

Abstract
Recent studies showed that specific isoprenoid modification may be critical for RhoB subcellular location and function. Therefore, we determined whether the function of the highly related RhoA protein is also critically dependent on specific isoprenoid modification: (a) in contrast to observations with RhoB or Ras proteins, where farnesylated and geranylgeranylated versions showed differences in subcellular location, both prenylated versions of RhoA showed the same plasma membrane and cytosolic location; (b) a farnesylated version of activated RhoA(63L) retained the same diverse functions as the normally geranylgeranylated RhoA(63L) protein, and both proteins show indistinguishable abilities to stimulate gene expression, cause growth transformation of NIH 3T3 mouse fibroblasts, to stimulate the motility of T47D human breast epithelial cells, and to block HIV-1 viral replication and gene expression; and (c) cells expressing farnesylated RhoA retained sensitivity to the growth inhibition caused by inhibition of geranylgeranyltransferase I, indicating that other proteins are critical targets for inhibitors of geranylgeranylation.
Introduction
The Rho family of small GTPases constitutes a major branch of the Ras superfamily. As with Ras, Rho proteins function as regulated GTP/GDP switches and cycle between an active GTP-bound form and an inactive GDP-bound form (reviewed in Ref. (1). To date, at least 18 distinct mammalian Rho family GTPases (2) have been identified and include RhoA, RhoB, RhoC, RhoD, RhoE/Rnd3, Rnd1, Rnd2, RhoG, Rac1, Rac2, Rac3, Cdc42, TTF, Chp (3), TCL (4), and Rif (5). Of these, RhoA, Rac1, and Cdc42 have been the most intensively studied.
Rho family GTPases are mediators of diverse cellular functions:
They are regulators of actin cytoskeletal organization. Whereas Cdc42 promotes filopodia formation, Rac1 promotes formation of lamellipodia or membrane ruffles, and RhoA triggers actin stress fiber and focal adhesion formation (reviewed in Refs. 1, 2). In contrast, RhoE causes a disruption of stress fibers and cell rounding (6, 7). Consequently, Rho GTPase function can influence cell-cell and cell-substratum interactions, which in turn can modulate cell movement. For example, we showed that Rac1 and Cdc42 activation promoted the motility and invasion in vitro of T47D human breast epithelial cells (8). Activated Rac1 promoted T-cell invasion in vitro (9). Overexpression of RhoC has been shown to promote the metastatic properties of melanoma cells in vivo (10). Thus, inhibitors of Rho GTPases have been considered as novel anticancer drugs (11).
Rho GTPases are regulators of gene expression and stimulate the activity of various transcription factors, including NF-κB3 (12, 13), SRF (14), and c-Jun (15, 16).
Rho GTPases can regulate cell cycle progression and cellular proliferation. For example, constitutive activation of RhoA, Rac1, and Cdc42 has been shown to promote tumorigenic transformation of rodent fibroblasts, and the function of these GTPases is required for the transforming activity of Ras and other oncoproteins (reviewed in Ref. 2). Finally, among a diverse spectrum of other functions, we determined recently that RhoA activation antagonizes HIV-1 viral replication and gene expression (17, 18). In light of the diverse signaling and biological activities of RhoA, it is not surprising that a large spectrum of functionally diverse proteins have been identified as downstream effector targets of RhoA (1, 2, 19).
In addition to GDP/GTP cycling, Rho GTPase function is also believed to be critically dependent on posttranslational modification by isoprenoid lipids. Similar to Ras, Rho GTPases also terminate in a COOH-terminal CAAX tetrapeptide sequence motif (where C is cysteine, followed by two aliphatic amino acids, ending in a variant amino acid; reviewed in Refs. 20, 21). This sequence signals for covalent attachment of an isoprenoid lipid group to the cysteine of the CAAX sequence, followed by endoprotease removal of the -AAX residues and carboxymethylation of the now terminal prenylated cysteine. These modifications promote the association of Ras and Rho GTPases with plasma and intracellular membranes.
The CAAX motif of Ras and Rho GTPases signal for covalent modification by either of two types of isoprenoid lipids (reviewed in Refs. 20, 21). The FTase enzyme catalyzes the addition of a C15 farnesyl group to a subset of CAAX sequences, when X is serine, methionine, cysteine, alanine, or glutamine. For CAAX sequences where X is leucine or isoleucine, the GGTaseI enzyme catalyzes addition of the more hydrophobic C20 geranylgeranyl moiety. All isoforms of Ras are modified by farnesylation, whereas the closely related R-Ras and TC21/R-Ras2 proteins are modified by geranylgeranylation. A majority of Rho GTPases is modified by the geranylgeranyl group (e.g., RhoA, Rac, and Cdc42), whereas some are modified by a farnesyl group (e.g., RhoE/Rnd3; Ref. 22), and others can be modified by both isoprenoids (e.g., RhoB; Refs. 23, 24).
Prenylation may critically influence subcellular location of a protein as well as regulate its ability to interact with other proteins. The contribution of CAAX-signaled processing to protein function has been best studied with Ras proteins (25–27). Mutation of the CAAX motif of Ras proteins prevents prenylation and abolishes plasma membrane association, causing a complete loss of oncogenic Ras transforming activity. This information has led to the development of FTIs to block Ras processing and function, and hence, are being developed as anticancer drugs to treat ras mutation-positive human cancers. To address the importance of specific isoprenoid modification for Ras function, CAAX mutants of Ras that undergo modification by geranylgeranylation were generated. Although the subcellular location of these variants was altered, with localization to intramembrane compartments, geranylgeranylated versions of oncogenic Ras retained potent transforming activity (28, 29). Therefore, it appears that oncogenic Ras function can be facilitated by modification with either isoprenoid group.
The importance of protein prenylation for Rho GTPase function has been best evaluated with RhoB. Activated RhoB can promote growth transformation, and a nonprenylated version of activated RhoB showed a loss of transforming activity (30, 31). However, it retained the ability to stimulate SRF activation. Thus, some but not all RhoB function is dependent on prenylation. However, in contrast to the observations with Ras, RhoB function appears to be critically dependent on specific isoprenoid function. RhoB appears to be modified primarily by farnesylation in vivo. However, FTI treatment caused an accumulation of geranylgeranylated RhoB (32). Prendergast and colleagues (33, 34) showed that geranylgeranylated RhoB inhibited the growth of rodent and human cells. Thus, whereas farnesylated RhoB is a growth-promoting protein, geranylgeranylated RhoB is an apoptosis-inducing protein. Consistent with these opposing functions, farnesylated RhoB function was shown to be required for Ras transformation (30), whereas geranylgeranylated RhoB blocked Ras transforming activity (33). Consequently, the FTI-induced formation of inhibitory geranylgeranylated RhoB has been proposed as the basis for the antitumor activity of FTIs.
RhoA and RhoB share significant sequence identity (>85%). For example, they share complete identity in core effector domain sequences (residues 32–40) required for interaction with downstream effectors. RhoA and RhoB exhibit greatest sequence divergence in residues immediately NH2-terminal to the CAAX motif. Because these sequences contain elements that influence the targeting of Ras and Rho GTPases to specific membrane compartments, it is not surprising that RhoA and RhoB exhibit different subcellular locations. One study found that only a minor fraction of RhoA was associated with the plasma membrane, with the majority of the protein located in the cytosol (35). In contrast, RhoB was associated with early endosomes and a prelysosomal compartment. However, a recent analysis of GFP-tagged proteins in live cells determined that RhoB was located predominantly at the plasma membrane, and it was suggested that the earlier localization of RhoB to endosomes may be an artifact of fixation (36). In contrast, RhoA showed a predominantly cytosolic location.
RhoA is modified exclusively by geranylgeranylation. Whether all RhoA function is critically dependent on prenylation and whether modification by farnesylation can support RhoA function has not been determined. Additionally, pharmacological inhibitors of GGTaseI, the enzyme that modifies RhoA and other Rho GTPases, have been shown to possess antitumor activity, possibly by blocking the function of RhoA (37–40). RhoA function is mediated by interaction with a multitude of downstream effectors, and the precise location of RhoA may critically influence proper interaction with effectors (1, 2, 19). In the present study, we determined the requirement for modification by geranylgeranylation for RhoA function, whether modification by farnesylation could support the diverse biological functions RhoA, and whether RhoA is the critical target for GGTIs. We determined that RhoA function was dependent on prenylation, but that a farnesylated form of RhoA was unchanged in function and subcellular location. Thus, our observations contrast with those of RhoB and show that RhoA function is not dependent on modification by a specific isoprenoid. Finally, cells expressing farnesylated RhoA were still sensitive to growth inhibition by GGTI treatment, indicating that RhoA is not the only critical target for GGTIs.
Results
A Mutation in the CAAX Motif of RhoA Renders the Protein Sensitive to FTase Inhibitor Treatment but Does Not Alter Subcellular Location
RhoB has been found to exhibit distinct and opposing functions when modified by different isoprenoids (33, 34). This alteration in function has been attributed to a distinct subcellular location of the farnesylated and geranylgeranylated forms of RhoB. To evaluate the importance of prenylation for RhoA function, we generated two variants of the activated and transforming RhoA(63L) mutant protein:
The WT CAAX sequence (CLVL) was mutated to the CAAX sequence of the farnesylated H-Ras protein [CVLS; designated RhoA(63L)-CVLS]. This CAAX motif has been determined to be specific to signal for farnesylation as opposed to geranylgeranylation (21, 41).
We also generated a nonprenylated version by mutating the isoprenoid-accepting cysteine into a serine [SLVL; designated RhoA(63L)-SLVL]. We verified that the two mutant proteins were stable and could be expressed at levels comparable with that of the WT protein. Transiently or stably transfected NIH 3T3 cells showed that RhoA(63L) and RhoA(63L)-CVLS were expressed at comparable levels (Fig. 1A). Interestingly, in both transiently and stably transfected cells, we observed that the RhoA(63L)-SLVL was expressed at higher levels than RhoA(63L).
Fig. 1.
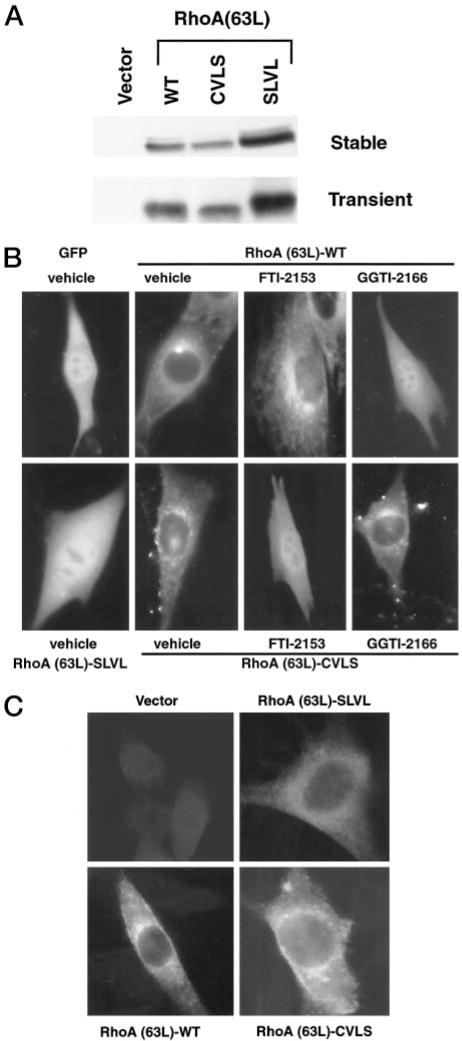
Substitution of the CAAX motif of RhoA(63L) with the CVLS motif from farnesylated H-Ras results in a protein that is not altered in subcellular location but is sensitive to FTase, but not GGTaseI, inhibitor treatment. A, lysates from NIH 3T3 cells stably or transiently expressing HA epitope-tagged RhoA(63L) proteins were normalized for total protein. The proteins were resolved by SDS-PAGE, and expression was determined by Western blot analysis using anti-HA epitope antibody. B, NIH 3T3 cells transiently expressing the indicated GFP-tagged RhoA(63L) proteins were cultured in growth medium supplemented with vehicle (DMSO), FTI-2153, or GGTI-2166 were analyzed as live cells. Cells were then visualized using a Axioskop 2 microscope and Openlab digital imaging software. C, alternatively prenylated, but not unprenylated, shows a similar subcellular location as geranylgeranylated RhoA(63L). NIH 3T3 cells stably expressing HA epitope-tagged RhoA(63L)-WT, RhoA(63L)-CVLS, and RhoA(63L)-SLVL were fixed, and the proteins were visualized by indirect immunofluorescence analyses using anti-HA epitope antibody and a FITC-conjugated antimouse secondary antibody. Data shown are representative of three independent experiments.
We next determined the subcellular location of the different CAAX variants of RhoA and then verified that the RhoA(63L)-CVLS protein was now modified by farnesylation. For these analyses, we generated GFP-tagged versions of each RhoA(63L) protein to evaluate subcellular localization in live cells. Similar to what has been described previously (35, 36), RhoA(63L)-WT showed both a punctate, perinuclear, and a plasma membrane staining distribution. GFP-tagged RhoA(63L)-CVLS showed a similar pattern and distribution (Fig. 1B). Thus, modification by a different isoprenoid did not cause a detectable change in subcellular location.
Because previous studies of RhoB localization found differences when evaluated in live cells when compared with fixed cells (36, 42), we also evaluated the location of HA epitope-tagged versions of these two proteins in fixed cells (Fig. 1C). Essentially similar results were seen, where both WT and CVLS versions of RhoA(63L) showed punctate, perinuclear staining patterns. In contrast, the nonprenylated RhoA(63L)-SLVL mutant showed a diffuse cytoplasmic localization. We also evaluated subcellular distribution by high-speed fractionation into cytosolic S100 soluble and membrane-containing P100 particulate fractions. Both RhoA(63L)-WT and RhoA(63L)-CVLS were found predominantly in the P100 fraction, although RhoA(63L)-CVLS showed a reproducibly greater percentage of protein in the S100 fraction (data not shown). Thus, in contrast to what has been described for RhoB, mutation of the CAAX motif to alter the specific isoprenoid modification did not cause a significant change in subcellular location.
We then determined whether the distribution of the CVLS mutant was sensitive to inhibition by a FTI. As expected, treatment with the FTI-2153 inhibitor caused a redistribution of RhoA(63L)-CVLS to a diffuse cytoplasmic and nuclear location, whereas the distribution of RhoA(63L)-WT was unchanged (Fig. 1B). Conversely, treatment with the GGTI-2166 GGTaseI inhibitor resulted in a diffuse distribution of RhoA(63L)-WT but did not alter the perinuclear distribution of RhoA(63L)-CVLS. Similar results were also seen with the HA epitope-tagged proteins, where RhoA(63L)-CVLS, but not RhoA(63L)-WT, was sensitive to FTI treatment, as measured by a change in mobility in SDS-PAGE (data not shown). We conclude that RhoA(63L)-CVLS is modified by farnesylation, but this alternative prenylation did not result in the significant change in subcellular location that has been described for the different prenylated forms of RhoB (32).
RhoA-mediated Transcriptional Activation of SRF, NF-κB, and the Cyclin D1 Promoter Is Dependent on Prenylation
Previously, it has been shown that RhoA can activate several transcription factors, such as SRF (14) and NF- κB (12), and can stimulate transcription from the cyclin D1 promoter (43). Therefore, to determine whether RhoA requires the modification by a specific prenyl group for activation of these factors, transient expression analyses were done using reporter plasmids, where expression of the luciferase gene is regulated by minimal promoters that contain SRF- or NF-κB-responsive elements or by the human cyclin D1 promoter. We found that RhoA(63L)-WT and RhoA(63L)-CVLS expression caused comparable activation of SRF (~ 10-fold), NF-κB (~11-fold), and cyclin D1 (~3-fold; Fig. 2). In contrast, the RhoA(63L)-SLVL mutant failed to cause significant stimulation in any assay. Thus, although prenylation is essential for the activation of these pathways, farnesylated and geranylgeranylated RhoA activate similar signaling pathways.
Fig. 2.
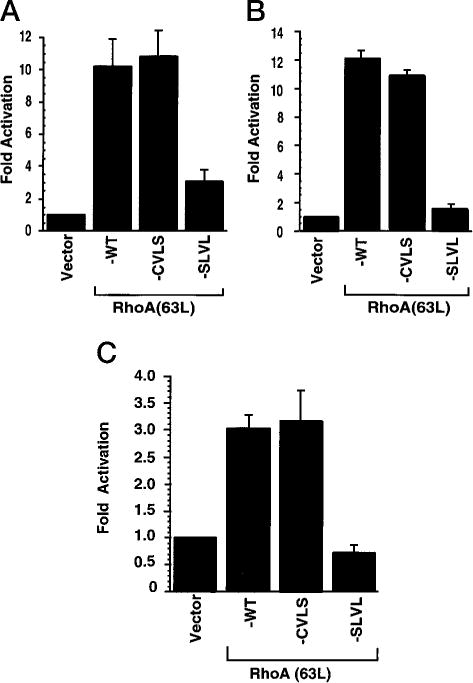
Farnesylated, but not unprenylated, RhoA(63L) retains the ability to stimulate transcriptional activation of SRF and NF-κB and the cyclin D1 promoter. 293T cells were transiently cotransfected with pcDNA3 expression plasmids encoding the indicated proteins (500 ng/30-mm dish) together with reporter plasmids, where the luciferase gene is under control of the fos minimal promoter containing either SRF (A)- or NF-κB (B)-responsive elements or the human cyclin D1 promoter (C). Forty-eight h after transfection, stimulation of transcriptional activity was determined by measuring luciferase activity of cell lysates. Fold activation was determined by the number of relative luciferase units relative to the number of units seen with the empty vector control. Data points are the means of three independent measurements; bars, SE. Data are representative of three independent assays.
Prenylation Is Essential for the Growth-promoting Properties of RhoA(63L) but Specific Isoprenoid Modification Is Not
To assess the importance of prenylation in RhoA transforming activity, we analyzed the activity of the two mutant proteins using two transformation assays. Because Rho family members are weakly transforming in traditional NIH 3T3 focus assays, we and others have used a cooperation focus formation assay to assess the transforming activity of Rho proteins and their activators (44–46). The cooperation assay allows for the synergistic enhancement of focus-forming ability when activated RhoA(63L) is co-expressed with a weakly transforming Raf-1 protein [Raf(340D)]. As we described previously, cultures transfected with the RhoA(63L) expression construct alone showed minimal transforming activity (< 10 foci/dish), whereas the coexpression of RhoA(63L) with Raf(340D) resulted in a 5-fold above additive increase in the appearance of transformed foci (Fig. 3; Ref. 47). The farnesylated RhoA(63L)-CVLS mutant showed a comparable level of focus-forming activity, whereas the unprocessed RhoA(63L)-SLVL mutant showed greatly impaired focus-forming activity.
Fig. 3.
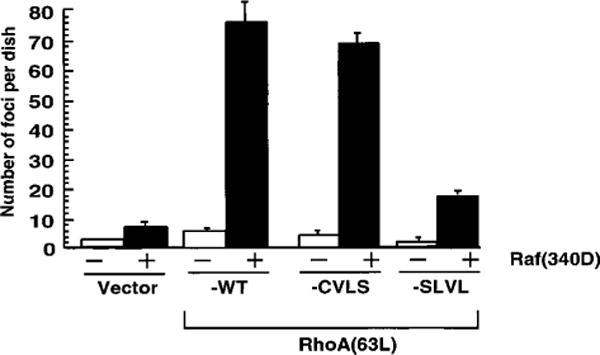
Farnesylated, but not unprenylated, RhoA(63L) retains potent focus-forming activity. NIH 3T3 cells were cotransfected with pZIP-NeoSV(x)1 expression plasmids encoding the indicated RhoA(63L) proteins, either together with the empty pZIP-NeoSV(x)1 vector (–) or with pZIP-raf(340D) (1 μg/60-mm dish). Seventeen to 21 days after transfection, the number of foci of transformed cells was quantified. Data points are the means of three independent measurements; bars, SE. Data are representative of three independent assays.
Next, we determined the ability of the prenylation mutants to promote growth factor-independent cell proliferation. Previously, we showed that RhoA(63L)-expressing NIH 3T3 cells continue to proliferate in growth medium supplemented with low serum (44). Both the RhoA(63L)- and RhoA(63L)-CVLS-expressing cells retained the ability to grow in 0.5% serum after 21 days (Fig. 4A) and retained a refractile morphology and formed multilayered cultures (Fig. 4B). In contrast, both the empty vector- and RhoA(63L)-SLVL-transfected cells had become quiescent and formed a monolayer of non-refractile cells. These results demonstrate that prenylation is crucial for the growth-promoting property of RhoA(63L), but either isoprenoid modification could support this activity.
Fig. 4.
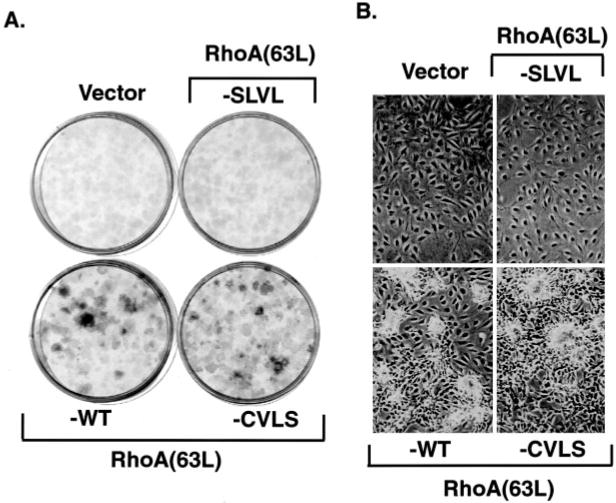
Farnesylated, but not unprenylated, RhoA(63L) retains the ability to promote growth in low serum. NIH 3T3 cells stably expressing RhoA(63L)-WT, RhoA(63L)- CVLS, or RhoA(63L)-SLVL were plated at 103 cells/dish and allowed to adhere in DMEM supplemented with 10% calf serum. Untransformed cells stably transfected with the empty pZIP-NeoSV(x)1 plasmid (Vector) were included as a control. After 24 h, the medium was replaced with DMEM supplemented with 0.5% calf serum. Twenty-one days after plating, the cells were fixed and stained with 0.4% crystal violet (A) to better visualize the cells. On day 20, the cells were photographed (B) to demonstrate the different morphology of the cells.
Prenylation Is Important for the Ability of RhoA to Promote Motility of T47D Cells
We showed previously that activated Rac1 or Cdc42 could promote increased motility and invasive properties of T47D human breast epithelial cells in vitro (8). Recently, we have also determined that activated RhoA(63L) can also cause these same changes (48).4 We therefore examined the importance of prenylation modification in mediating this RhoA function. For these analyses, we established mass populations of T47D cells stably transfected with expression vectors encoding each of the RhoA(63L) proteins. A comparable level of expression of each RhoA(63L) protein was verified by Western blot analyses (data not shown). As described previously, we used a haptotactic assay where transwells were coated on the underside of the filter with collagen. Each cell population was then plated on the transwell filter and allowed to migrate for 24 h through the pores of the transwell to the collagen-coated underside. RhoA(63L)- and RhoA(63L)-CVLS-expressing cells both caused an enhancement of motility across the transwell as compared with the vector control (Fig. 5). This enhancement was not seen with cells expressing the unprenylated SLVL mutant. Therefore, similar to our observations with the NIH 3T3 transformation assays, activated RhoA(63L)-stimulated migration of T47D cells was also dependent on prenylation but could be facilitated by either farnesylation or geranylgeranylation.
Fig. 5.
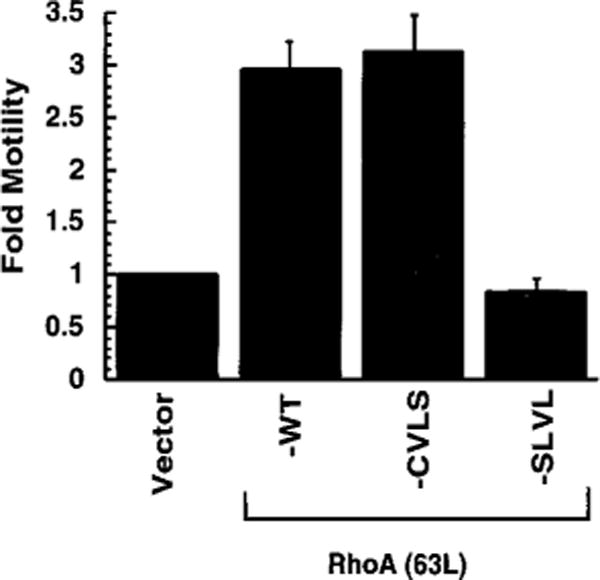
RhoA(63L)-induced migration of T47D human breast carcinoma cells is dependent on prenylation, but not specific prenylation. T47D cells stably expressing the indicated protein were analyzed for their ability to migrate through the pores of a transwell to the underside. Fold activation was determined by the number of migrating cells relative to the number of migrating vector-transfected cells. Western blot analyses with anti-HA epitope antibody was done to verify equivalent expression of RhoA(63L) protein in each cell population; although WT and CVLS versions of RhoA(63L) showed comparable levels of expression, the SLVL mutant consistently showed slightly higher levels of expression in this and other analyses (not shown). Data points are the means of two independent measurements; bars, SE. Data shown are representative of three independent assays.
Suppression of HIV-1 Replication by RhoA Requires Prenylation
We next wanted to determine whether geranylgeranylation of RhoA was crucial for RhoA inhibition of HIV-1 replication (18). For these analyses, we used two transient expression assays to evaluate the ability of the mutant RhoA(63L) proteins to block HIV-1 function:
We observed previously that the RhoA(63L) mutant inhibited HIV-1 replication, as measured by the production of infectious virus and of virion-associated reverse transcriptase activity (80 and 50% reduction, respectively). A similar level of inhibition was seen with the RhoA(63L)-CVLS mutant in both assays (80 and 60% reduction), whereas RhoA(63L)-SLVL was impaired significantly (30% reduction) in this ability (Fig. 6A).
We measured the ability of the prenylation mutants to block gene expression from the HIV-1 genome. These analyses used a recombinant HIV-1 virus where the luciferase gene is inserted in the HIV-1 Nef open reading frame (49). Similar to RhoA(63L), RhoA(63L)-CVLS also greatly impaired luciferase gene expression, whereas RhoA(63L)-SLVL showed a complete loss of this inhibitory activity (Fig. 6B). This suggests the importance of RhoA prenylation, but not the specific isoprenoid modification, in mediating inhibition of HIV-1 function.
Fig. 6.
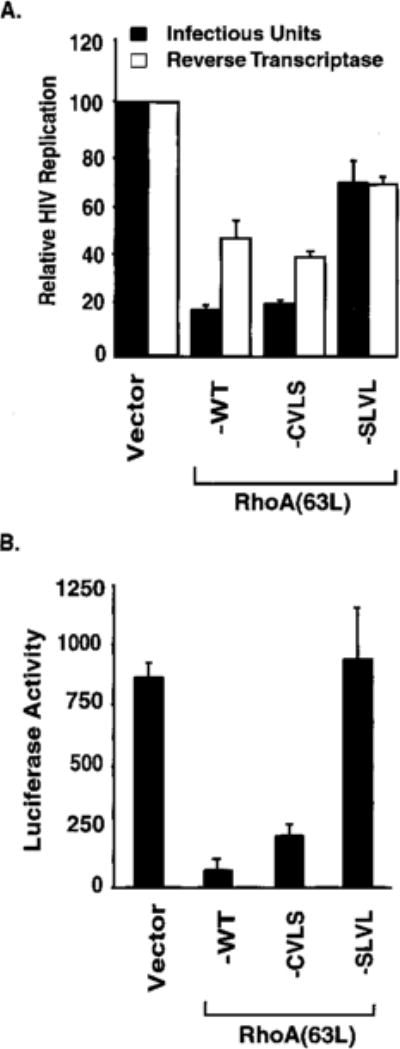
Farnesylated, but not unprenylated, RhoA(63L) retains the ability to antagonize HIV-1 replication and gene expression. A, the ability of the RhoA(63L) protein to block the transient replication of HIV-1 was assessed by cotransfecting the HIV-1 provirus pNL4–3 with pcDNA3 expression plasmids encoding the indicated proteins in 293T cells. At 40–50 h after transfection, HIV-1 virions in the culture supernatant were measured by reverse transcriptase activity (□) or by quantification of infectious units (■). The relative HIV-1 replication is presented as a percentage of vector controls (100%). B, the ability of RhoA(63L) protein to inhibit gene expression was assessed by cotransfecting pNL4–3-Luciferase plasmid with pcDNA3 expression plasmids encoding the RhoA proteins in Jurkat T cells. The level of luciferase expression (relative light units) was measured 48 h after transfection. Data points are the means of three independent measurements; bars, SE. Data shown are representative of two independent experiments.
Farnesylated RhoA Does Not Protect Cells from Growth Inhibition Caused by Inhibition of GGTaseI
Pharmacological inhibition of GGTaseI has been shown to inhibit normal and tumor cell proliferation, in part by blocking progression through G1 of the cell cycle (50). Rho GTPases are substrates for GGTaseI and have been shown to be required for proliferation through G1 (51, 52). Hence, geranylgeranylated Rho GTPases constitute logical targets for the inhibitory action of GGTIs. One approach for evaluating this possibility is to determine whether variants of Rho GTPases that are insensitive to GGTIs can protect cells from GGTI growth inhibition. In this study, we have shown that farnesylation can support the diverse signaling and biological functions of RhoA. Thus, this farnesylated variant of RhoA can serve as a useful reagent to evaluate whether RhoA is a critical target of GGTIs.
To determine whether RhoA is an important target for the inhibitory activity of GGTIs, we compared the sensitivity of NIH 3T3 cells stably expressing RhoA(63L)-WT and RhoA(63L)-CVLS. Similar to what has been described previously, treatment of cells expressing RhoA(63L)-WT with GGTI-2166 caused a dose-dependent inhibition of proliferation. RhoA(63L)-WT expressing cells cultured in growth medium supplemented with 3 μm GGTI-2166 showed an ~30% reduction in cell number and an ~80% reduction at 10 μm GGTI-2166 (Fig. 7). Essentially identical levels of growth inhibition was seen with RhoA(63L)-CVLS-expressing cells. Thus, the growth-inhibitory activity caused by GGTI treatment is not attributable solely, if at all, to the loss of RhoA function.
Fig. 7.
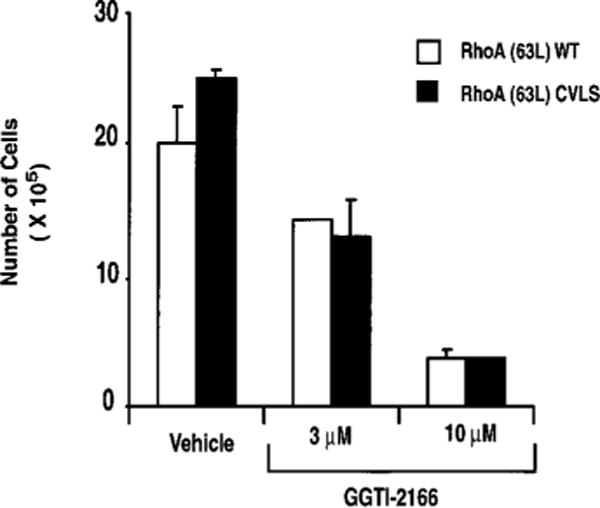
Cells expressing farnesylated RhoA are still sensitive to growth inhibition by inhibition of GGTaseI. Cultures of NIH 3T3 cells stably expressing RhoA(63L)-WT or RhoA(63L)-CVLS were incubated with growth medium supplemented with 3 or 10 μm GGTI-2166. Cell number was determined on days 3, 5, 6, 8, 10, and 12, with data shown for day 12, are the average of duplicate dishes and are representative of three independent experiments; bars, SE.
Discussion
Ras and Rho small GTPases undergo posttranslational modification by either the C15 farnesyl or C20 geranylgeranyl isoprenoid groups in reactions that are catalyzed by distinct but structurally related enzymes, FTase and GGTaseI, respectively (21, 25, 41). Whether protein function is uniquely influenced by modification by a specific isoprenoid remains incompletely understood. Recent studies suggest that RhoB function is greatly altered by the type of isoprenoid modification it undergoes (33, 34). Whereas farnesylated RhoB exhibits a growth-promoting function, geranylgeranylated RhoB exhibited a growth-inhibitory, apoptotic activity. These observations prompted us to evaluate whether the function of the closely related RhoA protein is also greatly influenced by specific isoprenoid modification. We found that a farnesylated version of activated RhoA(63L) retained the abilities to cause growth transformation of NIH 3T3 cells, to stimulate transcription factor activity, to promote T47D breast carcinoma migration, and to block HIV-1 replication and gene expression. Conversely, a nonprenylated form of RhoA(63L) showed a loss of all function. Thus, we conclude that RhoA function is dependent on prenylation but independent of specific isoprenoid modification. Additionally, because cells expressing farnesylated RhoA retained sensitivity to growth inhibition by GGTaseI inhibition, other GGTaseI substrates must also be important targets for GGTaseI inhibitors.
Our analyses of the role of isoprenoid modification on RhoA function extend on previous studies that address whether specific isoprenoid modification is critical for protein function. Approximately 0.5% of mammalian proteins undergo posttranslational modification by isoprenoid lipids, and of the two types of prenylation, geranylgeranylation is the predominant modification (21, 25). Interestingly, proteins that are both structurally and functionally related can be modified by different isoprenoid moieties. For example, the three Ras isoforms (H-Ras, K-Ras, and N-Ras) are all modified by farnesylation, whereas the closely related R-Ras1 and R-Ras2/TC21 proteins are modified by geranylgeranylation. However, despite differences in isoprenoid modification, constitutively activated mutants of Ras and R-Ras isoforms all cause growth transformation. Similarly, whereas RhoA is modified by geranylgeranylation, the related RhoE protein is modified by farnesylation (22). In this case, though, these proteins have opposing functions, with RhoA promoting actin stress fiber formation and RhoE causing a disruption of stress fibers. Whether the different prenylated nature of these two proteins accounts for their opposing functions has not been determined. The γ subunits of heterotrimeric G proteins are also modified by prenylation. Among the 11 known γ subunits, γ1, γ8, and γ11 are farnesylated, whereas the remaining subunits are modified by geranylgeranylation (53). When analyzed, no striking isoprenoid-specific differences in γ subunit function has been found. Thus, to date, studies evaluating a role for specific isoprenoid modification in protein function have determined surprisingly subtle or no differences in isoprenoid regulation of protein function (29, 54, 55).
Our observations that RhoA transforming activity could be mediated by geranylgeranylation as well as farnesylation contrasts sharply with the observations made with RhoB. Whereas farnesylated RhoB was found to exhibit a growth-promoting activity that was required for Ras transformation, geranylgeranylated RhoB instead showed a proapoptotic function that antagonized Ras transformation (33, 34). These strikingly different consequences of different isoprenoid modifications on RhoA and RhoB function are unexpected in light of their strong structural (>85% amino acid sequence identity) and functional (e.g., promotion of actin stress fibers) similarities. However, RhoA and RhoB diverge significantly in their COOH-terminal sequences and consequently do localize to very distinct regions in the cell. This may account for the different consequences of isoprenoid modification on RhoA and RhoB biological activity.
Our observations with RhoA transformation are similar to previous observations that geranylgeranylated versions of oncogenic H-Ras proteins also retained the ability to cause transformation (29). However, we also found that a geranylgeranylated mutant of WT H-Ras was growth inhibitory. This result suggests that specific isoprenoid modification is important for the normal function of H-Ras. Whether normal RhoA function is also dependent of specific isoprenoid modification remains to be determined. Finally, it will be important to determine whether the function of other Rho GTPases will be dependent (i.e., RhoB) or independent (i.e., RhoA) of specific isoprenoid modification.
RhoA function is mediated by interaction with a large number of functionally diverse effectors. The precise effector(s) important for mediating the ability of RhoA to cause growth transformation of NIH 3T3 cells, motility of T47D cells, and to block HIV-1 function remain to be determined. However, our analyses of a panel of effector domain mutants of activated RhoA(63L),5 which are impaired differentially in effector interactions, indicate that these three RhoA functions are initiated through the use of different effectors. Our observation that nonprenylated RhoA(63L) was deficient in all three activities indicates that membrane association is essential for RhoA interaction with the effectors involved in these processes. Finally, we found that RhoA activation of SRF was also dependent on prenylation. This result contrasts with the observation that nonprenylated RhoB can still activate SRF and further suggests that the distinct subcellular locations of RhoA and RhoB must greatly influence their functions (31).
During the course of our studies, Allal et al. (56) published a study showing that the function of activated RhoA was also independent of the type of isoprenoid modification. They determined that farnesylated RhoA(14V) was unchanged in subcellular location, interaction with RhoGDI, actin reorganization, growth transformation of NIH 3T3 cells, suppression of p21CIP1 gene expression, and stimulation of SRF. Thus, our observations are in agreement with and extend their observations to other biological functions of RhoA. We showed additionally that farnesylation can support two other diverse RhoA-mediated functions, induction of breast tumor cell migration in vitro and RhoA inhibition of HIV-1 function. Hence, it appears that the majority of RhoA effector function can be supported by either isoprenoid modification. Because a goal of our studies was to use farnesylated RhoA to evaluate the mechanism of GGTI-mediated growth inhibition, a complete evaluation of whether diverse functions of RhoA can be supported by alternative prenylation is critical. Additionally, because there are currently conflicting observations regarding whether RhoB function is dependent on a specific isoprenoid (33, 57), independent studies that show that RhoA function can be supported by modification by either isoprenoid is important.
Pharmacological inhibitors of GGTaseI have been developed as potential anticancer drugs (11) and have shown potent antitumor activity in cell culture and animal preclinical studies. The mechanism of growth inhibition of GGTIs has been ascribed, in part, to inhibition of progression through G1 of the cell cycle (50). Because RhoA, a GGTaseI substrate, has been shown to be critical for G1 progression (51, 52), it represents a possible target for GGTI-induced growth inhibition. In the present study, we established that farnesylated RhoA retained all signaling and biological functions of the authentic geranylgeranylated RhoA protein. This provided important validation that farnesylated RhoA will be a useful RhoA variant to determine whether RhoA is indeed the critical target for GGTIs. However, we found that NIH 3T3 cells stably expressing farnesylated RhoA showed the same sensitivity to GGTI-induced growth inhibition. Although this result does not eliminate RhoA as a critical target, it does indicate that other GGTaseI substrates must also be involved in GGTI growth inhibition. Logical candidates include two other Rho GTPases, Rac1 and Cdc42, that are substrates for GGTaseI and have also been shown to be required for G1 progression. Because loss of function of RhoA, Rac1, or Cdc42 alone was sufficient to block G1 progression, perhaps GGTI-mediated loss of function of all three Rho GTPases is responsible for GGTI growth inhibition. To evaluate this possibility, it must first be determined whether farnesylated versions of Rac1 and Cdc42 also retain the same functions as their authentic geranylgeranylated counterparts. If so, then it will be important to determine whether coexpression of farnesylated RhoA, Rac1, and Cdc42 can reduce the sensitivity of cells to GGTI-induced growth inhibition. If not, then other GGTaseI substrates must be targeted by GGTIs to cause growth inhibition. The identity of these geranylgeranylated targets will be important for the further development of GGTIs as anticancer drugs.
Materials and Methods
Molecular Constructs
To generate cDNA sequences encoding mutants of activated RhoA(63L) that undergo modification by a farnesyl isoprenoid [designated RhoA(63L)-CVLS] or is nonprenylated [designated RhoA(63L)-SLVL], we introduced missense mutations into the sequence that encodes the COOH-terminal CAAX motif. cDNA sequences encoding the CAAX mutations (CVLS and SLVL) were generated by using the QuikChange Site-Directed Mutagenesis kit (Stratagene) and the cDNA sequence for RhoA(63L) in the pBS(HA)rhoA(63L) plasmid as a template for mutagenesis. RhoA(63L)-CVLS terminates with the CAAX tetrapeptide sequence of the farnesylated H-Ras protein. RhoA(63L)-SLVL contains a cysteine-to-serine substitution in the RhoA CAAX motif to remove the site of prenylation addition. The template pBS(HA)rhoA(63L) was constructed first by inserting a DNA linker that encoded the HA epitope (YPYDVPDYA) into the BamHI site of pBluescript II(KS) (Stratagene), resulting in pBS(HA). The linker also contained an internal NdeI site in-frame and 3’ of the HA epitope tag. The NdeI-EcoRI fragment from pcDNA3rhoA(63L) (Ref. 43; which also contains a BamHI site 3’ of the coding sequence) was inserted into the NdeI-EcoRI sites of pBS(HA). All constructs were then confirmed by sequencing. The BamHI fragment from each of the mutated pBS(HA)rhoA(63L) constructs was inserted into the BamHI site of the pZIP-NeoSV(x)1 retrovirus mammalian expression vector (58) and the pcDNA3 mammalian expression vector (Invitrogen). The pZIP-raf(340D) plasmid encodes a weakly activated mutant of human Raf-1 and has been described previously (44). Mammalian expression vectors encoding GFP-tagged fusion proteins of RhoA(63L)-WT, RhoA(63L)-CVLS, and RhoA(63L)-SLVL were generated by insertion of the BamHI fragment from the pBS(HA)rhoA(63L) constructs into the BamHI site of pEGFP-C1 (Clontech).
Cell Culture, Transfection, and Transformation Assays
NIH 3T3 mouse fibroblasts were maintained in DMEM supplemented with 10% calf serum (Hyclone). Human embryonic kidney 293T cells and the HeLa-MAGI human cervical carcinoma cells were maintained in DMEM supplemented with 10% FCS (Life Technologies, Inc.). T47D human breast epithelial cells were grown in RPMI supplemented with 10% FCS and 0.2 units/ml insulin. For cooperation focus formation assays with Raf(340D), NIH 3T3 cells were transfected by the calcium phosphate precipitation along with a glycerol shock as described previously (47). For these transfections, 1 μg of the pZIP-rhoA and 1 μg of pZIP-raf(340D) plasmid DNA were cotransfected into NIH 3T3 cells. Twenty-one days after transfection, the dishes were stained with crystal violet, and the appearance of foci of transformed cells was quantitated by visual inspection.
To establish cell lines stably expressing WT or mutant RhoA(63L) protein, NIH 3T3 were transfected by calcium phosphate precipitation as above with 500 ng of the pZIP-NeoSV(x)1 retrovirus expression vector constructs. Three days after transfection, the transfected cultures were subcultured at a 1:5 split ratio into growth medium supplemented with 400 μg/ml of G418. Mass populations of G418-resistant colonies were pooled together to establish cell lines that were then used for GGTI sensitivity experiments, growth transformation assays, and immunofluorescence analysis for determination of protein location. Western blot analyses with anti-HA antibody (Covance) were done to verify that there was comparable expression of each RhoA(63L) protein in the established cell lines.
To evaluate the ability of each cell line to proliferate in growth medium supplemented with low serum, three dishes were plated with 103 cells/60-mm dish and were allowed to attach in growth medium. After 16 h, the growth medium was replaced with DMEM supplemented with 0.5, 2, or 10% calf serum. The dishes were cultured for 21 days, and the appearance of colonies of proliferating cells was visualized by staining with 0.4% crystal violet.
To evaluate the ability of each cell line to proliferate in growth medium supplemented with GGTI-2166 GGTI (FTI-277, FTI-2153, GGTI-298, and GGTI-2166 provided by Said Sebti and Andrew Hamilton; Ref. 59), NIH 3T3 cells stably expressing the WT and CVLS versions of RhoA(63L) proteins were plated at 103 cells/35-mm dishes in growth medium. After 18 h, normal growth medium was replaced with growth medium supplemented with either vehicle (DMSO), 3, or 10 μm GGTI-2166. Every 24 h, the medium was replaced with fresh vehicle/GGTI-containing medium. On days 3, 5, 6, 8, 10, and 12, the cells from two dishes were trypsinized and were counted on a hemacytometer for each condition.
Transient Expression Reporter Gene Assays
For the transcriptional activation assay, 293T cells were transfected by calcium phosphate coprecipitation as described above except that the glycerol shock was excluded. Five hundred ng of the pcDNA-rhoA plasmid DNA or pcDNA3 empty vector were cotransfected together with 2 μg of the luciferase gene reporter plasmids, where expression is controlled by a minimal promoter from the c-fos gene that contains multiple SRF (43) or NF-κB (60) responsive DNA elements. The cyclin D1-Luc plasmid contains the luciferase gene under the control of the human cyclin D1 promoter (61). Twenty-four h after transfection, the growth medium was replaced with DMEM supplemented with 0.5% FCS. After 16–20 h, the cells were lysed and then analyzed for luciferase activity using enhanced chemiluminescence and a Monolight 2010 luminometer (Analytical Luminescence).
Cell Migration Assays
Migration assays were performed as described previously (48). T47D cells were stably transfected with pZIP-NeoSV(x)1 plasmid DNA encoding the parental RhoA(63L) protein and the two CAAX mutants and selected in growth medium supplemented with 300 μg/ml G418. For the migration assay, transwell dishes (12-well cluster, 12 mm diameter, 12 μm pore size; Costar) were coated from the underside with 3 μg/ml collagen I in water (Collaborative Biomedical Products), and serum-free RPMI was placed in the lower chamber. Motility was assessed by adding 104 cells in the upper chamber of the transwell in RPMI supplemented with 5 mg/ml BSA. The transwells were incubated in a humidified CO2 incubator (10%) at 37°C for 16 h. The nonmotile cells were then removed from the upper chamber, and the transwell filter was fixed and stained with Wright-Giemsa stain (Diffquick; Baxter) and then removed and mounted on a microscope slide with Cytoseal (Baxter) solution and a coverslip. The number of remaining cells, which represent the motile cells, were counted on a Nikon microscope.
Localization Analyses
The subcellular locations of CAAX mutants of RhoA were determined in both live and fixed cells. For live cell analyses, NIH 3T3 cells plated on coverslips in 6-well dishes were transiently transfected (Lipofectamine-Plus; Life Technologies, Inc.) with expression vectors encoding GFP-tagged RhoA(63L) proteins. Twenty-four h after transfection, the coverslips were washed twice with PBS and inverted onto microscope slides. Live cells were examined with a Axioskop 2 fluorescence microscope (Zeiss) and Openlab digital imaging software (Improvision). To evaluate the sensitivity of prenylation-dependent subcellular localization, cells expressing RhoA(63L)-WT and RhoA(63L)-CVLS proteins were treated with FTase and GG-Tase inhibitors. For these analyses, cells transiently expressing the GFP-tagged proteins were incubated for 24 h with growth medium supplemented with either vehicle (DMSO), 10 μM FTI-2153, or 10 μm GGTI-2166.
For our second approach to evaluate subcellular location of the RhoA mutant proteins, NIH 3T3 cells stably expressing HA epitope-tagged WT and mutated RhoA(63L) proteins were plated onto coverslips and incubated overnight in growth medium. The cells were then fixed in 3.7% formaldehyde in PBS for 10 min and incubated for 30 min in PBS supplemented with 0.1% Triton X-100 and 5 mg/ml BSA. The HA epitope-tagged RhoA(63L) proteins were visualized by indirect immunofluorescence using an anti-HA antibody, followed by a FITC-conjugated goat antimouse secondary antibody (Jackson Immunoresearch Laboratories).
Analysis of HIV-1 Replication and Gene Expression
Inhibition of HIV-1 replication was assessed by cotransfection of the HIV-1 proviral genome pNL4–3 with each pcDNA-rhoA plasmid DNA into 293T cells using Effectene (Qiagen) as reported previously (18). Infectious units and virion-associated reverse transcriptase activity in culture supernatant were determined by titering on HeLa-MAGI cells at 48 h after transfection as described previously (62). A pAX142-lacZ reporter plasmid, where the lacZ gene is expressed constitutively from the EF1 α promoter, was also cotransfected, and β-galactosidase activity was measured to confirm similar transfection efficiency. Inhibition of the HIV-1 gene expression in human T cells (Jurkat) was analyzed by cotransfecting (Superfect; Qiagen) pcDNA-rhoA plasmid DNA together with pNL4-Luc (49), where the luciferase gene was under control of the HIV-1 long terminal repeat promoter. Forty-eight h after transfection, cells were lysed, and luciferase activity was determined as described (18).
Acknowledgments
We thank Que Lambert, Carol Martin, and Michael Townsend for valuable technical assistance and Misha Rand for figure and manuscript preparation. We also thank Said Sebti and Andrew Hamilton for providing the FTase and GGTase inhibitors and Adrienne Cox for helpful discussions.
Footnotes
Supported by NIH Grants CA42978, CA55008, and CA63071 (to C. J. D.) and AI48407 (to L. S.).
The abbreviations used are: NF-κB, nuclear factor-κB; SRF, serum response factor; FTase, farnesyltransferase; GGTaseI, geranylgeranyltransferase I; FTI, farnesyltransferase inhibitor; GGTI, geranylgeranyltransferase I inhibitor; HA, hemagglutinin; GFP, green fluorescent protein; SRF, serum response factor; WT, wild type.
P. J. Keely, manuscript in preparation.
P. A. Solski and C. J. Der, manuscript in preparation.
References
- 1.Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 2.Zohn IM, Campbell SL, Khosravi-Far R, Rossman KL, Der CJ. Rho family proteins and Ras transformation: the RHOad less traveled gets congested. Oncogene. 1998;17:1415–1438. doi: 10.1038/sj.onc.1202181. [DOI] [PubMed] [Google Scholar]
- 3.Han JS, Kim JH, Kim JG, Park JB, Noh DY, Lee KH. Molecular cloning and sequencing of rat Cdc42 GTPase cDNA. Exp Mol Med. 2000;32:115–119. doi: 10.1038/emm.2000.20. [DOI] [PubMed] [Google Scholar]
- 4.Vignal E, De Toledo M, Comunale F, Ladopoulou A, Gauthier-Rouviere C, Blangy A, Fort P. Characterization of TCL, a new GTPase of the Rho family related toTC10and Cdc42. J Biol Chem. 2000;275:36457–36464. doi: 10.1074/jbc.M003487200. [DOI] [PubMed] [Google Scholar]
- 5.Ellis S, Mellor H. The novel Rho-family GTPase Rif regulates coordinated actin-based membrane rearrangements. Curr Biol. 2000;10:1387–1390. doi: 10.1016/s0960-9822(00)00777-6. [DOI] [PubMed] [Google Scholar]
- 6.Guasch RM, Scambler P, Jones GE, Ridley AJ. RhoE regulates actin cytoskeleton organization and cell migration. Mol Cell Biol. 1998;18:4761–4771. doi: 10.1128/mcb.18.8.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nobes CD, Lauritzen I, Mattei MG, Paris S, Hall A, Chardin P. A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol. 1998;141:187–197. doi: 10.1083/jcb.141.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keely PJ, Westwick JK, Whitehead IP, Der CJ, Parise LV. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature (Lond) 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- 9.Michiels F, Habets GG, Stam JC, van der Kammen RA, Collard JG. A role for Rac in Tiam1-induced membrane ruffling and invasion. Nature (Lond) 1995;375:338–340. doi: 10.1038/375338a0. [DOI] [PubMed] [Google Scholar]
- 10.van Golen KL, Wu ZF, Qiao XT, Bao LW, Merajver SD. RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res. 2000;60:5832–5838. [PubMed] [Google Scholar]
- 11.Cox AD, Toussaint LG, III, Fiordalisi JJ, Rogers-Graham K, Der CJ. Farnesyltransferase and geranylgeranyltransferase inhibitors: the saga continues. In: Sebti SM, Hamilton A, editors. Farnesyltransferase and Geranylgeranyltransferase Inhibitors: The Saga Continues. Totowa, NJ: Humana Press; 2001. pp. 255–273. [Google Scholar]
- 12.Perona R, Montaner S, Saniger L, Sanchez-Perez I, Bravo R, Lacal JC. Activation of the nuclear factor-κB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11:463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- 13.Sulciner DJ, Irani K, Yu ZX, Ferrans VJ, Goldschmidt-Clermont P, Finkel T. Rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-κB activation. Mol Cell Biol. 1996;16:7115–7121. doi: 10.1128/mcb.16.12.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1 and Cdc42Hs regulate transcriptional activation by SRF. Cell. 1995;81:1159–1170. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- 15.Coso OA, Chiariello M, Yu J-C, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 16.Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Wang L, Kao S, Whitehead IP, Hart MJ, Liu B, Duus K, Burridge K, Der CJ, Su L. Functional interaction between the cytoplasmic leucine-zipper domain of HIV-1 gp41 and p115-RhoGEF. Curr Biol. 1999;9:1271–1274. doi: 10.1016/s0960-9822(99)80511-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Zhang H, Solski PA, Hart MJ, Der CJ, Su L. Modulation of HIV-1 replication by a novel RhoA effector activity. J Immunol. 2000;164:5369–5374. doi: 10.4049/jimmunol.164.10.5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348(Pt 2):241–255. [PMC free article] [PubMed] [Google Scholar]
- 20.Cox AD, Der CJ. Protein prenylation: more than just glue? Curr Opin Cell Biol. 1992;4:1008–1016. doi: 10.1016/0955-0674(92)90133-w. [DOI] [PubMed] [Google Scholar]
- 21.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 22.Foster R, Hu KQ, Lu Y, Nolan KM, Thissen J, Settleman J. Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in vivo farnesylation. Mol Cell Biol. 1996;16:2689–2699. doi: 10.1128/mcb.16.6.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post-translational modifications of p21rho proteins. J Biol Chem. 1992;267:20033–20038. [PubMed] [Google Scholar]
- 24.Baron R, Fourcade E, Lajoie-Mazenc I, Allal C, Couderc B, Barbaras R, Favre G, Faye JC, Pradines A. RhoB prenylation is driven by the three carboxyl-terminal amino acids of the protein: evidenced in vivo by an anti-farnesyl cysteine antibody. Proc Natl Acad Sci USA. 2000;97:11626–11631. doi: 10.1073/pnas.97.21.11626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox AD, Der CJ. Farnesyltransferase inhibitors and cancer treatment: targeting simply Ras? Biochim Biophys Acta. 1997;1333:F51–F71. doi: 10.1016/s0304-419x(97)00011-5. [DOI] [PubMed] [Google Scholar]
- 26.Gibbs JB, Oliff A. The potential of farnesyltransferase inhibitors as cancer chemotherapeutics. Annu Rev Pharmacol Toxicol. 1997;37:143–166. doi: 10.1146/annurev.pharmtox.37.1.143. [DOI] [PubMed] [Google Scholar]
- 27.Sebti SM, Hamilton AD. Inhibition of Ras prenylation: a novel approach to cancer chemotherapy. Pharmacol Ther. 1997;74:103–114. doi: 10.1016/s0163-7258(97)00014-4. [DOI] [PubMed] [Google Scholar]
- 28.Hancock JF, Cadwallader K, Paterson H, Marshall CJ. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 1991;10:4033–4039. doi: 10.1002/j.1460-2075.1991.tb04979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox AD, Hisaka MM, Buss JE, Der CJ. Specific isoprenoid modification is required for function of normal, but not oncogenic, Ras protein. Mol Cell Biol. 1992;12:2606–2615. doi: 10.1128/mcb.12.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prendergast GC, Khosravi-Far R, Solski PA, Kurzawa H, Lebowitz PF, Der CJ. Critical role of RhoB in cell transformation by oncogenic Ras. Oncogene. 1995;10:2289–2296. [PubMed] [Google Scholar]
- 31.Lebowitz PF, Du W, Prendergast GC. Prenylation of RhoB is required for its cell transforming function but not its ability to activate serum response element-dependent transcription. J Biol Chem. 1997;272:16093–16095. doi: 10.1074/jbc.272.26.16093. [DOI] [PubMed] [Google Scholar]
- 32.Lebowitz PF, Casey PJ, Prendergast GC, Thissen J. A Farnesyltransferase inhibitors alter the prenylation and growth-stimulating function of RhoB. J Biol Chem. 1997;272:15591–15594. doi: 10.1074/jbc.272.25.15591. [DOI] [PubMed] [Google Scholar]
- 33.Du W, Lebowitz PF, Prendergast GC. Cell growth inhibition by farnesyltransferase inhibitors is mediated by gain of geranylgeranylated RhoB. Mol Cell Biol. 1999;19:1831–1840. doi: 10.1128/mcb.19.3.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu AX, Du W, Liu JP, Jessell T, Prendergast GC. RhoB alteration is necessary for apoptotic and antineoplastic responses to farnesyltransferase inhibitors. Mol Cell Biol. 2000;20:6105–6113. doi: 10.1128/mcb.20.16.6105-6113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adamson P, Paterson HF, Hall A. Intracellular localization of the p21rho proteins. J Cell Biol. 1993;119:617–627. doi: 10.1083/jcb.119.3.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michaelson D, Silletti J, Murphy G, D’Eustachio P, Rush M, Philips MR. Differential Localization of Rho GTPases in live cells. Regulation by hypervariable regions and rhogdi binding. J Cell Biol. 2001;152:111–126. doi: 10.1083/jcb.152.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miquel K, Pradines A, Sun J, Qian Y, Hamilton AD, Sebti SM, Favre G. GGTI-298 induces G0-G1 block and apoptosis whereas FTI-277 causes G2-M enrichment in A549 cells. Cancer Res. 1997;57:1846–1850. [PubMed] [Google Scholar]
- 38.Vogt A, Sun J, Qian Y, Hamilton AD, Sebti SM. The geranylgeranyltransferase-I inhibitor GGTI-298 arrests human tumor cells in G0/G1 and induces p21WAF1/CIP1/SDI1 in a p53-independent manner. J Biol Chem. 1997;272:27224–27229. doi: 10.1074/jbc.272.43.27224. [DOI] [PubMed] [Google Scholar]
- 39.Sun J, Qian Y, Hamilton AD, Sebti SM. Both farnesyltrans-ferase and geranylgeranyltransferase I inhibitors are required for inhibition of oncogenic K-Ras prenylation but each alone is sufficient to suppress human tumor growth in nude mouse xenografts. Oncogene. 1998;16:1467–1473. doi: 10.1038/sj.onc.1201656. [DOI] [PubMed] [Google Scholar]
- 40.Sun J, Qian Y, Chen Z, Marfurt J, Hamilton AD, Sebti SM. The geranylgeranyltransferase I inhibitor GGTI-298 induces hypophosphorylation of retinoblastoma and partner switching of cyclin-dependent kinase inhibitors. A potential mechanism for GGTI-298 antitumor activity. J Biol Chem. 1999;274:6930–6934. doi: 10.1074/jbc.274.11.6930. [DOI] [PubMed] [Google Scholar]
- 41.Casey PJ, Seabra MC. Protein prenyltransferases. J Biol Chem. 1996;271:5289–5292. doi: 10.1074/jbc.271.10.5289. [DOI] [PubMed] [Google Scholar]
- 42.Lebowitz PF, Davide JP, Prendergast GC. Evidence that farnesyltransferase inhibitors suppress Ras transformation by interfering with Rho activity. Mol Cell Biol. 1995;15:6613–6622. doi: 10.1128/mcb.15.12.6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Westwick JK, Lambert QT, Clark GJ, Symons M, Van Aelst L, Pestell RG, Der CJ. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol Cell Biol. 1997;17:1324–1335. doi: 10.1128/mcb.17.3.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khosravi-Far R, Solski PA, Clark CJ, Kinch MS, Der CJ. Activation of Rac and Rho, and mitogen activated protein kinases, are required for Ras transformation. Mol Cell Biol. 1995;15:6443–6453. doi: 10.1128/mcb.15.11.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiu RG, Chen J, McCormick F, Symons M. A role for Rho in Ras transformation. Proc Natl Acad Sci USA. 1995;92:11781–11785. doi: 10.1073/pnas.92.25.11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature (Lond) 1995;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- 47.Solski PA, Abe K, Der CJ. Analyses of transforming activity of Rho family activators. Methods Enzymol. 2000;325:425–441. doi: 10.1016/s0076-6879(00)25463-3. [DOI] [PubMed] [Google Scholar]
- 48.Keely PJ. Ras and Rho protein induction of motility and invasion in T47D breast adenocarcinoma cells. Methods Enzymol. 2001;333:256–266. doi: 10.1016/s0076-6879(01)33061-6. [DOI] [PubMed] [Google Scholar]
- 49.He J, Landau NR. Use of a novel human immunodeficiency virus type 1 reporter virus expressing human placental alkaline phosphatase to detect an alternative viral receptor. J Virol. 1995;69:4587–4592. doi: 10.1128/jvi.69.7.4587-4592.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vogt A, Qian Y, McGuire TF, Hamilton AD, Sebti SM. Protein geranylgeranylation, not farnesylation, is required for the G1 to S phase transition in mouse fibroblasts. Oncogene. 1996;13:1991–1999. [PubMed] [Google Scholar]
- 51.Yamamoto M, Marui N, Sakai T, Morii N, Kozaki S, Ikai K, Imamura S, Narumiya S. ADP-ribosylation of the rhoA gene product by botulinum C3 exoenzyme causes Swiss 3T3 cells to accumulate in the G1 phase of the cell cycle. Oncogene. 1993;8:1449–1455. [PubMed] [Google Scholar]
- 52.Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac and Cdc42 GTPases in cell cycle progression through G1. Science (Wash DC) 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- 53.Casey PJ. Lipid modifications of G proteins. Curr Opin Cell Biol. 1994;6:219–225. doi: 10.1016/0955-0674(94)90139-2. [DOI] [PubMed] [Google Scholar]
- 54.Myung CS, Yasuda H, Liu WW, Harden TK, Garrison JC. Role of isoprenoid lipids on the heterotrimeric G protein γ subunit in determining effector activation. J Biol Chem. 1999;274:16595–16603. doi: 10.1074/jbc.274.23.16595. [DOI] [PubMed] [Google Scholar]
- 55.Matsuda T, Hashimoto Y, Ueda H, Asano T, Matsuura Y, Doi T, Takao T, Shimonishi Y, Fukada Y. Specific isoprenyl group linked to transducin γ-subunit is a determinant of its unique signaling properties among G-proteins. Biochemistry. 1998;37:9843–9850. doi: 10.1021/bi973194c. [DOI] [PubMed] [Google Scholar]
- 56.Allal C, Favre G, Couderc B, Salicio S, Sixou S, Hamilton AD, Sebti SM, Lajoie-Mazenc I, Pradines A. RhoA prenylation is required for promotion of cell growth and transformation and cytoskeleton organization but not for induction of serum response element transcription. J Biol Chem. 2000;275:31001–31008. doi: 10.1074/jbc.M005264200. [DOI] [PubMed] [Google Scholar]
- 57.Chen Z, Sun J, Pradines A, Favre G, Adnane J, Sebti SM. Both farnesylated and geranylgeranylated RhoB inhibit malignant transformation and suppress human tumor growth in nude mice. J Biol Chem. 2000;275:17974–17978. doi: 10.1074/jbc.C000145200. [DOI] [PubMed] [Google Scholar]
- 58.Cepko CL, Roberts B, Mulligan RC. Construction and applications of a highly transmissible murine retrovirus shuttle vector. Cell. 1984;37:1053–1062. doi: 10.1016/0092-8674(84)90440-9. [DOI] [PubMed] [Google Scholar]
- 59.Lerner EC, Qian Y, Blaskovich MA, Fossum RD, Vogt A, Sun J, Cox AD, Der CJ, Hamilton AD, Sebti SM. Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J Biol Chem. 1995;270:26802–26806. doi: 10.1074/jbc.270.45.26802. [DOI] [PubMed] [Google Scholar]
- 60.Galang CK, Der CJ, Hauser CA. Oncogenic Ras can induce transcriptional activation through a variety of promoter elements, including tandem c-Ets-2 binding sites. Oncogene. 1994;9:2913–2921. [PubMed] [Google Scholar]
- 61.Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- 62.Kimpton J, Emerman M. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated β-galactosidase gene. J Virol. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]