated regions (UTRs) that could possibly reduce the five unsolved cases. However, there are other factors that can initiate RB, such as promoter methylation of RB1 gene, and MYCN gene amplification [30]. In fact, we found MYCN amplification in tumor sample of a unilateral patient RB30 (data not shown). Hence, we propose that NGS panel for RB should include MYCN gene along with RB1.
Overall, targeted NGS approach is becoming more feasible and efficient in clinical settings, especially for cancer and can potentially identify germline and somatic variants comprehensively. However, we still suggest conventional methods for validation of the variants as we are in the initial phase of developing NGS methods for the diagnosis of RB. Further studies are necessary for the establishment of this approach in terms of cost-effectiveness.
Conclusions
This is the first such study (to the best of our knowledge) using multiplexed targeted NGS approach to detect pathogenic variants in the RB1 gene. We reported here that this approach with bioinformatics pipeline could detect germline and somatic variants including novel pathogenic variants. We demonstrated for the first time that this approach could detect copy number variations (CNVs) in RB1 gene. This comprehensive approach reduces the time and number of assays required for the detection of pathogenic variants by conventional methods. Our approach is sensitive (0.97) and efficient for RB1 screening.
Acknowledgements
We are grateful to the family members who participated in this study. We thank Aravind Eye Foundation (USA) and Aravind Research Medical Foundation (AMRF) for the financial support. We thank Dr. Arupa Ganguly, Department of Genetics and Dr. Tapan Ganguly, Penn Genomic Analysis Core at University of Pennsylvania for their help in generating and analysing the Ion Torrent PGM data. We acknowledge Indo-US Science and Technology Forum for the fellowship to Dr. Vanniarajan to carry out the NGS study at University of Pennsylvania.
Abbreviations
- RB
Retinoblastoma
- SNV
Single nucleotide variant
- InDel
Small insertions/deletions
- CNV
Copy number variation
- LOH
Loss of heterozygosity
- MLPA
Multiplex ligation-dependent probe amplification
- NGS
Next generation sequencing
- HGMD
Human gene mutation database
- COSMIC
Catalogue of somatic mutations in cancer
- dbSNP
Database for single nucleotide polymorphisms
- ESP
Exome sequencing project
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BD and LP developed bioinformatics pipeline and analysed NGS data. AV performed the Ion-Torrent PGM experiments and analysed the data. TRK, AAA and AV performed the molecular genetics studies. UK performed the clinical examination of the patients. VM conceived and oversaw the study, and helped in preparation of manuscript. BD and AV participated in study design and co-wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Bharanidharan Devarajan, Email: bharanid@gmail.com.
Logambiga Prakash, Email: loga2719@gmail.com.
Thirumalai Raj Kannan, Email: nigira85@gmail.com.
Aloysius A Abraham, Email: aaloci@gmail.com.
Usha Kim, Email: usha@aravind.org.
Veerappan Muthukkaruppan, Email: muthu@aravind.org.
Ayyasamy Vanniarajan, Email: vanniarajan@aravind.org.
References
- 1.Aerts I, Lumbroso-Le Rouic L, Gauthier-Villars M, Brisse H, Doz F, Desjardins L. Retinoblastoma. Orphanet J Rare Dis. 2006;1:31. doi: 10.1186/1750-1172-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Draper GJ, Sanders BM, Brownbill PA, Hawkins MM. Patterns of risk of hereditary retinoblastoma and applications to genetic counselling. Br J Cancer. 1992;66:211–9. doi: 10.1038/bjc.1992.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valverde JR, Alonso J, Palacios I. RB1 gene mutation update. A meta-analysis based on 932 reported mutations available in searchable database. BMC Genet. 2005;6:53. doi: 10.1186/1471-2156-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.AyariJeridi H, Bouguila H, Ansperger-Rescher B, Baroudi O, Mdimegh I, Omran I, et al. Genetic testing in Tunisian families with heritable retinoblastoma using a low cost approach permits accurate risk prediction in relatives and reveals incomplete penetrance in adults. Exp Eye Res. 2014;124:48–55. doi: 10.1016/j.exer.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Richter S, Vandezande K, Chen N, Zhang K, Sutherland J, Anderson J, et al. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet. 2003;72:253–69. doi: 10.1086/345651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–5. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levin JZ, Berger MF, Adiconis X, Rogov P, Melnikov A, Fennell T, et al. Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol. 2009;10:R115. doi: 10.1186/gb-2009-10-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonnefond A, Durand E, Sand O, De Graeve F, Gallina S, Busiah K, et al. Molecular diagnosis of neonatal diabetes mellitus using next-generation sequencing of the whole exome. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neveling K, Collin RW, Gilissen C, van Huet RA, Visser L, Kwint MP, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat. 2012;33:963–72. doi: 10.1002/humu.22045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Audo I, Bujakowska KM, Léveillard T, Mohand-Saïd S, Lancelot ME, Germain A, et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J Rare Dis. 2012;7:8. doi: 10.1186/1750-1172-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shanks ME, Downes SM, Copley RR, Lise S, Broxholme J, Hudspith KA, et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur J Hum Genet. 2013;21:274–80. doi: 10.1038/ejhg.2012.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Zhang VW, Feng Y, Tian X, Li FY, Truong C, et al. Dependable and efficient clinical utility of target capture-based deep sequencing in molecular diagnosis of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2014;55:6213–23. doi: 10.1167/iovs.14-14936. [DOI] [PubMed] [Google Scholar]
- 13.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.FastQC [http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc]
- 15.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a map reduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sathirapongsasuti JF, Lee H, Horst BA, Brunner G, Cochran AJ, Binder S, et al. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011;27:2648–54. doi: 10.1093/bioinformatics/btr462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klambauer G, Schwarzbauer K, Mayr A, Clevert DA, Mitterecker A, Bodenhofer U, et al. cn. MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 2012;40:e69. doi: 10.1093/nar/gks003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 24.Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37 doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J ComputBiol. 2004;11:377–94. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 26.Nichols KE, Houseknecht MD, Godmilow L, Bunin G, Shields C, Meadows A, et al. Sensitive multistep clinical molecular screening of 180 unrelated individuals with retinoblastoma detects 36 novel mutations in the RB1 gene. Hum Mutat. 2005;25:566–74. doi: 10.1002/humu.20184. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z, Moran K, Richards-Yutz J, Toorens E, Gerhart D, Ganguly T, et al. Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat. 2014;35:384–91. doi: 10.1002/humu.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–81. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 30.Thériault BL, Dimaras H, Gallie BL, Corson TW. The genomic landscape of retinoblastoma: a review. Clin Experiment Ophthalmol. 2014;42:33–52. doi: 10.1111/ceo.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]