

Abstract
Objectives
To use a systems biology approach to integrate genotype and protein-protein interaction (PPI) data to identify disease network modules associated with chronic obstructive pulmonary disease (COPD) and to perform traditional pathway analysis.
Methods
We used a standard gene-set association approach (FORGE) using gene-based association analysis and gene-set definitions from the molecular signatures database (MSigDB). As a discovery step we analyzed GWAS results from two well-characterized COPD cohorts, COPDGene and GenKOLS. We used a third well-characterized COPD case-control cohort for replication, ECLIPSE. Next, we used dmGWAS, a method that integrates GWAS results with PPI, to identify COPD disease modules.
Results
No gene-sets reached experiment-wide significance in either discovery population. We identified a consensus network of 10 genes identified in modules by integrating GWAS results with PPI that replicated in COPDGene, GenKOLS, and ECLIPSE. Members of four gene-sets were enriched among these 10 genes: (i) lung adenocarcinoma tumor sequencing genes, (ii) IL7 pathway genes, (iii) kidney cell response to arsenic, and (iv) CD4 T cell responses. Further, several genes have also been associated with pathophysiology relevant to COPD including KCNK3, NEDD4L and RIN3. In particular, KCNK3 has been associated with pulmonary arterial hypertension, a common complication in advanced COPD.
Conclusion
We report a set of new genes that may influence the etiology of COPD that would not have been identified using traditional GWAS and pathway analyses alone.
Keywords: COPD, genome-wide scan, protein-protein interaction network, gene-set association, pathway association, disease module
Introduction
Genome-wide association studies (GWAS) have been extremely successful at identifying novel genomic regions associated with complex traits. In the past 7 years, the use of this approach has led to identification of approximately 2,000 genetic associations for 300 complex traits[1]. However, in a typical GWAS, millions of genetic markers are tested and only a small number reach a genome-wide level of statistical significance (GWS). This is not surprising, since most of the genetic variants in the genome are likely not associated with disease, and for those that are associated, there is low power to detect valid genetic effects with the complex disease phenotype[2]. One strategy for overcoming this power limitation is to focus on investigating the joint effects of multiple functionally related genes, termed genesets or pathways. In addition to the potential for providing valuable biological insights into the etiology of complex disease[3], these methods may have increased power to detect disease-related genes. Further, recent developments have enabled the integration of data from GWAS with the protein-protein interaction (PPI) network. Using such approaches, genes and proteins involved in the same disease have been shown to cluster together when centrality is measured within the PPI[4]. These observations led to development of the term “disease module” to describe this phenomenon[4,5].
In this manuscript, we applied both gene-set association analysis and a method integrating GWAS results with PPI, called dmGWAS[6], in two GWAS of well-characterized Chronic Obstructive Pulmonary Disease (COPD) case-control studies. A third COPD case-control study was included for replication of our gene-set and network results. Several genetic loci have been associated with COPD susceptibility phenotypes using GWAS including FAM13A[7,8], HHIP[8], CHRNA3/CHRNA5/IREB2 locus on chromosome 15[9], a multi-gene locus on chromosome 19[10] (harboring RAB4B, EGLN2, MIA and CYP2A6), TGFB2[11], MMP3/MMP12[11] and RIN3[11]. These results demonstrate the utility of GWAS data to discover novel genes involved in the etiology of COPD. In the most recent meta-analysis of four COPD case-control studies from our research group, applying gene-based analysis to the meta-analysis results provided additional support for novel genomic regions associated with COPD[11]. We hypothesized that applying both pathway and network approaches to GWAS data would identify additional genes involved in COPD pathogenesis.
Methods
Ethics Statement
Studies were approved by Institutional Review Boards at Partners Healthcare and all participating centers.
GWAS
Non-Hispanic White COPDGene participants (Ncases=2812, Ncontrols=2534) were genotyped on the Illumina Human OmniExpress chip. Norwegian GenKOLS participants (Ncases=863, Ncontrols=808) and ECLIPSE participants (Ncases=1764, Ncontrols=178) were genotyped on the Illumina HumanHap 550 array. GWAS data quality control, including both genotyped and imputed SNPs, was performed separately for COPDGene, GenKOLS and ECLIPSE participants adjusting for population substructure, smoking history and age of enrollment, as previously detailed[9,11].
Briefly, COPDGene is an observational study which enrolled COPD cases and control smokers at 21 U.S. clinical centers[12]. Enrolled subjects were self-identified Non-Hispanic White or Non-Hispanic African-American aged 45–80 with at least 10 pack-years of lifetime smoking history. Subjects with other diagnosed lung diseases except asthma and subjects with a first or second-degree relative enrolled in the study were excluded. GenKOLS is a COPD case-control study of enrolled subjects from Bergen, Norway[13]. Subjects in GenKOLS had at least 2.5 pack-years of smoking history. COPD subjects and controls aged 45–75 years with a history of smoking ≥ 10 pack-years from 46 centers across 12 countries were recruited as part of the ECLIPSE (Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints) study (SCO104960, NCT00292552, www.eclipse-copd.com)[14].
Using FORGE for gene-based and gene-set association testing
Gene-based analysis of the three study populations (COPDGene, GenKOLS and ECLIPSE) was conducted separately using FORGE[15]. The analyses focused on genomic sequences that included both the genes themselves and a 20kb window on either side of the genes to account for important regulatory regions. Along with the summary statistics for the two studies, genotype data from the European HapMap 2 samples were used (CEU). Details about the program and the test statistic used to calculate the gene-based p-values (fixed-effects Z score method) are provided elsewhere[16]. The gene level p-values for both studies were subsequently subjected to gene-set level analyses using the same software (FORGE). We used the molecular signatures database (MSigDB v4.0[17]) to retrieve information on gene-set collections. This included the pathways from BioCarta (217 gene-sets successfully analyzed), Chemical and Genomic Perturbations (3398 gene-sets), REACTOME (674 genesets), MicroRNA Targets (221 gene-sets), Transcription Factor Targets (615 gene-sets), and immunologic signatures (1910 gene-sets). In addition, information was obtained for Gene Ontology (GO; 1454 GO terms) terms and the Kyoto Encyclopedia of Genes and Genomes (KEGG, 186 KEGG pathways). A total of 8,675 pathways were included. More information on the procedure for gene-set analyses in FORGE can be found elsewhere[16]. The gene-set association analysis was run using the gene-based results from COPDGene for discovery and GenKOLS for replication. Weighted meta-analysis p-values were generated using the R library SEQSTAR and using an effective sample size (2*Ncases*Ncontrols)/(Ncases + Ncontrols) between COPDGene and GenKOLS[11,18,19]. Replication in the second replication population, ECLIPSE, was defined as P<0.05 for both gene and gene-set analyses.
Using dmGWAS to define disease modules by integrating GWAS and PPI data
We used the R library dmGWAS version 2.4 to integrate GWAS findings with PPI data [6]. We downloaded human PPI data from PINA[20], which encompasses collected and annotated data from 6 public databases: MINT, IntAct, DIP, BioGRID, HPRD and MIPS/MPact, on March 25, 2013. Briefly, dmGWAS uses the PPI data from PINA to create a network search space. In our analysis, each node in the dmGWAS search space also corresponds to a FORGE permuted gene-based result. We used the gene-based results from FORGE as inputs to dmGWAS, as this method corrects for both the number of SNPs and the size of the genomic region being considered in addition to generating a permuted p-value. Version 2.4 of dmGWAS does not provide such a method within the library. The dmGWAS algorithm considers every gene in the GWAS results as an initial seed module. It then tests all PPI neighbors within a step size defined by d to expand the seed modules. A PPI neighbor is added to the seed module if its distance to any node in the module is equal to or less than d and it increases the module test statistic, where k is number of genes in the module, by a factor of r. We used d=2 and r=0.1 as recommended by the dmGWAS developers[6]. In order to compare modules with different numbers of genes, dmGWAS generates a normalized module score, Zn, by standardizing the module’s Zm to a distribution of module test statistics generated by randomly selecting the same number of genes in the module from the entire network 100,000 times. This analysis was performed both in COPDGene and GenKOLS separately. We then used the dmGWAS function ‘dualEval()’ with COPDGene as the discovery dataset and GenKOLS as the replication dataset in order to combine the results. This function calculates test statistics for the modules defined in the discovery dataset and then for the same modules in the replication dataset. If a module is in the top 5% of the discovery dataset dmGWAS modules and also in the top 5% of the replication dataset dmGWAS modules, the module is considered to have replicated. We then assessed whether these modules replicated nominally (P<0.05) in a second replication population: ECLIPSE. To further assess the effect of network topology, we shuffled the FORGE gene-based p-values 10 times in both COPDGene and GenKOLS to assess the influence of network topology on dmGWAS. We then ran dmGWAS 10 times on COPDGene and GenKOLS with COPDGene as the discovery population and GenKOLS as the replication population. In order to further characterize the network modules that were obtained by usage of dmGWAS, we used GSEA[17] (http://www.broadinstitute.org/gsea) selecting the same MSigDB gene-sets as detailed in the FORGE analysis above to search for gene-set enrichment.
Results
FORGE Gene-Based Results
In order to perform pathway and network-based analyses using gene-based p-values for association with COPD, FORGE was used to generate gene-based association results for COPD affection status separately in COPDGene and GenKOLS. The top ten FORGE gene-based COPDGene results are listed in Table 1. In COPDGene, there were four genes that met gene-based genome-wide levels of statistical significance (p<2.65×10−6). All four of these genes (IREB2, FAM13A, CHRNB4 and RIN3) are located within previously reported regions of genome-wide significant associations to COPD. In the GenKOLS gene-based analysis, six of the top ten genes replicated nominally (P<0.05). In the combined results between COPDGene and GenKOLS, IREB2, FAM13A, CHRNA3 and CHRNB4 withstood correction for multiple testing. Only IREB2 also replicated nominally in ECLIPSE. In addition to previously identified COPD genes, one novel gene nearly met gene-based genome-wide significance in the analysis of the combined COPDGene and GenKOLS results (EEFSEC, P=5.4×10−6) but did not replicate nominally in ECLIPSE.
Table 1.
Top 10 FORGE gene-based results for COPD affection status in COPDGene and corresponding results in GenKOLS.
| Gene | Chr | Start | End | Discovery | Replication | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| COPDGene | GenKOLS | Combined Gene P |
ECLIPSE | ||||||||||
| Min P | Gene P | N | Min P | Gene P | N | Min P | Gene P | N | |||||
| IREB2 | 15 | 78729773 | 78793798 | 3.3E-07 | 1.0E-06 | 69 | 1.5E-04 | 0.00019 | 69 | 4.1E-09 | 0.0033 | 0.030 | 69 |
| FAM13A | 4 | 89647106 | 90032549 | 1.9E-08 | 1.0E-06 | 357 | 4.2E-04 | 0.0020 | 357 | 2.8E-08 | 7.7E-04 | 0.11 | 360 |
| CHRNB4 | 15 | 78916461 | 79012628 | 1.3E-08 | 1.0E-06 | 85 | 7.1E-04 | 0.0080 | 86 | 1.0E-07 | 0.16 | 0.52 | 86 |
| RIN3 | 14 | 92980118 | 93155339 | 1.7E-06 | 2.0E-06 | 186 | 0.0048 | 0.96 | 185 | 0.0014 | 0.041 | 0.21 | 189 |
| EEFSEC | 3 | 127872297 | 128127485 | 4.1E-06 | 1.0E-05 | 190 | 0.011 | 0.045 | 191 | 5.4E-06 | 0.035 | 0.13 | 192 |
| SULT1C3 | 2 | 108863651 | 108881807 | 8.3E-06 | 1.1E-05 | 80 | 0.040 | 0.14 | 80 | 2.4E-05 | 0.043 | 0.50 | 80 |
| CHRNA3 | 15 | 78885394 | 78913637 | 1.3E-08 | 1.7E-05 | 60 | 3.8E-04 | 0.0027 | 60 | 6.6E-07 | 0.059 | 0.17 | 60 |
| CHSY1 | 15 | 101715928 | 101792137 | 1.3E-05 | 2.1E-05 | 105 | 4.3E-04 | 0.012 | 104 | 2.9E-06 | 0.067 | 0.97 | 108 |
| SLC22A10 | 11 | 62905339 | 63137190 | 0.00017 | 2.9E-05 | 119 | 0.023 | 0.13 | 121 | 5.1E-05 | 0.061 | 0.57 | 121 |
| GRASP | 12 | 52400724 | 52409673 | 5.0E-05 | 3.5E-05 | 15 | 0.11 | 0.69 | 15 | 0.0012 | 0.15 | 0.82 | 15 |
‘Min P’ denotes the most significant single SNP association test p-value in that gene. ‘Gene P’ denotes the FORGE gene-wise p-value and ‘N’ denotes the number of SNPs evaluated in the gene. ‘Combined Gene P’ denotes fixed weighted meta-analysis gene-wise p-value between COPDGene and GenKOLS results.
Gene-Set Association Results
None of the 8,675 tested pathways were associated with COPD case-control status at a level that reached statistical significance in COPDGene as the discovery population. Out of 616 (5.77%) nominally significant gene-sets from the COPDGene analysis, 42 (6.82%) also reached nominal significance in the GenKOLS analysis. A list of all 42 of these replicated gene-sets is provided in Supplementary Table 1. The top COPDGene gene-set that also was significant using a level of nominal significance in GenKOLS is called REACTOME PRESYNAPTIC NICOTINIC ACETYLCHOLINE RECEPTORS (PCOPDGene=3.2×10−4, PGENKOLS=0.0066, Pcombined=2.8×10−5). This gene-set is comprised of 12 genes (Supplementary Table 2) including CHRNA3, CHRNA5, and CHRNB4 which have been previously associated with COPD in case-control GWAS. We also ran the gene-set association analysis for all gene-sets listed in Supplementary Table 1 excluding genes known to be associated with COPD (CHRNA3, CHRNA5, CHRNB4, IREB2, FAM13A and HHIP). Results excluding genes known to be associated with COPD were no longer as significant when the known COPD genes were removed. Along with the separate p-values for all three cohorts, p-values are also provided for the combined analyses between COPDGene and GenKOLS (Supplementary Table 1). None of the 42 gene-sets showed an experiment-wide significant p-value after correcting for multiple statistical testing (Bonferonni P<5.8×10−6).
Integrating COPD GWAS results with PPI data using dmGWAS
Using the FORGE gene-based results, disease modules were generated from COPDGene and GenKOLS data using dmGWAS. A total of 12,378 modules were generated for COPDGene and 12,385 modules were generated for GenKOLS. The normalized module score ranged from 6.0 (P=9.7×10−10) to 8.0 (P=6.7×10−16) for the COPDGene analysis and from 0.56 (P=2.9×10−1) to 6.8 (P=6.7×10−12) for the GenKOLS analysis. There was a much wider range in module scores for the GenKOLS analysis due to three modules with P>0.23. These three modules contained the same set of 5 genes where only one gene had a nominally significant result and the rest of the genes had P>0.17. The average module size was 13.4 +/− 2.1 genes for COPDGene and 12.3 +/− 1.9 genes for GenKOLS. By default, dmGWAS only reports modules containing at least 5 genes. The smallest module generated for COPDGene had 8 genes whereas the smallest module generated for GenKOLS had 5 genes. A total of 15 modules were significant in both COPDGene and GenKOLS dmGWAS analyses when COPDGene was used as the discovery dataset. These 15 modules are comprised of 55 non-redundant genes (Supplementary Figure 1a). A total of 12 of these 15 modules reached nominal statistical significance in ECLIPSE (Figure 1a; Table 2), comprising 50 non-redundant genes. In Figure 1a, the consensus module highlights UBC, which has an extremely high degree of connectivity (degree 7,872). To determine whether these results highlighting UBC might be biased due to its extreme connectivity, despite the permutation implemented in dmGWAS, we performed node-based permutation by shuffling the FORGE gene p-values in both COPDGene and GenKOLS. We then repeated the analyses using COPDGene and GenKOLS to generate consensus networks. In every permutation, UBC was a hub in the consensus modules (Supplementary Figure 2).
Figure 1.
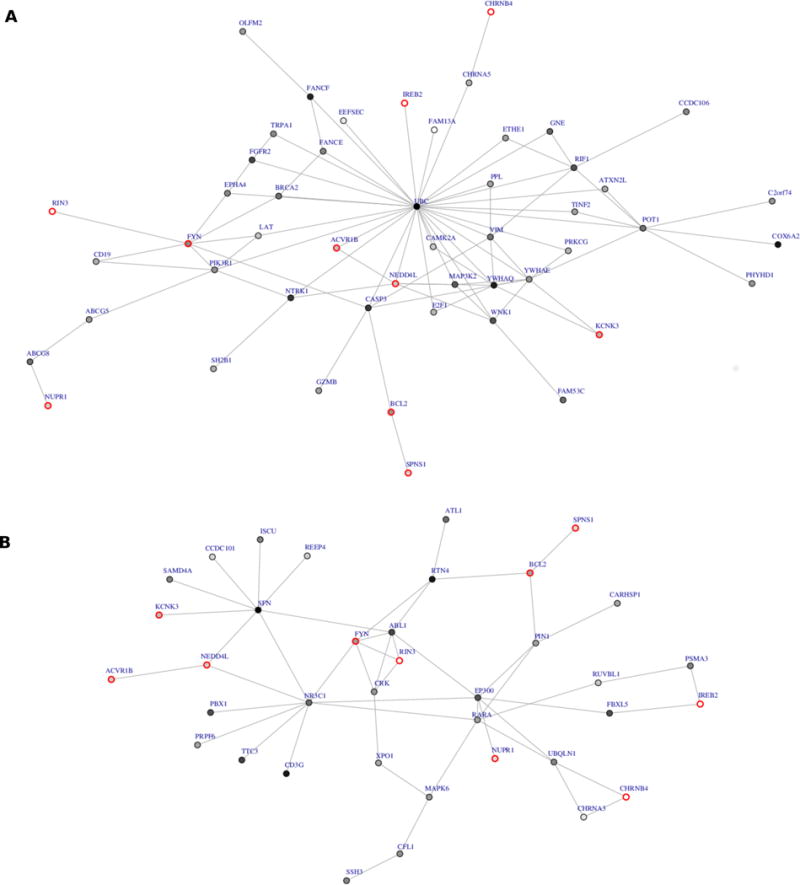
a. dmGWAS consensus module generated using the COPDGene results that replicated in GenKOLS and ECLIPSE. Node color is proportional to p-value significance where the lighter the node shading the smaller the p-value from the gene-based analysis in FORGE. b. dmGWAS analysis repeated excluding UBC.
Table 2. The 15 significant modules in the dmGWAS analysis of COPDGene and GenKOLS.
Modules shaded in grey did not replicate in ECLIPSE (P<0.05). Zn – dmGWAS normalized module score.
| COPDGene | GenKOLS | ECLIPSE | ||||
|---|---|---|---|---|---|---|
| Module Seed Gene | Zn | Pvalue | Zn | Pvalue | Zn | Pvalue |
| FANCE | 7.87 | 1.8E-15 | 3.77 | 8.2E-05 | 2.53 | 0.0057 |
| EPHA4 | 7.87 | 1.8E-15 | 3.88 | 5.3E-05 | 2.48 | 0.0065 |
| FANCF | 7.78 | 3.6E-15 | 4.13 | 1.9E-05 | 2.45 | 0.0071 |
| FGFR2 | 7.85 | 2.0E-15 | 3.81 | 7.0E-05 | 2.31 | 0.011 |
| GNE | 7.69 | 7.3E-15 | 3.82 | 6.6E-05 | 2.03 | 0.021 |
| NTRK1 | 7.68 | 8.0E-15 | 3.64 | 1.3E-04 | 1.96 | 0.025 |
| ABCG8 | 7.81 | 2.9E-15 | 3.84 | 6.2E-05 | 1.93 | 0.027 |
| YWHAQ | 7.78 | 3.8E-15 | 4.27 | 9.9E-06 | 1.87 | 0.030 |
| CASP3 | 7.78 | 3.7E-15 | 4.09 | 2.2E-05 | 1.83 | 0.034 |
| FAM53C | 7.79 | 3.2E-15 | 4.73 | 1.2E-06 | 1.79 | 0.037 |
| COX6A2 | 7.68 | 7.9E-15 | 3.69 | 1.1E-04 | 1.70 | 0.045 |
| MAP3K2 | 7.78 | 3.7E-15 | 4.60 | 2.1E-06 | 1.69 | 0.045 |
|
| ||||||
| WNK1 | 7.75 | 4.4E-15 | 4.62 | 1.9E-06 | 1.45 | 0.074 |
| STAC | 7.81 | 2.9E-15 | 4.19 | 1.4E-05 | 0.94 | 0.17 |
| RGS3 | 7.75 | 4.8E-15 | 3.77 | 8.3E-05 | 0.63 | 0.26 |
In order to address the concern that the top dmGWAS modules were driven by the high degree of UBC, we repeated the dmGWAS analysis detailed above, including replication in ECLIPSE, without UBC (Figure 1b, Supplementary Figure 1b). We found that several of the same genes were present in this consensus module generated with COPDGene as the discovery population that replicated with GenKOLS. Ten of the 50 genes in the original consensus network (Figure 1a) were also in the new consensus network (Figure 1b). These genes are listed in Table 3. The degree of connectivity of the 10 genes in the consensus modules ranged from 5 to 298.
Table 3.
The 10 genes in the consensus module of the dmGWAS analysis performed in COPDGene and GenKOLS, with replication in ECLIPSE, that were also in the consensus module when dmGWAS was run again excluding UBC.
| METAINFO | Discovery | Replication | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| COPDGene | GenKOLS | Combined Gene P |
ECLIPSE | |||||||||||
| GENE | CHR | START | STOP | Degree | Min P | Gene P | N | Min P | Gene P | N | Min P | Gene P | N | |
| ACVR1B | 12 | 52345451 | 52390862 | 24 | 5.0E-05 | 3.4E-04 | 25 | 0.11 | 0.34 | 23 | 1.5E-03 | 0.058 | 0.30 | 25 |
| BCL2 | 18 | 60790579 | 60987361 | 85 | 2.1E-04 | 7.2E-03 | 229 | 9.6E-04 | 0.41 | 235 | 2.5E-02 | 0.0020 | 0.16 | 234 |
| CHRNB4 | 15 | 78916461 | 79012628 | 5 | 1.3E-08 | 1.0E-06 | 85 | 7.1E-04 | 0.0080 | 86 | 1.0E-07 | 0.16 | 0.52 | 86 |
| FYN | 6 | 111981535 | 112194655 | 298 | 0.017 | 0.012 | 292 | 0.0024 | 0.0032 | 293 | 9.7E-04 | 0.039 | 0.32 | 294 |
| IREB2 | 15 | 78729773 | 78793798 | 7 | 3.3E-07 | 1.0E-06 | 69 | 1.5E-04 | 1.9E-04 | 69 | 4.1E-09 | 0.0033 | 0.030 | 69 |
| KCNK3 | 2 | 26915619 | 26956288 | 9 | 6.1E-04 | 0.0011 | 32 | 7.3E-04 | 3.9E-04 | 32 | 1.6E-05 | 0.014 | 0.35 | 32 |
| NEDD4L | 18 | 55711599 | 56068772 | 132 | 0.0023 | 2.0E-04 | 437 | 0.0019 | 0.0043 | 437 | 1.3E-05 | 0.0094 | 0.27 | 436 |
| NUPR1 | 16 | 28548606 | 28550495 | 8 | 1.0E-05 | 7.2E-05 | 14 | 0.044 | 0.42 | 14 | 6.5E-04 | 0.017 | 0.020 | 14 |
| RIN3 | 14 | 92980118 | 93155339 | 15 | 1.7E-06 | 2.0E-06 | 186 | 0.0048 | 0.96 | 185 | 1.4E-03 | 0.041 | 0.21 | 189 |
| SPNS1 | 16 | 28985542 | 28995869 | 6 | 3.1E-04 | 7.6E-05 | 21 | 0.44 | 0.78 | 15 | 3.5E-03 | 0.15 | 0.19 | 22 |
METAINFO: GENE – Hugo gene name, CHR – chromosome, START – start position in base pairs from the start of the chromosome, STOP – stop position in base pairs from the start of the chromosome; Degree – connectivity of the gene in the protein-protein interaction network; COPDGene/GenKOLS: N – number of SNPs annotated to this gene for the respective study, Min P – p-value for the SNP with the lowest p-value (of all SNPs annotated to the gene), Gene P – gene-based p-value for this gene from FORGE analyses. ‘Combined Gene P’ denotes fixed weighted meta-analysis gene-wise p-value.
Probing the Overlap of dmGWAS and Gene-Set Association Results
For additional comparison purposes with the gene-set association analysis, we performed gene-set enrichment analysis (GSEA) of the 10 genes in common between the two consensus dmGWAS networks (Table 3). We used the same MSigDB pathways as the gene-set association analysis. Enrichment was found for gene-sets comprising lung adenocarcinoma tumor sequencing project genes (P=2.25×10−6, FDR q-value=1.69×10−2), the Biocarta IL-7 signal transduction pathway (P=4.63×10−6, FDR q-value=1.69×10−2), kidney cell response to sodium arsenite (P=5.21×10−6, FDR q-value=1.69×10−2), and genes up-regulated in CD4 T cells versus untreated CD8 T cells (P=6.69×10−6, FDR q-value=1.69×10−2).
Discussion
In this manuscript, we report pathway and network-based association analysis of COPD in two large, well-characterized COPD case-control populations, and identify 10 genes of interest for COPD pathogenesis by integrating GWAS and PPI data. These 10 genes include both genes previously known to be associated with COPD and also novel candidates for further investigation. Our gene-set analysis did not identify any significant gene-sets; the top gene-set identified in COPDGene, that replicated nominally in GenKOLS, contained a number of nicotinic receptor genes that have been associated with COPD in other studies. In contrast, when we used GSEA to examine the list of genes generated as a network incorporating GWAS and PPI, lung cancer, IL-7 signaling pathway, response to arsenic, and CD4 T cell regulatory genes were enriched in the networks in addition to several other genes that have been associated with disease processes relevant to COPD such as KCNK3, NEDD4L and RIN3. The majority of the genes highlighted through our network approach would not have been identified using a traditional GWAS approach. Thus, these results shed light on other genes that we would have not considered had the analysis been restricted to a standard GWAS.
Our gene-based association analysis with FORGE revealed a potential new candidate gene for COPD, EEFSEC. When we previously performed a gene-based analysis on the meta-analysis results from several COPD case-control GWAS[11], EEFSEC had a gene-based result near genome-wide significance of P=1.6×10−5 (result not published). As the goal of the current manuscript was to investigate genetic pathways and networks influencing the etiology of COPD, we performed gene-wise analyses within each study and then searched for significant genetic pathways and networks using COPDGene for discovery and GenKOLS for replication. In the current analysis, EEFSEC showed suggestive evidence for association for the meta-analysis of COPDGene and GenKOLS samples (P=5.4×10−6), as well as in COPDGene (P=1×10−5) but did not withstand correction for multiple testing (P<2.65×10−6). For EEFSEC, there was also nominal evidence for replication in GenKOLS (P<0.05) and a trend for association in ECLIPSE (P=0.13). EEFSEC (MIM 60795), which codes for Eukaryotic Elongation Factor for Selenocysteine-tRNA-specific, is necessary for the incorporation of selenocysteine in proteins. As there is an increased oxidant burden in smokers who develop COPD[21], the antioxidant enzymes that require trace elements such as selenium may be involved in COPD pathogenesis. Our gene-based analysis also highlighted several genes that are known to be associated with COPD including FAM13A[7,8], IREB2[9], and CHRNA3[9]. We also found an association with CHRNB4, which resides in the CHRNA3/CHRNA5/IREB2 locus on chromosome 15[9] and has also been associated with age of onset of daily smoking and habitual smoking[22].
The list of 10 genes that was generated from the genes common to the dmGWAS networks with and without UBC indicated that several gene-sets were enriched including genes associated with lung adenocarcinoma, IL-7 signaling pathway, kidney cell response to arsenic, and CD4 T cell responses. Smoking is a risk factor for both lung cancer and COPD. The prevalence of COPD is higher in lung cancer cases than the general population independent of age, sex and smoking history[23]. Thus, enrichment of genes (FYN and ACVR1B) known to be enriched with mutations in lung cancer cases in the consensus network associated with COPD case-control status is intriguing. IL-7 is an important cytokine for B and T cell development and as such has a role in the inflammatory response. Thymic stromal lymphopoietin (TSLP), an IL-7 orthologue that is expressed in airway biopsies, has been shown to mediate the effects of cigarette smoke-induced airway smooth muscle contractility in COPD cases [24,25]. Further, variants near TSLP have been associated with asthma, which is also characterized by airflow obstruction and has been suggested to share overlapping genetic determinants with COPD[26]. Additional genes from the consensus list that were annotated to the IL-7 pathway were FYN and Bcl-2; however, IL-7 and IL-7RA were not found in our consensus list. Of interest, TSLP has been shown to induce the expression of Bcl-2 in Th2 cells [27]. Moreover, PIK3R1 has been suggested as a drug target for COPD [26,28]. Arsenic is found in cigarette smoke[29] and as such it is intriguing that we identified genes (Bcl-2 and KCNK3) associated with kidney cell response to arsenic in the consensus network. Further, several of the consensus network genes (FYN, ACVR1B and Bcl-2) are known to be upregulated in comparison of untreated CD4 with untreated CD8 cells[30]. Lung CD4+ T cells are known to accumulate in the lungs of COPD cases and recently distinct polarization profiles of CD4+ T cells were correlated with degree of emphysema and reduction in spirometry[31]. It was only by taking a network approach that these pathways were highlighted in our results.
Within the list of 10 genes common to the dmGWAS networks with and without UBC are other genes of interest, such as KCNK3, NEDD4L and RIN3. The KCNK3 gene, which codes for potassium channel subfamily K member 3, has been recently associated with pulmonary arterial hypertension [32]; secondary pulmonary arterial hypertension is a known complication in advanced COPD. Knockout of NEDD4L in lung epithelia causes a cystic fibrosis-like disease [33], and low expression of the gene has been implicated as a prognostic marker for non-small cell lung cancer[34]. Recently, our group has demonstrated genetic association of variants near RIN3 with COPD and also that RIN3 gene expression in lung tissue is reduced in COPD cases compared to controls[11]. Thus, we have highlighted a panel of genes involved in the etiology of COPD using a systems biology approach; some of these genes would have been overlooked using traditional approaches.
Although gene-set association methods have been successfully used to highlight important pathways and gene-sets in other diseases, there are some limitations to consider when discussing our results. None of the tested gene-sets in our analyses were significantly associated with COPD case-control status using a Bonferonni corrected level of statistical significance. The top gene-set results, as listed in Supplementary Table 1, include many genes already known to be associated with COPD, and thus it is difficult to conclude that these gene-set results contribute additional insights. When we ran the gene-set association analysis for all gene-sets listed excluding genes known to be associated with COPD, the results were no longer as significant. Gene-set association analyses, although less demanding in terms of power requirements compared to GWAS, are still dependent on adequate sample size in the analyses. Although our discovery population, COPDGene, is the largest COPD case-control genetic study performed to date, it may have not been large enough to detect all gene-sets annotated in MSigDB that are associated with COPD. Further, gene-set definitions tend to be incomplete and rely on established biological hypotheses. For this reason, we elected to use dmGWAS to search for modules without a priori definitions of gene-sets.
One of the main disadvantages of using PPI information generated from yeast-two-hybrid and affinity-tag purification experiments is that the laboratory conditions may not correctly model biologically relevant conditions or states[35]. Further, much of the interactome has not been characterized, and efforts are continuing to be directed at remedying this limitation[25,36,37]. A specific limitation of approaches such as dmGWAS is that there are currently no methods for calculating power to assess if our study was large enough for the analysis. However, using this method is likely be more powerful than traditional GWAS as it provides a means for incorporating information from GWAS (i.e., results with P > 5×10−8) that may have been overlooked and also other sources of biological information[38]. It is important to note that many network analyses involving the interactome generate a network that is pejoratively termed a “hairball” due to its complexity[39]. In contrast, Figure 1a presents a somewhat simple network centered on a hub gene, UBC. This gene codes for ubiquitin C, a polyubiquitin precursor, and a key component of the ubiquitin-mediated protein degradation pathway controlling almost every process in cells[40]. By permuting P-value assignments, we demonstrated that UBC was present as a hub gene in every consensus network—suggesting that the high connectivity of this gene led to its inclusion in our COPD network. This motivated our group to run dmGWAS without UBC in an attempt to gain insight into how the topology of the PPI network may be influencing the results. This led to the compilation of a list of 10 genes that were present in both networks that are enriched with genes involved in lung cancer, IL-7 signaling, response to arsenic, and CD4 T cell response. Other than UBC, the three most connected nodes in the PPI network used to run dmGWAS were ELAVL1 (degree=1416), SUMO2 (degree=1142) and CUL3 (degree=1009). These three genes were not present in the list of 10 genes, giving some indication that this list of genes may be robust to the influence of hubs in the network topology we initially observed with UBC.
It has been suggested by network science investigators that hub genes are less likely to carry highly detrimental mutations due to their vital role in the viability of a cell [41]. Peripheral nodes are more likely to contain genetic variants influencing a complex disease, and these genetic variants may be of modest effect size. One of the most attractive features of dmGWAS is that even results that do not reach GWS are incorporated in the analysis, thereby maximizing the utility of the data. It is a versatile R library that only requires a PPI network as input and results formatted as a gene name and a test statistic. Given the flexibility in input framework, the algorithm could be applied to any set of experiments that produces a test statistic for each gene such as gene expression. Recently, Han et al used dmGWAS to define modules associated with alcohol dependence[42]. In contrast to our present report, they used the smallest p-value per gene as input to dmGWAS. It has been reported that using a gene-wise p-value as input to dmGWAS may be more robust to biases generated by gene length, SNP density, and/or linkage disequilibrium [42,43]. However, some SNPs are located in non-coding regulatory regions that may not act on the nearest gene[44,45,46,47]. As such, these situations would not be adequately addressed by assigning SNPs to genes based on distance alone. Thus, there is much room to develop algorithms such as dmGWAS to address issues such as accounting for the influence of gene size, the influence of non-coding regulatory regions on distant genes, the cell specificity of gene regulation, and the impact of network topology on consensus modules.
In sum, we have presented the application of gene-set association analysis and the integration of GWAS data with PPI in two well-characterized COPD cohorts. We have probed the landscape between these methods in performing GSEA on results generated from the integration of GWAS findings with a PPI network using the same gene-sets as our gene-set association. Methods integrating GWAS data and PPI networks, such as dmGWAS, are the first steps towards maximizing the use of genomic, transcriptomic and proteomic data to gain insight into human disease.
Supplementary Material
Acknowledgments
Funding: NHLBI support from NHLBI R01HL089856, R01HL089897, P01 HL105339, R01 HL113264, R01 HL111759 and R01 HL075478. GenKOLS and ECLIPSE were funded by GlaxoSmithKline.
References
- 1.Manolio TA. Bringing genome-wide association findings into clinical use. Nature Reviews Genetics. 2013;14:549–558. doi: 10.1038/nrg3523. [DOI] [PubMed] [Google Scholar]
- 2.Silverman EK, Loscalzo J. Network medicine approaches to the genetics of complex diseases. Discovery medicine. 2012;14:143–152. [PMC free article] [PubMed] [Google Scholar]
- 3.Herold C, Mattheisen M, Lacour A, Vaitsiakhovich T, Angisch M, et al. Integrated genome-wide pathway association analysis with INTERSNP. Human Heredity. 2012;73:63–72. doi: 10.1159/000336196. [DOI] [PubMed] [Google Scholar]
- 4.Goh KI, Cusick ME, Valle D, Childs B, Vidal M, et al. The human disease network. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loscalzo J, Barabasi AL. Systems biology and the future of medicine. Wiley interdisciplinary reviews Systems biology and medicine. 2011;3:619–627. doi: 10.1002/wsbm.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia P, Zheng S, Long J, Zheng W, Zhao Z. dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics (Oxford, England) 2011;27:95–102. doi: 10.1093/bioinformatics/btq615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho MH, Boutaoui N, Klanderman BJ, Sylvia JS, Ziniti JP, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nature Genetics. 2010;42:200–202. doi: 10.1038/ng.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hancock DB, Eijgelsheim M, Wilk JB, Gharib SA, Loehr LR, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nature Genetics. 2010;42:45–52. doi: 10.1038/ng.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pillai SG, Ge D, Zhu G, Kong X, Shianna KV, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genetics. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho MH, Castaldi PJ, Wan ES, Siedlinski M, Hersh CP, et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Human molecular genetics. 2012;21:947–957. doi: 10.1093/hmg/ddr524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho MH, McDonald ML, Zhou X, Mattheisen M, Castaldi PJ, et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. The Lancet Respiratory medicine. 2014;2:214–225. doi: 10.1016/S2213-2600(14)70002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, et al. Genetic epidemiology of COPD (COPDGene) study design. COPD. 2010;7:32–43. doi: 10.3109/15412550903499522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu G, Warren L, Aponte J, Gulsvik A, Bakke P, et al. The SERPINE2 gene is associated with chronic obstructive pulmonary disease in two large populations. American journal of respiratory and critical care medicine. 2007;176:167–173. doi: 10.1164/rccm.200611-1723OC. [DOI] [PubMed] [Google Scholar]
- 14.Vestbo J, Anderson W, Coxson HO, Crim C, Dawber F, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE) The European respiratory journal: official journal of the European Society for Clinical Respiratory Physiology. 2008;31:869–873. doi: 10.1183/09031936.00111707. [DOI] [PubMed] [Google Scholar]
- 15.Pedroso I, Breen G. Gene set analysis and network analysis for genome-wide association studies. Cold Spring Harbor protocols. 2011;2011 doi: 10.1101/pdb.top065581. [DOI] [PubMed] [Google Scholar]
- 16.Pedroso I, Lourdusamy A, Rietschel M, Nothen MM, Cichon S, et al. Common genetic variants and gene-expression changes associated with bipolar disorder are over-represented in brain signaling pathway genes. Biological Psychiatry. 2012;72:311–317. doi: 10.1016/j.biopsych.2011.12.031. [DOI] [PubMed] [Google Scholar]
- 17.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Belle G. Statistical rules of thumb. xviii. New York: Wiley-Interscience; 2002. p. 221. [Google Scholar]
- 19.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu J, Vallenius T, Ovaska K, Westermarck J, Makela TP, et al. Integrated network analysis platform for protein-protein interactions. Nature methods. 2009;6:75–77. doi: 10.1038/nmeth.1282. [DOI] [PubMed] [Google Scholar]
- 21.Macnee W, Rahman I. Oxidants and antioxidants as therapeutic targets in chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. 1999;160:S58–65. doi: 10.1164/ajrccm.160.supplement_1.15. [DOI] [PubMed] [Google Scholar]
- 22.Kapoor M, Wang JC, Bertelsen S, Bucholz K, Budde JP, et al. Variants located upstream of CHRNB4 on chromosome 15q25.1 are associated with age at onset of daily smoking and habitual smoking. PloS One. 2012;7:e33513. doi: 10.1371/journal.pone.0033513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young RP, Hopkins RJ, Christmas T, Black PN, Metcalf P, et al. COPD prevalence is increased in lung cancer, independent of age, sex and smoking history. The European respiratory journal. 2009;34:380–386. doi: 10.1183/09031936.00144208. [DOI] [PubMed] [Google Scholar]
- 24.Smelter DF, Sathish V, Thompson MA, Pabelick CM, Vassallo R, et al. Thymic stromal lymphopoietin in cigarette smoke-exposed human airway smooth muscle. Journal of immunology. 2010;185:3035–3040. doi: 10.4049/jimmunol.1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodge S, Hodge G, Holmes M, Reynolds PN. Increased peripheral blood T-cell apoptosis and decreased Bcl-2 in chronic obstructive pulmonary disease. Immunology and cell biology. 2005;83:160–166. doi: 10.1111/j.1440-1711.2005.01317.x. [DOI] [PubMed] [Google Scholar]
- 26.Barnes PJ. New anti-inflammatory targets for chronic obstructive pulmonary disease. Nature reviews Drug discovery. 2013;12:543–559. doi: 10.1038/nrd4025. [DOI] [PubMed] [Google Scholar]
- 27.Kitajima M, Lee HC, Nakayama T, Ziegler SF. TSLP enhances the function of helper type 2 cells. European journal of immunology. 2011;41:1862–1871. doi: 10.1002/eji.201041195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito K, Caramori G, Adcock IM. Therapeutic potential of phosphatidylinositol 3-kinase inhibitors in inflammatory respiratory disease. The Journal of pharmacology and experimental therapeutics. 2007;321:1–8. doi: 10.1124/jpet.106.111674. [DOI] [PubMed] [Google Scholar]
- 29.Pappas RS, Fresquez MR, Martone N, Watson CH. Toxic metal concentrations in mainstream smoke from cigarettes available in the USA. Journal of analytical toxicology. 2014;38:204–211. doi: 10.1093/jat/bku013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pearl JI, Lee AS, Leveson-Gower DB, Sun N, Ghosh Z, et al. Short-term immunosuppression promotes engraftment of embryonic and induced pluripotent stem cells. Cell stem cell. 2011;8:309–317. doi: 10.1016/j.stem.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freeman CM, McCubbrey AL, Crudgington S, Nelson J, Martinez FJ, et al. Basal Gene Expression by Lung CD4+ T Cells in Chronic Obstructive Pulmonary Disease Identifies Independent Molecular Correlates of Airflow Obstruction and Emphysema Extent. PloS One. 2014;9:e96421. doi: 10.1371/journal.pone.0096421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma L, Roman-Campos D, Austin ED, Eyries M, Sampson KS, et al. A novel channelopathy in pulmonary arterial hypertension. The New England Journal of Medicine. 2013;369:351–361. doi: 10.1056/NEJMoa1211097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura T, Kawabe H, Jiang C, Zhang W, Xiang YY, et al. Deletion of the ubiquitin ligase Nedd4L in lung epithelia causes cystic fibrosis-like disease. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3216–3221. doi: 10.1073/pnas.1010334108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakashita H, Inoue H, Akamine S, Ishida T, Inase N, et al. Identification of the NEDD4L Gene as a Prognostic Marker by Integrated Microarray Analysis of Copy Number and Gene Expression Profiling in Non-small Cell Lung Cancer. Annals of surgical oncology. 2013 doi: 10.1245/s10434-013-3059-6. [DOI] [PubMed] [Google Scholar]
- 35.Beltrao P, Ryan C, Krogan NJ. Comparative interaction networks: bridging genotype to phenotype. Advances in experimental medicine and biology. 2012;751:139–156. doi: 10.1007/978-1-4614-3567-9_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujimori S, Hirai N, Ohashi H, Masuoka K, Nishikimi A, et al. Next-generation sequencing coupled with a cell-free display technology for high-throughput production of reliable interactome data. Scientific reports. 2012;2:691. doi: 10.1038/srep00691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dreze M, Monachello D, Lurin C, Cusick ME, Hill DE, et al. High-quality binary interactome mapping. Methods in enzymology. 2010;470:281–315. doi: 10.1016/S0076-6879(10)70012-4. [DOI] [PubMed] [Google Scholar]
- 38.Sun YV. Integration of biological networks and pathways with genetic association studies. Human Genetics. 2012;131:1677–1686. doi: 10.1007/s00439-012-1198-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lander AD. The edges of understanding. BMC biology. 2010;8:40. doi: 10.1186/1741-7007-8-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Komander D, Rape M. The ubiquitin code. Annual review of biochemistry. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 41.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nature Reviews Genetics. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han S, Yang BZ, Kranzler HR, Liu X, Zhao H, et al. Integrating GWASs and Human Protein Interaction Networks Identifies a Gene Subnetwork Underlying Alcohol Dependence. American Journal of Human Genetics. 2013;93:1027–1034. doi: 10.1016/j.ajhg.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jia P, Wang L, Fanous AH, Chen X, Kendler KS, et al. A bias-reducing pathway enrichment analysis of genome-wide association data confirmed association of the MHC region with schizophrenia. Journal of Medical Genetics. 2012;49:96–103. doi: 10.1136/jmedgenet-2011-100397. [DOI] [PubMed] [Google Scholar]
- 44.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.