

Abstract
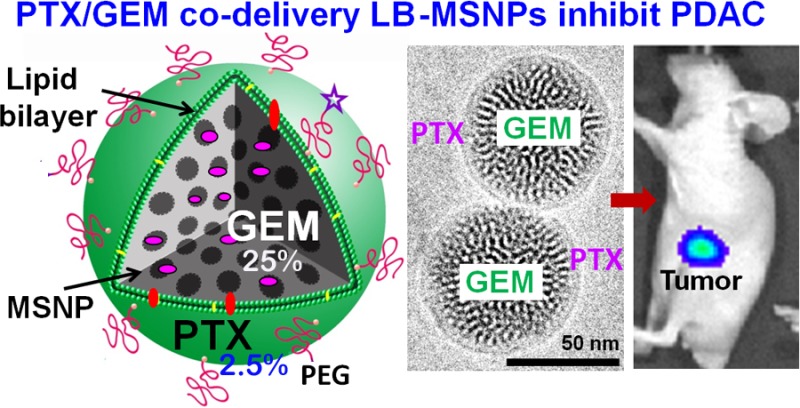
Recently, a commercial albumin-bound paclitaxel (PTX) nanocarrier (Abraxane) was approved as the first new drug for pancreatic ductal adenocarcinoma in almost a decade. PTX improves the pharmaceutical efficacy of the first-line pancreatic cancer drug, gemcitabine (GEM), through suppression of the tumor stroma and inhibiting the expression of the GEM-inactivating enzyme, cytidine deaminase (CDA). We asked, therefore, whether it was possible to develop a mesoporous silica nanoparticle (MSNP) carrier for pancreatic cancer to co-deliver a synergistic GEM/PTX combination. High drug loading was achieved by a custom-designed coated lipid film technique to encapsulate a calculated dose of GEM (40 wt %) by using a supported lipid bilayer (LB). The uniform coating of the 65 nm nanoparticles by a lipid membrane allowed incorporation of a sublethal amount of hydrophobic PTX, which could be co-delivered with GEM in pancreatic cells and tumors. We demonstrate that ratiometric PTX incorporation and delivery by our LB-MSNP could suppress CDA expression, contemporaneous with induction of oxidative stress as the operating principle for PTX synergy. To demonstrate the in vivo efficacy, mice carrying subcutaneous PANC-1 xenografts received intravenous (IV) injection of PTX/GEM-loaded LB-MSNP. Drug co-delivery provided more effective tumor shrinkage than GEM-loaded LB-MSNP, free GEM, or free GEM plus Abraxane. Comparable tumor shrinkage required coadministration of 12 times the amount of free Abraxane. High-performance liquid chromatography analysis of tumor-associated GEM metabolites confirmed that, compared to free GEM, MSNP co-delivery increased the phosphorylated DNA-interactive GEM metabolite 13-fold and decreased the inactivated and deaminated metabolite 4-fold. IV injection of MSNP-delivered PTX/GEM in a PANC-1 orthotopic model effectively inhibited primary tumor growth and eliminated metastatic foci. The enhanced in vivo efficacy of the dual delivery carrier could be achieved with no evidence of local or systemic toxicity. In summary, we demonstrate the development of an effective LB-MSNP nanocarrier for synergistic PTX/GEM delivery in pancreatic cancer.
Keywords: gemcitabine, paclitaxel, co-delivery, synergy, ratiometric, mesoporous silica nanoparticle, pancreatic cancer
The 5 year survival rate of pancreatic ductal adenocarcinoma has remained unchanged despite significant progress for other major cancers.1 One of the contributing reasons of the poor prognosis of pancreatic cancer is the resistance to chemotherapeutic agents, including gemcitabine (GEM), which is frequently used as first-line therapy.2,3 This resistance could result from acquired and/or intrinsic pathways, a dense dysplastic stroma that acts as a barrier to vascular perfusion and drug permeability, as well as unfavorable GEM pharmacokinetics as a result of insufficient activation or rapid inactivation.3 Recent progress in pancreatic cancer therapy has been the introduction of stromal-directed agents that obliterate the dense stromal microenvironment and improve chemotherapy drug delivery. For example, an ongoing clinical trial has demonstrated that the combination of GEM with PEGylated hyaluronidase can overcome stromal resistance, allowing chemotherapeutic drug access to the cancer site.4 In addition to the use of small-molecule therapeutics, nanotechnology is poised to make an impact on pancreatic cancer treatment, including the delivery of stromal-targeting agents. One example is the recent FDA approval for the albumin-bound paclitaxel nanocomplex, Abraxane, which extends the survival outcome of GEM by a few months upon coadministration.5,6 The proposed mechanism of Abraxane action is the suppression of stromal density as well as reduced expression of cytidine deaminase (CDA), an enzyme that is responsible for the metabolic inactivation of GEM, including at stromal and tumor sites.5−7
While drug delivery by a nanocarrier could make a big impact on cancer treatment, most therapeutic nanocarriers currently being used in clinical trials are monocarriers, which are primarily designed for passive drug delivery and/or reducing drug toxicity.8 Carrier design often does not address optimal drug loading, synergistic drug combinations at controlled ratio, or overcoming tumor-specific impediments, such as the stromal barrier or unfavorable pharmacokinetics of cancer drugs.9 Our aim is to advance pancreatic cancer nanotherapy by our multifunctional mesoporous silica nanoparticle (MSNP) platform,10−17 which can be designed to circumvent the stromal barrier, improve GEM pharmacokinetics, and allow contemporaneous delivery of synergistic drug combinations, such as illustrated by the clinical paclitaxel (PTX)/GEM example. However, while we have previously demonstrated that GEM delivery to human pancreatic cancer xenografts can be improved by a copolymer-designed MSNP carrier that releases a small-molecule TGF-β receptor kinase inhibitor, it was not possible to achieve high GEM loading capacity in this platform due to the inability of the copolymer to securely seal pores and rapidly entrap the drug.17 It was also difficult to load PTX in the same carrier in a controlled fashion for synergistic drug co-delivery. We therefore contemplated the design of a novel carrier that can achieve simultaneous PTX and GEM loading by a MSNP carrier, which also considers the chemical structure and hydrophobicity of these drugs, their combination at an optimal dose ratio, drug loading capacity, and rapid and high entrapment efficiency. Since these design characteristics cannot be achieved by our copolymer-designed MSNPs, we investigated the use of a supported lipid bilayer (LB) as proposed by Sackmann et al.,18 with adaptation to amorphous and mesoporous silica nanoparticles.19−28
Our original attempt to obtain GEM entrapment by a supported LB that forms when MSNPs are co-incubated with liposomes was met with limited success due to the lack of uniform coating. This is likely due to inefficiency in the stepwise liposomal procedure, which requires absorption to the particle surface, disruption, and electrostatic fusion with the particle surface; moreover, it is often required that defects in this incomplete LB be filled by the use of a second wave of liposomes.28 This inefficiency prompted us to develop a custom-designed procedure in which we use a lipid-film-coated on a round-bottomed glass surface for overlay with drug-soaked MSNPs, which could subsequently be rapidly sealed through energy input and with the ability to entrap a high GEM load. This lipid-film-coating procedure also allowed us to introduce, in a ratiometric approach, co-delivery of the hydrophobic drug, PTX, which is incorporated in the LB. We define a ratiometric approach as the in vivo release of a drug combination from a nanocarrier, with the purpose of providing a fixed drug ratio at the target site.29 Following demonstration of in vitro drug synergy, we asked whether the dual delivery MSNP could also lead to a synergistic outcome by treating mice with established human xenograft and orthotopic pancreatic tumors. We demonstrate the efficacy of our dual delivery carrier versus the use of free GEM, combined with Abraxane in the same animal models.
Results
Use of a Supported LB To Develop MSNPs for Synergistic GEM/PTX Co-delivery
We have previously demonstrated the use of MSNPs as a multifunctional carrier for delivery of chemotherapeutic agents to human tumors in nude mice.10,12,13,17 This includes the use of a PEI/PEG-coated MSNP for stromal targeting of human pancreatic tumors; this carrier enables the delivery of a small-molecule TGF-β receptor kinase inhibitor to interfere in pericyte-mediated stromal vascular obstruction, thereby improving access to second wave therapeutic carriers, such as GEM-delivering liposomes.17 While an ideal delivery system would be to combine the therapeutics in a single carrier, copolymer-functionalized MSNPs cannot effectively entrap a sufficient GEM load to make therapeutic delivery possible. Although we have developed a series of nanovalves for drug encapsulation,14 this approach requires multistep synthesis and proved to be inefficient for loading a high dose of GEM, a nucleoside analogue. This prompted us to consider alternative entrapment procedures for GEM drug delivery. In this regard, it has previously been reported that the electrostatic attachment of zwitterionic liposomes to the MSNP surface, followed by vesicle rupture, can form a supported LB that leads to pore sealing and drug entrapment.27,28 However, this synthesis method also requires several steps and only leads to effective pore sealing if the LB is complete. This was illustrated by our inability to encapsulate a high-dose GEM by the liposomal approach in addition to problems with nonuniform particle coating, leakiness, nanoparticle aggregation, and insufficient batch sizes for use in animal experimentation. This prompted us to develop an alternative sealing method to rapidly attach a supported LB that can be used for high drug loading and therapeutic-scale drug delivery. A supported LB could also facilitate coentrapment of hydrophobic drugs such as PTX.
Instead of the multistep liposomal approach, we developed a coated lipid film method in which GEM-soaked MSNP suspensions were added to a continuous lipid film coated onto a round-bottom glass surface, allowing uniform particle coating upon controlled energy input. This leads to instantaneous and complete particle coating by an intact LB, providing effective drug sealing and loading without the necessity to perform multiple washing procedures (Figure 1A). Briefly, nanoporous silica particles of a uniform particle size (65 nm) were synthesized according to our standard sol/gel procedure, in which tetraethyl orthosilicate (TEOS) was used as the silica precursor and cetyltrimethylammonium chloride (CTAC) as the structure-directing agent. Brunauer–Emmett–Teller characterization showed a total surface area of 850 m2/g, from which we could calculate a pore volume of ∼0.7 cm3/g (see the theoretical calculations in the Supporting Information S1). Figure 1B shows transmission electron microscopy (TEM) and cryoEM images of the GEM-soaked particles. These high-resolution pictures show that the particle cores represent highly ordered helical hexagonal pore arrangements with pore sizes of ∼2.75 nm in diameter.
Figure 1.

Synthesis, physicochemical characterization, and in vitro effects of PTX/GEM-loaded LB-MSNP. (A) Scheme depicting the procedure for LB-MSNP synthesis as a carrier for delivery of GEM only or a combination of GEM and PTX. MSNPs were synthesized by a sol/gel method. We further developed a coated lipid film procedure in which GEM-soaked MSNP suspensions were added to a continuous lipid film coated onto a glass surface, allowing uniform particle coating upon sonication. The coated lipid film was developed by mixing dipalmitoylphosphatidylcholine (DPPC)/cholesterol/1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE)-PEG at a 77.5:20:2.5 molar ratio. After sonication, the particle suspension was purified by centrifugation and washed to separate the coated particles from liposomes and free drug. LB coating also allowed copackaging of the hydrophobic drug, PTX, with GEM trapped in the pores. This allowed ratiometric incorporation of 25 wt % GEM combined with 0–5 wt % PTX. (B) TEM (left) and cryoEM (right) images of GEM-soaked MSNPs before LB coating. The MSNP cores show ordered helical hexagonal pore arrangements. (C) TEM (left) and cryoEM (right) images of GEM-loaded LB-coated MSNPs before purification. The images demonstrate a particle size of 75 nm with uniform and intact LB, with 7 nm thickness; this is equivalent to the thickness of the lipid bilayer in liposomes (7.1 nm) that coform in the synthesis process. (D) CryoEM picture of GEM LB-MSNP after centrifugation, purification, and washing. (E) Ratiometric design of PTX/GEM co-delivery. (E1) Series of GEM/PTX combinations at different mixing ratios (100:1 to 0.2:1) were prepared as described in the Supporting Information S2. Each mixture was considered as a starting concentration to make a series of dilutions. Using the samples to conduct MTS experiments, we calculated the 50% killing concentrations of these mixtures as shown in the top panel in E1. To keep the dose of PTX in the low range, we decided on using a ratio of 10:1 in further experimentation. At this ratio, the free PTX concentration is 0.25 μg/mL, and that of GEM is 2.5 μg/mL (E, top panel). CompuSyn software was used to calculate combination index (CI). We showed a CI of 0.5 at a 10:1 ratio, which is indicative of strong synergy. The additional evaluation of the GEM/PTX synergy of the carrier is performed. A series of GEM/PTX LB-MSNPs were prepared using different encapsulation ratios (100:1, 10:1, and 5:1). Each particle type was used to assess cytotoxicity and determine the concentration of each drug in the mixture for a 50% killing effect (see E1, bottom panel). At an encapsulation ratio of 10:1, we reduced the GEM concentration required for 50% cell killing from 5.2 to 2.3 μg/mL. This occurred in the presence of a PTX dose of 0.23 μg/mL, which is nontoxic. (E2) Independent cell killing (MTS) experiments using free PTX or PTX-only LB-MSNP in PANC-1 cells. (F) TEM (left) and cryoEM (right) images of the ratiometric-designed PTX/GEM LB-MSNP, which at the ratio of 10:1 was used in cellular and animal studies. (G) Detection of CDA and heme oxygenase 1 (HO-1) expression by PTX/GEM LB-MSNP using Western blotting. PANC-1 cells were treated with PTX/GEM LB-MSNP (particle dose = 25 μg/mL; GEM = 6.25 μg/mL; PTX = 0.625 μg/mL) in complete DMEM medium for 0–24 h. The CDA and HO-1 expression was determined by immunoblotting. The relative density of the protein bands were determined by ImageJ software. Representative immunoblot bands at 0 and 24 h are shown in the inset. (H) Pretreatment of PANC-1 cells using N-acetylcysteine at 1.5 mg/mL interfered with the effects of dual drug delivery on CDA and HO-1 expression.
To obtain the most efficient LB-coating procedure and drug-loading/release profiles, we used an iterative scheme for nanocarrier design. This includes (i) determination of optimal LB composition (e.g., different types of lipid, lipid ratios, adjusting the cholesterol concentration, etc.), (ii) experimenting with different drug concentrations in the loading buffer, (iii) varying the loading time, (iv) experimenting with different drug/particle/LB ratios, and (v) varying the sonication conditions and power output, etc. In the optimized protocol, nanoparticle coating was achieved by soaking 200 mg of MSNP in an ethanol/water (7:3, v/v) solution containing 20 mg/mL of GEM. After centrifugation at 15 000 rpm for 20 min, the pellets were quickly resuspended in 5 mL of saline by sonication and immediately added on top of a continuous lipid film and coated on a round-bottom flask with a 6 cm diameter (total surface area of ∼75 cm2). The optimal coated lipid film composition was obtained through iterative rounds of experimentation to obtain the most stable bilayer (defined as the absence of leakage and physical evidence of an intact LB for at least 1 month at 4 °C). The most stable coated lipid film used a DPPC/cholesterol/DSPE-PEG mixture that contains the various lipids at a molar ratio of 77.5:20:2.5. Probe sonication at a power output of 32.5 W for 20 min yielded 5 mL particle suspensions, which were further characterized by TEM (Figure 1C, left) and cryoEM (Figure 1C, right) to assess the morphology and surface coating. High-magnification cryoEM images demonstrated primary particle sizes of ∼75 nm, showing uniform coating of the surfaces with an intact LB of 7 nm. This is close to the thickness of the bilayer in a population of adjacent liposomes (7.1 nm), which are formed during the synthesis process. Because of differences in their density, the free liposomes and non-encapsulated GEM could easily be separated from the GEM-encapsulated LB-MSNPs by centrifugation at 15 000 rpm for 10 min. Following particle washing in saline, we obtained a population of uniformly coated and well-suspended GEM-loaded particles, as demonstrated by cryoEM (Figure 1D).
We used high-performance liquid chromatography (HPLC) analysis and UV spectroscopy analysis in a microplate reader to determine the drug loading capacity of our particles. This involved subtraction analysis (see Supporting Information S1 for detailed description), according to which “loading capacity = [the total amount of GEM introduced for particle soaking – (non-entrapped GEM in the supernatant before pore sealing + GEM in the washings after pore sealing)]/[the total amount of MSNP] × 100%. This yielded a GEM loading capacity of 40 wt % (drug: MSNP), which is ∼10-fold higher than the amount of drug trapped in the corresponding liposomes.9,17 While it was not possible to characterize the exact physicochemical state of GEM trapped in the porous interior, we suspect that the enhanced loading capacity of the MSNPs is due to drug interaction with the walls of the pores through hydrogen bonding, van der Waals interactions, and electrostatic binding.
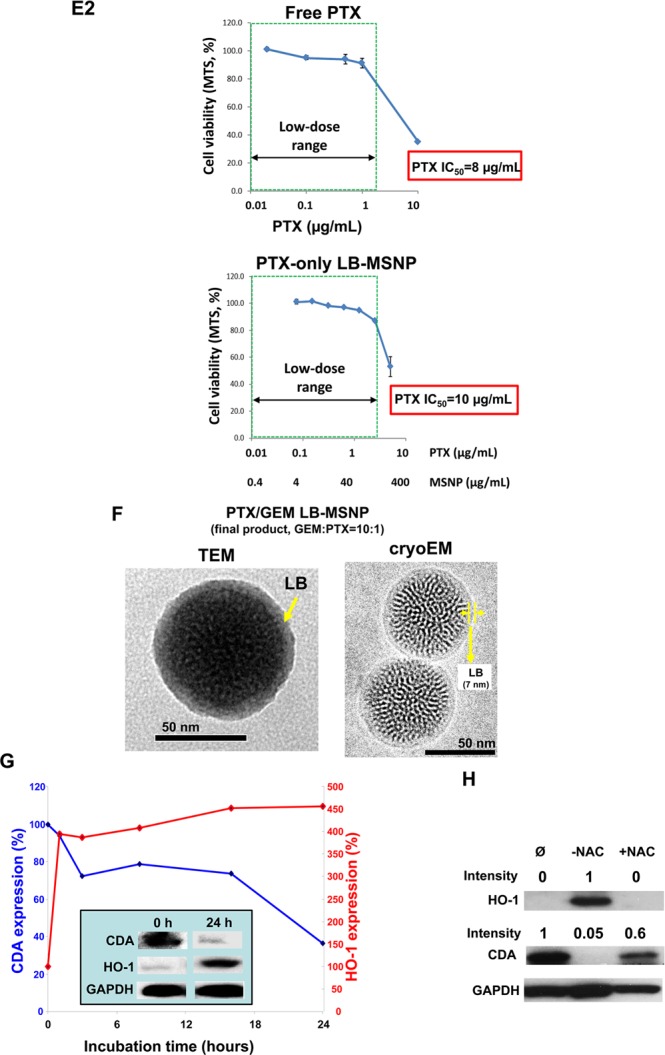
As mentioned above, a supported LB can theoretically be used to copackage a hydrophobic drug, such as PTX, which could act synergistically with GEM, as demonstrated by the Abraxane experience. To demonstrate the synergy in vitro, we initially established the IC50 of free GEM and PTX in PANC-1 cells, obtaining values of 18.2 μg/mL (Figure 1E1, top left) and 8 μg/mL (Figure 1E2, top panel), respectively. We further determined the synergy of a free GEM/PTX mixture in PANC-1 cells. The experimental details are described in the Supporting Information S2. A series of GEM/PTX mixtures were prepared in which we initially used a fixed amount of GEM plus different amounts of PTX to yield GEM/PTX ratios over the range of 100:1 to 0.2:1. Each mixture was considered as a starting concentration to make a series of dilutions. Using these samples to conduct cell viability (MTS) experiments, we were able to calculate free GEM/PTX concentrations in the mixtures providing 50% killing (Figure 1E1, top panel). To keep the dose of PTX in the low range, we decided to use a ratio of 10:1 for further experimentation. At this ratio, the free PTX concentration is 0.25 μg/mL and that of GEM is 2.5 μg/mL (Figure 1E, top panel). Using CompuSyn software to calculate drug synergy, we obtained a combination index (CI) of 0.5; the instruction software suggests a synergistic effect when CI < 1 and a strong synergy if CI ≤ 0.5. We used the free drug data as a reference point to design the dual delivery LB-MSNP, including a 10:1 ratio, which was compared against 100:1 and 5:1. The additional evaluation of the GEM/PTX synergy of the carrier is necessary because the nanoparticle-mediated drug combination is more complex than a physical drug mixture. In addition to drug ratio, the cytotoxic effect of the dual delivery nanoparticle may also be influenced by particle properties (e.g., size, shape, surface, and release profile, etc.) as well as the rate and abundance of cellular internalization. A series of GEM/PTX LB-MSNPs were prepared in which we used a fixed amount of GEM (25%, w/w) in the presence of 0.25–5 wt % PTX to yield GEM/PTX ratios of 100:1, 10:1, and 5:1. Each particle type was used to assess cytotoxicity and to determine the concentration of each drug in the mixture for a 50% killing effect (Figure 1E1, bottom panel). At an encapsulation ratio of 10:1, we reduced the GEM concentration required for 50% cell killing from 5.2 μg/mL (using GEM-only particle) to 2.3 μg/mL (using dual delivery particle). This occurred in the presence of a PTX dose of 0.23 μg/mL (Figure 1E1, bottom panel). Please notice that this falls into the low or nontoxic dose of PTX, as shown by an independent experiment using PTX-only LB-MSNP (Figure 1E2, bottom panel). CompuSyn software analysis showed a CI of 0.5 for this ratio. The particle using an encapsulation ratio of 5:1 also showed potent killing, but we did not want to use this mixture due to the high dose of PTX (0.48 μg/mL), which was relatively close to a PTX cytotoxic dose, as we show in Figure 1E2. The particles with the 100:1 encapsulation ratio were not effective at killing and had a CI of ∼1, which means an additive effect. Thus, co-delivery of PTX/GEM by LB-MSNP could allow an optimal ratio (10:1) to be obtained, which indicates that the major mechanism of action of Abraxane in combination with GEM is not its chemotherapeutic activity, but induction of oxidative stress (see data in Figures 1G,H and 2D,E).5,6 All considered, the 10:1 GEM/PTX encapsulation ratio was used in subsequent in vitro and in vivo experiments.
Figure 2.


Growth inhibition of PANC-1 subcutaneous xenografts in nude mice. (A) PANC-1 cells were subcutaneously injected 14 days before commencing treatment (gray boxes). These animals received six intravenous injections (pink boxes) every 3–8 days (green boxes) for 38 days. Comparisons of the tumor inhibition effect of PTX/GEM LB-MSNP versus other treatment groups, including saline, free GEM, Abraxane, GEM-loaded particles without PTX, and mixtures of GEM plus Abraxane at 1× and 12× doses, are shown. Tumor size was measured 1–2 times per week, and tumor weight was calculated by the formula: tumor weight (mg) = (length in mm) × (width in mm)2/2; *p < 0.05. Biodistribution was studied using IVIS optical imaging (Xenogen) of NIR-labeled MSNPs (50 mg/kg) injected into the nude mice. At the end of the experiment, the animals were sacrificed and tumor tissues and major organs were collected for the measurement of the Si content using inductively coupled plasma optical emission spectroscopy (ICP-OES). Biodistribution of MSNPs was expressed as % of total particle load distributing to the individual organs. T, tumor; S, spleen. (B) Photographs of excised tumors in each treatment group are shown at the same magnification. Quantitative analysis of TUNEL-positive cells for each treatment group. At least three fields were counted to estimate the % of TUNEL-positive cells. The statistical analysis was performed by Excel software using a t test (*p < 0.05). (C) Stromal collagen deposition in the tumor tissue, determined by Masson’s trichrome staining. The blue color represents collagen staining. Total collagen content of the tumor tissue was determined using the Sircol soluble collagen assay kit (Biocolor Ltd., Carrickfergus, UK); *p < 0.05 compared with saline control. (D) Immunohistochemistry staining to determine the expression of CDA (red) in relation to the oxidative stress marker, HO-1 (green). Pieces of the tumor tissue from the saline, GEM LB-MSNP, PTX/GEM LB-MSNP, and GEM/Abraxane (12×) groups were used to make sections that were overlaid with anti-CDA and anti-HO-1 primary antibodies and subsequently visualized by a rhodamine isothiocyanate-conjugated and fluorescein isothiocyanate-conjugated secondary antibodies, respectively. Slides were visualized under a fluorescence microscope at 100× (Zeiss, Germany). (E) Quantitative comparison of tumor CDA expression in the dual delivery group compared to controls using ImageJ software; *p < 0.05.
Physicochemical characterization of the dual-drug-loaded MSNPs demonstrated that coencapsulation of PTX did not affect the particle structure or stability of the LB. TEM (Figure 1F, left) as well as cryoEM (Figure 1F, right) images demonstrated the presence of uniform and complete coating of each particle by an intact 7 nm LB bilayer. We also assessed the hydrodynamic particle size and ζ-potential in saline and saline plus 5% serum, yielding sizes of 101 and 112 nm, respectively. The slightly bigger size during dynamic light scattering analysis in aqueous suspension reflects the contribution of the electrical double layer on the particles as well as the presence of PEG, which is not visible by electron microscopy. PTX/GEM-loaded LB-MSNPs exhibited negative ζ-potentials of −27.2 and −5.4 mV in saline and saline plus 5% serum, respectively.
We also assessed the GEM encapsulation stability and release profile of LB-MSNPs in the absence or presence of PTX loading (Figure S3). In both particle types, GEM encapsulation was stable in phosphate-buffered saline (PBS) (pH 7.4) for 48 h at 37 °C, with premature release of <3.6%. GEM LB-MSNPs and PTX/GEM LB-MSNPs also showed comparable GEM release kinetics under abiotic conditions using simulated lysosomal fluid with a pH of 5. This resulted in the release of 25.3 and 22.9% GEM, respectively, from LB-MSNP with PTX and without PTX after 48 h (Figure S3).
Time- and dose-dependent studies were undertaken in PANC-1 cells to determine the effect of PTX/GEM co-delivery on CDA and heme oxygenase 1 (HO-1) expression. HO-1 is a biomarker of oxidative stress, as previously demonstrated in experimental Abraxane studies to explain synergistic interaction with GEM.5,6 When used at a particle dose of 25 μg/mL (that includes 0.6 and 6 μg/mL PTX and GEM, respectively) for 0–24 h, dual drug delivery by LB-MSNP could be seen to induce a progressive decrease in CDA expression, accompanied by a rapid and sustained increase in HO-1 expression over time (Figure 1G). Please notice that the PTX dose of ∼0.6 μg/mL has a negligible killing effect, as shown in Figure 1E, suggesting a noncytotoxic mechanism of action. We also demonstrated dose-dependent (0–25 μg/mL) CDA and HO-1 effects over a 24 h observation period (Figure S4). These findings are compatible with the hypothesis that the major pharmacological action of PTX is delivery of an oxidative stress stimulus, which leads to the decline in CDA expression. This was confirmed by the ability of N-acetylcysteine, a thiol antioxidant and radical scavenger, to negate the particle effects on CDA and HO-1 expression (Figure 1H).
PTX/GEM Co-delivery Synergizes in Inhibiting PANC-1 Tumor Growth in a Xenograft Model
PANC-1 cells, stably transfected with a luciferase reporter gene, were used to grow subcutaneous xenografts in nude mice for 14 days. The tumor-bearing mice were intravenously (IV) injected with PTX/GEM-loaded LB-MSNPs every 3–8 days for 38 days (Figure 2A). Each animal received a particle dose of 250 mg/kg (GEM, 100 mg/kg; PTX, 10 mg/kg) per injection. The controls included animals receiving IV injection with saline, free GEM (100 mg/kg), Abraxane (PTX dose, 10 mg/kg), GEM-loaded LB-MSNPs (GEM, 100 mg/kg; particle, 250 mg/kg), as well as a mixture of free GEM (100 mg/kg) plus Abraxane (PTX dose, 10 mg/kg) (we will refer to this as the “1× dose”). For comparative purposes, we also included a treatment regimen previously reported for preclinical Abraxane studies, in which PTX is IV injected at 120 mg/kg (“12× dose”) together with the intraperitoneal (IP) free GEM at 100 mg/kg.6 When comparing the effects on tumor growth, PTX/GEM LB-MSNP showed a significantly higher rate of tumor shrinkage than the negative control (saline), free GEM, Abraxane alone, GEM LB-MSNP, and free GEM plus the 1× Abraxane dose (Figure 2A). A comparable degree of tumor shrinkage to the GEM/PTX-loaded particles could be obtained with a 12× Abraxane dose. In addition to the impact on tumor size, we also assessed the percent apoptotic cells in the excised xenografts by TUNEL staining (Figure 2B). This demonstrated that the % TUNEL-positive cells was significantly higher (p < 0.05) for PTX/GEM co-delivery by LB-MSNPs (36%) compared to the mice treated with free GEM (16%), GEM plus a 1× dose of Abraxane (17%), or particles delivering GEM only (23%). However, the co-delivery of GEM and the 12× Abraxane dose resulted in approximately the same number of apoptotic cells (35%). These results demonstrate the efficacy of PTX/GEM co-delivery by our ratiometric-designed LB-MSNPs.
Our particle synthesis is done with 2.5% DSPE-PEG, which leads to decreased reticuloendothelial system (RES) uptake and an increased circulatory half-life. This resulted in passive retention (a.k.a. an enhanced permeability and retention effect) of 6.5% of the administered particle dose at the tumor site, as determined by quantification of the Si content in harvested organs using ICP-OES (Figure 2A). The ICP data were further confirmed by live imaging of the xenograft site in mice injected intravenously with 50 mg/kg near-infrared (NIR)-labeled PTX/GEM LB-MSNP. Pronate views of the animals demonstrated a strong nanocarrier signal at the tumor site, which was maintained for at least 28 h. We also observed considerable particle distribution to the spleen without evidence of systemic toxicity (see the biocompatibility study in Table S1).
Since previous studies have shown that Abraxane co-delivery leads to stromal depletion in a pancreatic cancer murine model,6 Masson’s trichrome staining was performed to analyze the impact of different treatments on stromal abundance (Figure 2C). In contrast to the well-organized and dense desmoplastic stroma in the saline-treated group, treatment with PTX/GEM LB-MSNP resulted in clear evidence of stromal disruption and a significant decrease in the collagen content in the xenografts. This finding was corroborated by quantification of the biochemical collagen content using a Sircol assay. This demonstrated a significant reduction in collagen content in the tumors of animals receiving PTX/GEM co-delivery by LB-MSNP or GEM plus 12× Abraxane. Other treatments had no effect (Figure 2C).
PTX Co-delivery by LB-MSNP Suppresses CDA Expression and Changes GEM Metabolic Profiling in PANC-1 Xenografts
Figure 1G and Supporting Information Figure S4 demonstrate a dose- and time-dependent effect on CDA and HO-1 expression in PANC-1 cells treated with an optimal GEM/PTX ratio. Similar assessment was performed in excised tumor tissue, using immunohistochemistry (IHC) staining by rhodamine isothiocyanate-tagged (red fluorescence) anti-CDA or fluorescein isothiocyanate-tagged (green fluorescence) anti-HO-1 antibodies, respectively (Figure 2D). In animals treated with particles delivering PTX plus GEM, we observed a clear decrease in CDA expression, in parallel with a significant increase in HO-1 staining. This is in contrast to significantly less prominent effect on HO-1 and CDA expression in tumors of animals treated with GEM-loaded LB-MSNP in the absence of PTX. Use of 12× Abraxane plus GEM had the same effect on HO-1 induction and CDA inhibition as the dual delivery particle. No change of expression was observed in the saline control. Use of ImageJ software analysis to quantify CDA expression demonstrated significant reduced expression in response to the dual delivery LB-MSNP as well as the free GEM plus 12× Abraxane (Figure 2E).
Deamination of GEM, a nucleoside pro-drug, by CDA yields an inactive metabolite, difluorodeoxyuridine (dFdU). However, cellular uptake of GEM and drug phosphorylation leads to the formation of an active metabolite, GEM triphosphate (dFdCTP) (Figure 3A).30,31 This explains why CDA down-regulation by PTX could exert a significant effect on the pharmacokinetics and half-life of GEM.6 Increased bioavailability of GEM could lead to increased drug conversion (to dFdCTP) by deoxycytidine kinase (dCK) at the tumor site.6 HPLC analysis was used to quantify the levels of GEM, dFdCTP, and dFdU in tumor tissue obtained from mice, injected intravenously on a single occasion with the same amounts of free GEM, free GEM plus 1× Abraxane, GEM LB-MSNP, or PTX/GEM LB-MSNP as described in Figure 2A. Approximately 100 mg of tumor tissue was harvested after 48 h from each tumor site (n = 3) after animal sacrifice. HPLC analysis demonstrated that while GEM LB-MSNP or GEM plus 1× Abraxane leads to significant increases in total GEM, GEM/PTX co-delivery by LB-MSNP showed the highest total GEM (9-fold) and active metabolite (13-fold) concentrations compared to free GEM (Figure 3B). In contrast, while both free GEM plus 1× Abraxane and GEM LB-MSNP decreased the levels of the inactive metabolite, PTX/GEM co-delivery by LB-MSNP was more potent, that is reducing dFdU levels ∼5-fold (Figure 3B).
Figure 3.

Determination of GEM and GEM metabolites in PANC-1 xenografts. (A) Schematic to show the metabolism and activation of GEM in pancreatic cancer. After transport into the cells, GEM is phosphorylated by deoxycytidine kinase (dCK), leading to the formation of the active dFdCTP metabolite. Stromal and cellular CDA convert GEM to an inactive metabolite, dFdU. CDA leads to GEM inactivation at the tumor site, implying that interference in CDA expression could prolong GEM pharmacokinetics, leading to increased drug uptake at the tumor site. (B) HPLC and quantification of total GEM and GEM metabolite (dFdU and dFdCTP) concentrations in the tumor tissue. The HPLC experiment was carried out in a separate batch of animals that were IV injected with the same amounts free GEM, GEM LB-MSNP, and PTX/GEM LB-MSNP as described in Figure 2A; *p < 0.05 compared with free GEM, #p < 0.05 compared with GEM plus 1× dose Abraxane.
PTX/GEM Co-delivery by LB-MSNP Exerts a Synergistic Effect in an Orthotopic Pancreatic Cancer Model
The orthotopic model is considered to be a more clinically relevant model because it mimics more closely the human pancreatic cancer microenvironment and also includes the development of metastases. Thus, orthotopic pancreatic cancer models are more robust and provide a better prediction of treatment success in humans than xenograft models.29 For logistical reasons (i.e., the number of orthotopic implants that can be accomplished in one experiment), we reduced the animal number to three per group and limited the number of comparative treatments to saline, free GEM, PTX/GEM LB-MSNP, and free GEM plus 1× Abraxane (Figure 4A). We used PANC-1 cells, transfected with a luciferase gene, to perform noninvasive luminescence imaging of the orthotopic tumor site after tumor cell implantation in the tail of the pancreas. The treatment schedule (Figure 4A) shows that, 14 days after initial implantation, the tumor-bearing mice received four IV injections of PTX/GEM LB-MSNP (particles, 250 mg/kg; GEM, 100 mg/kg; PTX, 10 mg/kg, per injection) over a 3 week period. The same dosing schedule was used for the control groups, which were treated with saline, 100 mg/kg free GEM, or 100 mg/kg free GEM plus 1× Abraxane per injection. This was followed by a 19 day observation period before animal sacrifice. Bioluminescence signals in the tumor implants became visible after ∼14 days and remained localized to the pancreas tail region. At the conclusion of the observation period, the magnitude of the bioluminescence signals was quite extensive in the saline-treated group, with the tendency to spread beyond the primary tumor site (Figure 4B). While free GEM showed significant tumor shrinkage, one out of three animals had metastatic spread. While additional tumor shrinkage could be seen in animals treated by GEM plus 1× Abraxane, one animal had evidence of metastatic disease (Figure 4B). In animals treated with PTX/GEM LB-MSNP, only a faint luminescence signal could be seen at day 38, without any evidence of metastases (Figure 4B). To quantify the primary tumor size for each group, the bioluminescence signal intensity was analyzed by IVIS software, using the “operator defined region of interest” parameter. This demonstrated that PTX/GEM co-delivery by LB-MSNP provided the most effective shrinkage of the orthotopic cancer mass compared to other treatments (Figure 4C).
Figure 4.

Tumor growth inhibition in the orthotopic PANC-1 tumor model in nude mice. (A) Luciferase transfected PANC-1 cells were orthotopically implanted into the tail of the pancreas, 14 days before the commencement of treatment (gray boxes). These animals (n = 3) received four IV injections (pink boxes) every 3–6 days (green boxes), followed by a 19 day nonintervention time period. (B) IVIS optical imaging system was used to study the tumor growth in mice. To visualize tumor growth in vivo, anesthetized mice received IP injection of 75 mg/kg d-Luciferin, followed 8 min later by collecting bioluminescence data. Representative animal images, collected at day 38, are shown. (C) Bioluminescence intensity in the operator-defined region of interest (pancreas) was analyzed by Xenogen software and quantitatively expressed; *p < 0.05. (D) Representative photographs of animals in each group on which autopsy was performed. The primary tumor boundary is highlighted by a broken line: S, spleen; T, primary tumor; M, metastasis.
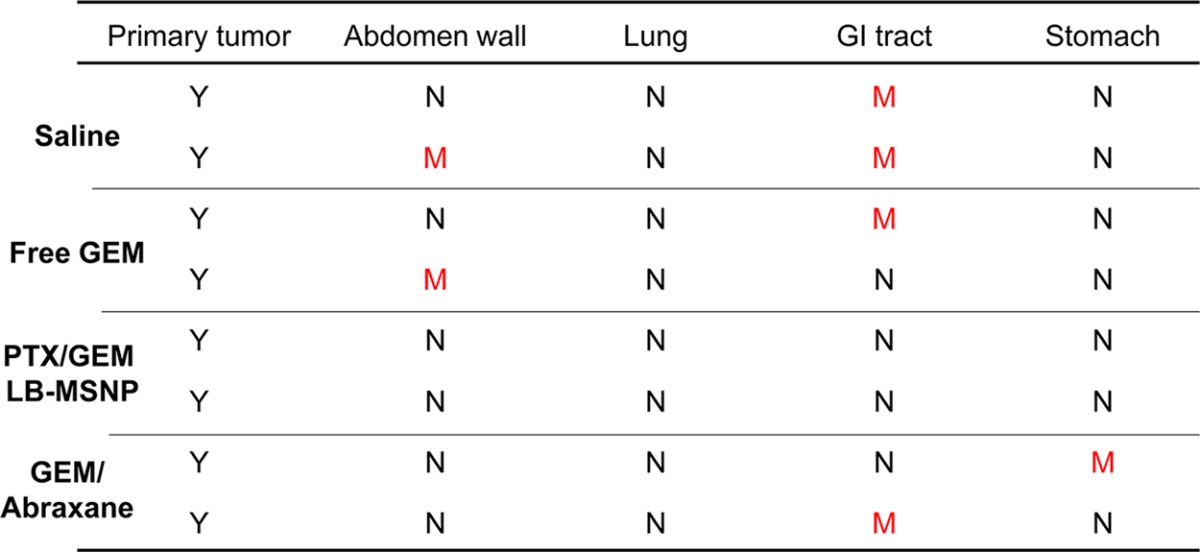
Animal autopsy at the conclusion of the experiment demonstrated the presence of large, locally invasive tumors with metastatic spread in the saline-treated group (Figure 4D). Other than the pancreas, the tumors also spread to the spleen and abdominal cavity. No visible infiltration was seen in the hearts, lungs, or the kidneys. While considerable tumor shrinkage was seen in mice treated with free GEM or GEM plus 1× Abraxane, macroscopically visible tumor metastasis could still be seen (Figure 4D, arrow pointed “M”). This contrasted with the treatment using PTX/GEM LB-MSNP, demonstrating an obviously smaller primary tumor with no sign of tumor metastasis. A summary of the autopsy results appears in Table 1.
Table 1. Ex vivo Examination of the Clinical Spectrum in PANC-1 Orthotopic Model after Various Treatments (M, Metastasis; N, Negative; Y, Detectable/Visible Primary Tumor).
Lack of Systemic Toxicity during Treatment with PTX/GEM Delivering LB-MSNPs
Overcoming or reducing chemotherapy side effects is an important consideration for cancer nanotherapeutics.10,32−34 We have previously demonstrated, in multiple MSNP platforms, that the carrier alone does not result in any systemic toxicity.12,13 Carrier safety is likely to be further enhanced by coating with a LB, which mimics a normal cell membrane. To assess the biosafety of the LB-MSNP platform, we compared the impact of the different treatment strategies on the general well-being, body weight, blood chemistry, and organ histology. Liver function studies showed a slight elevation (82.1 ± 18.4 U/L) of alkaline phosphatase (ALP) in mice receiving GEM/12× Abraxane, but this was not seen during co-delivery of PTX/GEM by LB-MSNP (36.9 ± 18.6 U/L) (Table S1). No statistically significant change in body weight was observed between groups. Histological examination of other major target organs, which can be damaged by chemotherapy, including the spleen, kidney, liver, or heart, failed to show any gross pathology in any of the treatment groups.
Discussion
Inspired by the success of combining Abraxane with GEM to achieve a synergistic outcome in pancreatic cancer as a result of improved pharmacokinetics of GEM, we designed a single carrier to co-deliver these drugs at an optimized drug ratio. The synthesis of a MSNP carrier was accomplished by highly efficient drug encapsulation, using a coated lipid film technique that leads to rapid entrapment of a high GEM drug load through pore sealing by a supported LB. Moreover, the LB also allowed the incorporation of an optimal ratio of the hydrophobic drug, PTX, which could be co-delivered with GEM in pancreatic cells and tumors. We demonstrated that synergistic PTX/GEM co-delivery led to dose- and time-dependent CDA inhibition and induction of oxidative stress in PANC-1 cells. To demonstrate the in vivo efficacy, mice carrying subcutaneous PANC-1 xenografts received IV injection of PTX/GEM-loaded LB-MSNP. Drug co-delivery by this carrier provided more efficient tumor killing than free GEM, GEM-loaded LB-MSNP without PTX, or free GEM plus Abraxane. Comparable tumor shrinkage required coadministration of 12 times the free Abraxane dose. HPLC analysis of GEM metabolites in tumor tissue confirmed that, compared to free GEM, LB-MSNP co-delivery increased the active GEM concentration 13-fold, while decreasing the concentration of the inactivated (deaminated) GEM ∼5-fold. The impact of co-delivery was also tested in a more rigorous PANC-1 orthotopic model, demonstrating that PTX/GEM LB-MSNP can effectively inhibit primary tumor growth, as well as eliminating metastatic tumor foci. In summary, we demonstrated the development of a highly effective LB-MSNP nanocarrier for systemic and synergistic PTX/GEM co-delivery to pancreatic cancer.
We define a ratiometric approach as the in vivo release of a drug combination from a nanocarrier, with the purpose of providing a fixed drug ratio at the target site.29,35 Combination chemotherapy regimens are typically developed for cancer treatment by establishing the recommended dose of one drug and then adding subsequent agents to the mix at incremental doses until the aggregate cytotoxicity effects become dose-limiting.29,35 This is done with the assumption that maximum therapeutic activity can be achieved with maximum dose intensity of all drugs in the mixture and may overlook the possibility that more subtle concentration-dependent drug interactions could achieve synergistic outcomes or, in some instances, antagonism. Not only is synergy dependent on the distinct pharmacological actions of each drug, but individual agents in a conventional anticancer drug combination are distributed and eliminated independently.35 This could turn out to be essential if the extent of synergy (or antagonism) depends on the ratio and concentrations of the drugs in the mixture. A ratiometric approach to drug delivery could overcome these problems,35−38 as we demonstrate for the delivery of a synergistic PTX/GEM combination in pancreatic cancer by a LB-MSNP carrier. A number of experimental methods have been developed for drug co-delivery by nanocarriers, including direct drug encapsulation into the particle interior, covalent conjugation of drug combinations to a carrier, surface attachment of drugs to an existing drug carrier, or supramolecular assembly of synergistic drug combinations into drug-carrying nanoparticles.35,39 Examples include liposome encapsulation of cytarabine and daunorubicin at molar ratio of 5:1 for synergistic treatment of leukemia in a mouse model,40 coencapsulation of irinotecan and floxuridine in a 1:1 molar ratio in a liposome for treating colorectal cancer,41 independent coupling of doxorubicin and camptothecin to a particle with a polymer backbone for treating human breast cancer,42 and ratiometric incorporation of GEM monophosphate and cisplatin drug precipitates into 120 nm PLGA particles at a 5:1 molar ratio for treatment of bladder cancer.43 While combined drug loading has been attempted for MSNPs (e.g., porous drug encapsulation with surface attachment of peptide/siRNA or porous encapsulation of paired small-molecule drugs),12,39 these studies have not attempted to achieve ratiometric control of the delivery to specific cancers. Moreover, we improved the synergy between GEM and PTX to the extent that we could achieve the same efficacy in an animal model as provided by 12 times of the Abraxane dose. In fact, ratiometric design allowed us to reduce the PTX dose of the carrier below the level that is required for cytotoxic killing. This can be explained by the pharmacological effect of PTX in inducing oxidative stress (HO-1 expression), which suppresses CDA expression.6,44 HO-1 expression at low levels (tier 1) of oxidative stress45 is achieved through the transcriptional activation and intranuclear release of the transcription factor, Nrf2.46 Future studies will explore whether transcriptional activation of the CDA promoter is governed by pathways that are sensitive to the biological effects of Nrf2, which also regulates cellular redox equilibrium. It is interesting that we could negate the PTX effect on HO-1 expression by the thiol antioxidant, NAC.
In this study, we achieved high loading of a water-soluble nucleoside analogue (GEM) into the porous interior of the MSNP while also able to incorporate the hydrophobic drug, PTX, into the coated bilayer. This required the development of coated lipid film technique that could overcome the problems posed by a liposome approach to achieve surface coating. Coating of solid surfaces with supported LBs commenced with the primary objective to obtain structural models to study the biophysics of lipid membranes or to provide biocompatible coatings for synthetic materials, such as silica.47 Bayerl et al. reported in 1990 the use of lipid vesicle fusion to coat the surface of silica particles (0.3–10 μm in size) with a unilamellar lipid bilayer that is structurally similar to the bilayer forming on planar SiO2 substrates.48 It was shown subsequently that coating of silica beads could support the formation of a LB that exhibits the same diffusion coefficient as natural bilayers.49 To address the difficulties of visualizing supported LBs, Mornet et al. provided the first direct approach to viewing the structural details of an LB attached to a 110 nm Stober silica nanoparticle surface, using cryoEM imaging.50 Recently, LB coating has been applied as a design feature of drug delivery carriers, including MSNP20,21,23−28,51,52 and chitosan particles.53 One approach has been to use MSNP, synthesized by an aerosol-assisted self-assembly method, for the sequential adhesion, rupture, and fusion of electrostatically charged liposomes to the particle surfaces.28 Another approach uses a solvent exchange reaction in which EtOH-dispersed lipid solutions are added to a pellet of centrifuged particles.20 Our attempts to coat different batches of MSNPs with presynthesized liposomes were inefficient for GEM entrapment, likely due to leakage of the incompletely sealed LBs as well as the multiple washing steps to prepare the particles.28 For ease of controlling the desired amount of PTX in the LB, we did not use EtOH dispersion, which could be another promising method to achieve LB coating.20 Instead, our procedure involves the use of a one-step coated lipid film technique, which leads to rapid pore sealing and complete surface coverage, beyond which washing and purification can be performed without jeopardy of drug loss. This allowed us to prepare 200 mg of LB-MSNP per batch, enough to perform comprehensive animal experiments. Although we suspect that electrostatic charge plays a role in LB adhesion to the particle surface, it is likely that van der Waals forces contribute to the rapid and complete coating of the MSNP surface.47 A better understanding of the mechanics and physical processes involved in surface coating by the LB necessitates further biophysical studies to look at the contribution of free energy change between the membrane and the particle surface, the thickness of the hydration layer, LB fluidity and stability, and diffusivity, etc.47 An additional advantage of our approach is the ability to introduce hydrophobic drug components that can incorporate into the LB, as we demonstrate for PTX.
Our study addresses the importance of designing the nanocarrier to address the specific biological and clinical challenges posed by each cancer type. In the case of pancreatic cancer, it is important to consider the role of a dysplastic stromal barrier, abnormal vascular perfusion, high pericyte coverage, early metastasis, and cancer-specific drug resistance among the challenges.3,54 Regarding drug resistance, for instance, the rapid deamination of GEM by CDA limits the circulatory half-life to 0.28 h,55 while intracellular activation is dependent on phosphorylation by the kinase, deoxycytidine kinase (dCK), for generating the active DNA-binding metabolite, dFdCTP.56 Thus, in addition to carrier design suppressing CDA activity by co-incorporation of PTX, it is also possible to consider the role of dCK as a rate-limiting step in GEM activation. This can be addressed by designing the carrier to deliver the prodrug, GEM-bisphosphonate, which bypasses dCK.43,56 This modification could be applicable to treating a subset of patients with GEM resistance due to low dCK expression and potentially discoverable by performing PET imaging with fluorinated GEM analogues.56
We have previously demonstrated how stromal vascular access in pancreatic cancer can be improved by a MSNP that delivers a small-molecule inhibitor to the TGF-β receptor kinase, which results in the detachment of stromal pericytes from endothelial cells.17 The improved vascular access allows increased GEM delivery to the tumor site by a liposomal carrier.17 We now demonstrate the design of an additional carrier that provides stromal targeting through co-delivery of PTX and could serve as a “second wave” drug carrier following the stromal delivery of a small-molecule TGF-β inhibitor by a “first wave” PEI/PEG-MSNP carrier.17 This is in line with our thinking of an “engineered approach” to pancreatic cancer by nanocarriers; we define an engineered approach as the dynamic integration of the drug delivery properties with additional nanocarrier properties that address tumor-specific challenges. We envisage further improvement of our platform through the addition of design features such as targeting, inclusion of activated GEM metabolites, or delivering additional synergistic drug combinations. This could include the co-delivery of GEM with erlotinib (a hydrophobic EGFR kinase inhibitor),57 ubiquitin ligase inhibitors,58 or incorporating oxidative-stress-inducing metal oxide nanoparticles that suppress CDA expression.59 We can also include Gd-based imaging components or up-conversion lanthanides,60 as well as targeting ligand such as iRGD peptides.61,62 However, it is important to consider the design complexity and the cost of each component in terms of clinical use potential. Minimally, the use of a supported LB introduces a modification that holds a lot of advantages, including high loading drug capacity and drug co-delivery. The LB also allows the addition of targeting ligands and imaging agents and contributes to the platform safety. Moreover, LB-MSNPs hold several advantages over liposomes, including drug loading capacity, stability, and the ease of achieving multifunctionality.
Conclusion
In conclusion, we have developed an effective procedure for rapid pore sealing and uniform surface coating of MSNPs by a LB, which allows high GEM loading and colloidal stability, with the ability to copackage PTX. PTX/GEM co-delivery by LB-MSNP acts synergistically in suppressing pancreatic cancer stromal volume and tumor size, outperforming the delivery of free GEM plus Abraxane in xenograft and orthotopic animal models.
Methods
LB-MSNP Synthesis and Drug Loading
MSNP cores were synthesized by slight modification of our sol/gel procedure. Five milliliters of CTAC (25%) was mixed with 15 mL of H2O and was stirred for 15 min at 75 °C at 350 rpm. This was followed by the addition of 0.8 mL of 10% TEA at 75 °C for an additional 15 min. The silica precursor, TEOS (1.5 mL), was added to the mixture dropwise at a rate of approximately 30 drops per minute. To achieve the synthesis of particles with a primary size of 60–70 nm, the solution was stirred at 350 rpm at 80 °C for 1 h. To remove the surfactant, the particles were washed by menthol and HCl (500:19, v/v) at room temperature for 24 h. The particles were centrifuged at 10 000 rpm for 60 min and washed three times in menthol.
To attach a surface LB coating, we developed a coated lipid film procedure in which GEM-soaked MSNP suspensions were added to a large lipid film surface, coated on a round-bottom flask. Using different lipid bilayer compositions, we performed a series of experiments to find a composition that provides rapid and uniform particle coating. Briefly, 200 mg of empty MSNPs was soaked in 10 mL of a GEM ethanol/water (7:3, v/v) solution for 48 h, with the GEM concentration kept at 20 mg/mL by gentle shaking. After centrifugation at 15 000 rpm for 20 min, the soaked pellet was presuspended in 5 mL of saline by 30 s sonication. The suspension was immediately added on top of the coated lipid film, which occupied a ∼75 cm2 surface area at the bottom of a round-bottom flask. After experimenting with different lipid mixes, we decided on a combination of DPPC/cholesterol/DSPE-PEG at a molar ratio of 77.5:20:2.5. These components were suspended in chloroform at 10 mg/mL. Lipid film formation was achieved by solvent evaporation over ∼1 h using a rotary evaporator connected to a vacuum system at room temperature. These films were placed in a chemical hood overnight to remove trace amounts of organic solvent impurities. At the time of coating, we used a MSNP/LB ratio of 1:1.1 (w/w). Following the addition of the 5 mL of particle suspension to the coated lipid film, probe sonication was used for 20 min with 15/15 s on/off working cycle at a power output of 32.5 W. Since the suspension contains coated particles, liposomes, and free drug, the particles were separated by centrifugation at 15 000 rpm for 10 min, followed by washing three times in saline. To achieve PTX/GEM co-delivery, the procedure was adapted to include 0.1–5 wt % PTX in a chloroform solution to the lipid mixture. After the formation of a PTX-containing lipid film, the GEM-soaked particle suspension was added to the flask, followed by rehydration, sonication, centrifugation, and washing, similar to what was described above.
Physicochemical Characterization
Uncoated and coated particles were characterized for morphology, size distribution, and surface charge. The shape and structure were characterized using regular TEM (JEOL 1200-EX) as well as in a cryo-electron microscope (cryoEM, TF20 FEI TecnaiG2). For TEM analysis, microfilms were made by placing a drop of the respective MSNP suspensions onto a 200 mesh copper TEM grid (Ted Pella, CA) and then drying at room temperature for 2 h. A minimum of three images for each sample was captured, and representative images were included in Figure 1. To visualize the LB coating with higher resolution and contrast in saline, we also performed cryoEM analysis. Each MSNP saline suspension was adsorbed onto a holey carbon-coated grid (Ted Pella, CA) and vitrified into liquid ethane at −178 °C. To preserve the vitrification of the saline sample, frozen grids were carefully transferred onto a cryoEM equipped with a cryo-holder. Electron micrographs were recorded at an accelerating voltage of 200 kV, keeping the sample at −175 °C through the use of liquid N2. CryoEM was also used to check the efficiency of liposome removal after centrifugation and washing steps. Particle size and ζ-potential in solution were measured by ZetaSizer Nano (Malvern Instruments Ltd., Worcestershire, UK). The measurements were performed with the nanoparticles suspended in saline plus 5% serum at 100 μg/mL nanoparticle concentration. The loading capacity was determined by a subtraction method. The detailed descriptions as well as a flowchart outlining the major steps are included in the Supporting Information S1. Briefly, the non-encapsulated GEM content in the supernatant before pore sealing (m1) and GEM content in the collected washing buffer during purification after pore sealing (m2) were determined by OD at 265 nm using a microplate reader (M5e, Molecular Device, USA) or HPLC (C18 column equipped with a K-2600 spectrophotometer). We defined loading capacity = [the total amount of GEM (m0) – (m1 + m2)]/[the total amount of MSNP (mNP)] × 100%. This experiment was repeated three times.
Cell Culture
PANC-1 cells were purchased from American Type Culture Collection (ATCC). The cells were cultured in Dulbecco’s modified Eagle medium (DMEM) (Carlsbad, CA), containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine. For tumor visualization in mice using optical imaging, permanent transfection was performed in the UCLA vector core facility using a luciferase-based lentiviral vector. Following a limiting dilution protocol to select single cell clones, the PANC-1-luc cell populations were used for creating subcutaneous xenograft and orthotopic models, which could be studied by bioluminescence imaging, as well as determining carrier biodistribution.
Assessment of Cytotoxicity in PANC-1 Cells Treated by PTX/GEM Co-delivery
Please see the detailed methods for the in vitro cytotoxicity assay, including the dosimetry calculation for free drug mixtures and dual delivery LB-MSNPs, that are provided in the Supporting Information S2.
Assessment of CDA and HO-1 Expression via Immunoblotting
PANC-1 cells were plated at 2 × 105 cells per well in 6-well plates. In one experiment, the cells were treated with a fixed dose of 25 μg/mL LB-MSNPs (GEM = 10 μg/mL; PTX = 1 μg/mL) in complete DMEM medium for 0–24 h. In a second experiment, the cells were treated with a 0–200 μg/mL dose of particles for 24 h. To assess CDA expression, the cells were washed in PBS and the pellets lysed in a buffer containing Triton X-100 and protease inhibitors. Following centrifugation, the protein content of the supernatants was determined by the Bradford method. Eighty micrograms of protein was electrophoresed by 12% SDS-PAGE and transferred to a PVDF membrane. After being blocked, the membranes were incubated with 1:1000 dilution of primary monoclonal antibody to CDA (anti-CDA, Abcam). The membranes were overlaid with secondary antibody (1:1000 dilution) before the addition of the HRP-conjugated streptavidin–biotin complex. The proteins were detected by the ECL reagent according to the manufacturer’s instructions. Using the same membrane, HO-1 protein was detected by anti-HO-1 monoclonal antibody (1:1000 dilution), followed by the incubation of rabbit anti-mouse HRP-conjugated antibody before the addition of ECL reagent. The signal intensity was calculated using ImageJ software.
Establishment of a PANC-1 Subcutaneous Xenograft Model
Athymic BALB/c nu/nu female mice (6–8 weeks) were purchased from the Charles River Laboratory and maintained under pathogen-free conditions. All animal experiments were performed using protocols approved by the UCLA Animal Research Committee. To grow subcutaneous tumor xenografts, female mice were implanted with 2 × 106 cells in Matrigel (BD Biosciences, 1:1 dilution) subcutaneously in the right flank. To determine treatment efficacy in the subcutaneous xenograft model, the mice received a series of treatments, starting 14 days after initial tumor implantation. To perform the imaging experiments, the tumor-bearing animals were used approximately 4 weeks after tumor implantation, by which time point the tumors had grown to a size of ∼0.8 cm.
Subcutaneous Xenograft Studies To Determine Treatment Efficacy of LB-MSNPs
Two weeks after tumor implantation, the PANC-1 tumor-bearing mice were randomly divided into seven groups to compare the treatment effects of saline, free GEM, free Abraxane, GEM loaded LB-MSNPs without PTX, PTX/GEM co-delivery by LB-MSNPs, and two mixtures of free GEM plus Abraxane. Each animal in the PTX/GEM LB-MSNP group received a particle dose of 250 mg/kg, which delivers 100 mg/kg GEM and 10 mg/kg PTX per IV injection. A total of six injections were performed over a 38 day time interval. Animals injected with free GEM or GEM-loaded LB-MSNP received 100 mg/kg GEM. For comparison, we included the group receiving 100 mg/kg GEM plus 10 mg/kg Abraxane (1× dose) IV, as well as a separate group receiving 100 mg/kg free GEM IP together with 120 mg/kg Abraxane (12× dose) IV, as reported in the literature.6 Saline-treated animals were regarded as a negative control. Animal body weights and tumor sizes were recorded 1–2 times per week. For tumor size measurement, the length and width axes of each tumor were measured by an electronic caliper to the nearest 0.1 mm. Tumor weight, assuming a tissue density of 1 mg/mm3, was calculated according to the formula tumor weight (mg) = (length in mm) × (width in mm)2/2.63 The statistical analysis of the differences between the groups was performed by using a t test, with a p level of 0.05 (Excel software, Microsoft).
After animal sacrifice, a piece of tumor tissue from each animal was used for TUNEL staining according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). Nuclear staining was performed with Hoechst 33342 dye (blue) to locate the cells, whereupon the percentage of TUNEL-positive (green stained) cells was obtained by fluorescence microscopy (200×). Separate tissue sections were stained with Masson’s trichrome to visualize the stromal elements, while pieces of homogenized tumor tissue (Fisher Scientific homogenizer) were used to assess the biochemical collagen content using a Sircol collagen assay.
Systemic Biodistribution
IVIS optical imaging (Xenogen) was used to study the biodistribution of NIR-labeled MSNPs in the PANC-1-luc subcutaneous xenograft model in nude mice. To visualize luciferase expression in the cancer cells, the anesthetized mice received d-Luciferin before collection of the bioluminescence images. Reference fluorescence images were captured before IV injection of 50 mg/kg NIR-labeled particles into the tumor-bearing mice. This was followed by the capture of another image at 28 h post-injection. At the end of the experiment, the animals were sacrificed and tumor tissues as well as major organs were collected for the measurement of the Si content using ICP-OES. The biodistribution of MSNPs was expressed as % of total particle load distributing to the individual organs.
HPLC Analysis
To determine the effectiveness of GEM delivery, its bioinactivation, and its bioactivation, HPLC analysis was used to quantify total GEM content as well as its metabolites (dFdU and dFdCTP) in excised xenograft tissue. The HPLC experiment was carried out using a separate batch of animals, which received IV injection of a single dose of free GEM, GEM LB-MSNP, and PTX/GEM LB-MSNP, as described in Figure 2A. The total dose of GEM was 100 mg/kg per injection. The tumor tissue was harvested 48 h after injection, weighed, and homogenized. GEM and GEM metabolites were extracted from the homogenates by the addition of 1.5 mL of methanol/acetonitrile (1:9, v/v). The samples were vortexed for 1 min and then centrifuged at 5000 rpm for 10 min. Supernatants were collected, and extraction was repeated three times. The supernatants were combined and evaporated to dryness under a gentle stream of nitrogen at 60 °C. The residue was dissolved in a 0.2 mL mobile phase (ammonium acetate buffered H2O/MeOH = 93:7, v/v, pH = 5.5) and then centrifuged at 15 000 rpm for 10 min before use. The HPLC system is a Knauer Smartline Pneumatic Pump, C18 column, and K-2600 spectrophotometer with data acquisition software. The flow rate of the mobile phase was 1.00 mL/min. Twenty microliter sample aliquots were used to measure drug absorption at 268 nm. Standard curves for GEM, dFdU, and dFdCTP were generated using pure, commercially available compounds. This allowed us to determine the retention time of each compound and calculate the area under the curve values for known quantities of standard materials. We also calculated the concentrations of the GEM and GEM metabolites in each tumor by regression analysis.
Blood Biochemistry To Assess the Toxicity of the Delivery Platform
Following animal sacrifice at the conclusion of the xenograft experiment on day 38, cardiac puncture was performed and serum collected for biochemical analysis by the UCLA Division of Laboratory Animal Medicine (DLAM) diagnostic laboratory services. Appropriate slices of tumor tissue, liver, kidney, spleen, and heart were fixed in 10% formalin, followed by paraffin embedding or direct freezing using the Tissue-Tek O.C.T. reagent. Tissue sections of 4 μm thickness were mounted on glass slides. The sections were stained with hematoxylin–eosin (H&E) and/or trichrome and examined by light microscopy.
Establishment of a PANC-1 Orthotopic Pancreatic Tumor Model
To grow orthotopic xenografts, the animals were anesthetized with isoflurane, followed by IP injection of 50 mg/kg ketamine and 10 mg/kg xylazine. The surgical site was sterilized by repetitive application of betadine and 70% ethanol. Animals were placed in an appropriate position for surgery on a water heating pad in the tissue culture hood, and the surgical site was draped with sterile gauze. A surgical incision of 1 cm was made in the left flank to expose the injection site, following which 50 μL of DMEM/Matrigel (1:1, v/v) containing 2 × 105 PANC-1 cells was injected into the tail of the pancreas by a 27 gauge needle. After orthotopic implantation, the fascial layers were closed with absorbent sutures (LOOK Plain Gut) and the skin was closed with nonabsorbent sutures (PROLENE polypropylene, Ethicon). The external sutures were removed after 7 days. The entire procedure was accomplished within 45 min for each animal. The animals were kept on the warming pads for the duration of recovery. Once fully recovered, animals were individually moved to clean cages and maintained in a pathogen-free facility.
Determination of Treatment Efficacy in the Orthotopic Tumor Model
Because of the labor intensiveness, cost, and limitation of the number of animals that can be handled in a single experiment, it was not possible to use the same treatment groups as in the xenograft model. Thus, we limited treatment to injection with saline, free GEM, PTX/GEM LB-MSNP, and free GEM plus Abraxane (1×). The tumor-bearing mice were randomly allocated to each of the four groups, which included three animals per group. Each animal in the PTX/GEM LB-MSNP group received IV injection of a particle dose of 250 mg/kg (100 mg/kg GEM and 10 mg/kg PTX) per injection. A total of four injections were administered over a 19 day time period, followed by a 19 day nonintervention period. The free GEM and GEM plus 1× Abraxane groups received the same GEM dose. Tumor visualization was accomplished by performing bioluminescence imaging on days 1 and 38 in anesthetized mice, after IP injection with 75 mg/kg d-luciferin. Eight minutes after injection, animals were placed in an IVIS imaging system and bioluminescence images were collected using an acquisition time of 10 s. To estimate the primary tumor size, bioluminescence intensity in the “operator defined region of interest” was quantified by IVIS software. Upon animal sacrifice at day 38, we also obtained ex vivo bioluminescence images of the primary tumor tissue as well as major organs such as liver, spleen, lung, kidney, GI tract, and heart, with a view to look for metastases. Pieces of the tissue samples were fixed for histological analysis.
Acknowledgments
This study was funded by the U.S. Public Health Service Grant RO1 CA133697. H.L. was partially supported by the National Natural Science Foundation of China No. 31271075.
Supporting Information Available
Additional figures, table, and results as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cancer Facts & Figures; American Cancer Society, 2014; http://www.cancer.org/acs/groups/content/@research/documents/webcontent/acspc-042151.pdf.
- Mini E.; Nobili S.; Caciagli B.; Landini I.; Mazzei T. Cellular Pharmacology of Gemcitabine. Ann. Oncol. 2006, 17, v7–v12. [DOI] [PubMed] [Google Scholar]
- Erkan M.; Hausmann S.; Michalski C. W.; Fingerle A. A.; Dobritz M.; Kleeff J.; Friess H. The Role of Stroma in Pancreatic Cancer: Diagnostic and Therapeutic Implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [DOI] [PubMed] [Google Scholar]
- Jacobetz M. A.; Chan D. S.; Neesse A.; Bapiro T. E.; Cook N.; Frese K. K.; Feig C.; Nakagawa T.; Caldwell M. E.; Zecchini H. I.; et al. Hyaluronan Impairs Vascular Function and Drug Delivery in a Mouse Model of Pancreatic Cancer. Gut 2012, 62, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff D. D.; Ervin T.; Arena F. P.; Chiorean E. G.; Infante J.; Moore M.; Seay T.; Tjulandin S. A.; Ma W. W.; Saleh M. N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel Plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese K. K.; Neesse A.; Cook N.; Bapiro T. E.; Lolkema M. P.; Jodrell D. I.; Tuveson D. A. Nab-Paclitaxel Potentiates Gemcitabine Activity by Reducing Cytidine Deaminase Levels in a Mouse Model of Pancreatic Cancer. Cancer Discovery 2012, 2, 260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi N.; Zhang C.; Schwarz A. M.; Hinz S.; Wang C.; Williams N. S.; Schwarz M. A.; Schwarz R. E. Comparative Benefits of Nab-Paclitaxel over Gemcitabine or Polysorbate-Based Docetaxel in Experimental Pancreatic Cancer. Carcinogenesis 2013, 34, 2361–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourzac K. Nanotechnology: Carrying Drugs. Nature 2012, 491, S58–S60. [DOI] [PubMed] [Google Scholar]
- Federico C.; Morittu V.; Britti D.; Trapasso E.; Cosco D. Gemcitabine-Loaded Liposomes: Rationale, Potentialities and Future Perspectives. Int. J. Nanomed. 2012, 2012, 5423–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai W. X.; Meng H. Mesoporous Silica Nanoparticles: A Multifunctional Nano Therapeutic System. Integr. Biol. 2013, 5, 19–28. [DOI] [PubMed] [Google Scholar]
- Meng H.; Liong M.; Xia T.; Li Z.; Ji Z.; Zink J. I.; Nel A. E. Engineered Design of Mesoporous Silica Nanoparticles To Deliver Doxorubicin and P-Glycoprotein siRNA To Overcome Drug Resistance in a Cancer Cell Line. ACS Nano 2010, 4, 4539–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H.; Mai W. X.; Zhang H.; Xue M.; Xia T.; Lin S.; Wang X.; Zhao Y.; Ji Z.; Zink J. I.; et al. Codelivery of an Optimal Drug/siRNA Combination Using Mesoporous Silica Nanoparticles To Overcome Drug Resistance in Breast Cancer in Vitro and in Vivo. ACS Nano 2013, 7, 994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H.; Xue M.; Xia T.; Ji Z.; Tarn D. Y.; Zink J. I.; Nel A. E. Use of Size and a Copolymer Design Feature To Improve the Biodistribution and the Enhanced Permeability and Retention Effect of Doxorubicin-Loaded Mesoporous Silica Nanoparticles in a Murine Xenograft Tumor Model. ACS Nano 2011, 5, 4131–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H.; Xue M.; Xia T.; Zhao Y.-L.; Tamanoi F.; Stoddart J. F.; Zink J. I.; Nel A. E. Autonomous In Vitro Anticancer Drug Release from Mesoporous Silica Nanoparticles by pH-Sensitive Nanovalves. J. Am. Chem. Soc. 2010, 132, 12690–12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H.; Xue M.; Zink J. I.; Nel A. E. Development of Pharmaceutically Adapted Mesoporous Silica Nanoparticles Platform. J. Phys. Chem. Lett. 2012, 3, 358–359. [DOI] [PubMed] [Google Scholar]
- Meng H.; Yang S.; Li Z.; Xia T.; Chen J.; Ji Z.; Zhang H.; Wang X.; Lin S.; Huang C.; et al. Aspect Ratio Determines the Quantity of Mesoporous Silica Nanoparticle Uptake by a Small GTPase-Dependent Macropinocytosis Mechanism. ACS Nano 2011, 5, 4434–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H.; Zhao Y.; Dong J.; Xue M.; Lin Y.-S.; Ji Z.; Mai W. X.; Zhang H.; Chang C. H.; Brinker C. J.; et al. Two-Wave Nanotherapy To Target the Stroma and Optimize Gemcitabine Delivery to a Human Pancreatic Cancer Model in Mice. ACS Nano 2013, 7, 10048–10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackmann E. Supported Membranes: Scientific and Practical Applications. Science 1996, 271, 43–48. [DOI] [PubMed] [Google Scholar]
- Linseisen F. M.; Hetzer M.; Brumm T.; Bayerl T. Differences in the Physical Properties of Lipid Monolayers and Bilayers on a Spherical Solid Support. Biophys. J. 1997, 72, 1659–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauda V.; Engelke H.; Sauer A.; Arcizet D.; Bräuchle C.; Rädler J.; Bein T. Colchicine-Loaded Lipid Bilayer-Coated 50 nm Mesoporous Nanoparticles Efficiently Induce Microtubule Depolymerization upon Cell Uptake. Nano Lett. 2010, 10, 2484–2492. [DOI] [PubMed] [Google Scholar]
- Ashley C. E.; Carnes E. C.; Epler K. E.; Padilla D. P.; Phillips G. K.; Castillo R. E.; Wilkinson D. C.; Wilkinson B. S.; Burgard C. A.; Kalinich R. M.; et al. Delivery of Small Interfering RNA by Peptide-Targeted Mesoporous Silica Nanoparticle-Supported Lipid Bilayers. ACS Nano 2012, 6, 2174–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyo C.; Weiss V.; Bräuchle C.; Bein T. Multifunctional Mesoporous Silica Nanoparticles as a Universal Platform for Drug Delivery. Chem. Mater. 2013, 26, 435–451. [Google Scholar]
- Yang Y.; Song W.; Wang A.; Zhu P.; Fei J.; Li J. Lipid Coated Mesoporous Silica Nanoparticles as Photosensitive Drug Carriers. Phys. Chem. Chem. Phys. 2010, 12, 4418–4422. [DOI] [PubMed] [Google Scholar]
- Mackowiak S. A.; Schmidt A.; Weiss V.; Argyo C.; von Schirnding C.; Bein T.; Bräuchle C. Targeted Drug Delivery in Cancer Cells with Red-Light Photoactivated Mesoporous Silica Nanoparticles. Nano Lett. 2013, 13, 2576–2583. [DOI] [PubMed] [Google Scholar]
- van Schooneveld M. M.; Vucic E.; Koole R.; Zhou Y.; Stocks J.; Cormode D. P.; Tang C. Y.; Gordon R. E.; Nicolay K.; Meijerink A.; et al. Improved Biocompatibility and Pharmacokinetics of Silica Nanoparticles by Means of a Lipid Coating: A Multimodality Investigation. Nano Lett. 2008, 8, 2517–2525. [DOI] [PubMed] [Google Scholar]
- Ashley C. E.; Carnes E. C.; Phillips G. K.; Padilla D.; Durfee P. N.; Brown P. A.; Hanna T. N.; Liu J.; Phillips B.; Carter M. B.; et al. The Targeted Delivery of Multicomponent Cargos to Cancer Cells by Nanoporous Particle-Supported Lipid Bilayers. Nat. Mater. 2011, 10, 476–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Stace-Naughton A.; Jiang X.; Brinker C. J. Porous Nanoparticle Supported Lipid Bilayers (Protocells) as Delivery Vehicles. J. Am. Chem. Soc. 2009, 131, 1354–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Jiang X.; Ashley C.; Brinker C. J. Electrostatically Mediated Liposome Fusion and Lipid Exchange with a Nanoparticle-Supported Bilayer for Control of Surface Charge, Drug Containment, and Delivery. J. Am. Chem. Soc. 2009, 131, 7567–7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer L.; Janoff A. Optimizing Combination Chemotherapy by Controlling Drug Ratios. Mol. Interventions 2007, 7, 216–223. [DOI] [PubMed] [Google Scholar]
- Baker J. A. R.; Wickremsinhe E. R.; Li C. H.; Oluyedun O. A.; Dantzig A. H.; Hall S. D.; Qian Y.-w.; Ring B. J.; Wrighton S. A.; Guo Y. Pharmacogenomics of Gemcitabine Metabolism: Functional Analysis of Genetic Variants in Cytidine Deaminase and Deoxycytidine Kinase. Drug Metab. Dispos. 2013, 41, 541–545. [DOI] [PubMed] [Google Scholar]
- Lin N.; Zeng S.; Ma S.; Fan Y.; Zhong H.; Fang L. Determination of Gemcitabine and Its Metabolite in Human Plasma Using High-Pressure Liquid Chromatography Coupled with a Diode Array Detector. Acta. Pharmacol. Sin. 2004, 12, 1584–1589. [PubMed] [Google Scholar]
- Ferrari M. Cancer Nanotechnology: Opportunities and Challenges. Nat. Rev. Cancer 2005, 5, 161–171. [DOI] [PubMed] [Google Scholar]
- Peer D.; Karp J. M.; Hong S.; Farokhzad O. C.; Margalit R.; Langer R. Nanocarriers as an Emerging Platform for Cancer Therapy. Nat. Nanotechnol. 2007, 2, 751–760. [DOI] [PubMed] [Google Scholar]
- Zhao F.; Meng H.; Yan L.; Wang B.; Zhao Y. Nanosurface Chemistry and Dose Govern the Bioaccumulation and Toxicity of Carbon Nanotubes, Metal Nanomaterials and Quantum Dots in Vivo. Sci. Bull. 2015, 60, 3–20. [Google Scholar]
- Hu C.; Aryal S.; Zhang L. Nanoparticle-Assisted Combination Therapies for Effective Cancer Treatment. Ther. Delivery 2010, 1, 323–334. [DOI] [PubMed] [Google Scholar]
- Parhi P.; Mohanty C.; Sahoo S. K. Nanotechnology-Based Combinational Drug Delivery: An Emerging Approach for Cancer Therapy. Drug Discovery Today 2012, 17, 1044–1052. [DOI] [PubMed] [Google Scholar]
- Ma L.; Kohli M.; Smith A. Nanoparticles for Combination Drug Therapy. ACS Nano 2013, 7, 9518–9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers T.; Storm G.; Kiessling F. Nanomedicine Formulations for Combination Therapies. Nano Rev. 2010, 10.3402/nano.v1i0.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal S.; Hu C.-M. J.; Zhang L. Combinatorial Drug Conjugation Enables Nanoparticle Dual-Drug Delivery. Small 2010, 6, 1442–1448. [DOI] [PubMed] [Google Scholar]
- Tardi P.; Johnstone S.; Harasym N.; Xie S.; Harasym T.; Zisman N.; Harvie P.; Bermudes D.; Mayer L. In Vivo Maintenance of Synergistic Cytarabine: Daunorubicin Ratios Greatly Enhances Therapeutic Efficacy. Leuk. Res. 2009, 33, 129–139. [DOI] [PubMed] [Google Scholar]
- Batist G.; Gelmon K. A.; Chi K. N.; Miller W. H.; Chia S. K. L.; Mayer L. D.; Swenson C. E.; Janoff A. S.; Louie A. C. Safety, Pharmacokinetics, and Efficacy of CPX-1 Liposome Injection in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2009, 15, 692–700. [DOI] [PubMed] [Google Scholar]
- Aryal S.; Hu C.-M. J.; Zhang L. Polymeric Nanoparticles with Precise Ratiometric Control over Drug Loading for Combination Therapy. Mol. Pharmaceutics 2011, 8, 1401–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao L.; Guo S.; Zhang J.; Kim W. Y.; Huang L. Nanoparticles with Precise Ratiometric Co-loading and Co-delivery of Gemcitabine Monophosphate and Cisplatin for Treatment of Bladder Cancer. Adv. Funct. Mater. 2014, 24, 6601–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff D. D.; Ramanathan R. K.; Borad M. J.; Laheru D. A.; Smith L. S.; Wood T. E.; Korn R. L.; Desai N.; Trieu V.; Iglesias J. L.; et al. Gemcitabine Plus Nab-Paclitaxel Is an Active Regimen in Patients with Advanced Pancreatic Cancer: A Phase I/II Trial. J. Clin. Oncol. 2011, 29, 4548–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H.; Xia T.; George S.; Nel A. E. A Predictive Toxicological Paradigm for the Safety Assessment of Nanomaterials. ACS Nano 2009, 3, 1620–1627. [DOI] [PubMed] [Google Scholar]
- Nel A.; Xia T.; Mädler L.; Li N. Toxic Potential of Materials at the Nanolevel. Science 2006, 311, 622–627. [DOI] [PubMed] [Google Scholar]
- Sackmann E.; Tanaka M. Supported Membranes on Soft Polymer Cushions: Fabrication, Characterization and Applications. Trends Biotechnol. 2000, 18, 58–64. [DOI] [PubMed] [Google Scholar]
- Bayerl T. M.; Bloom M. Physical Properties of Single Phospholipid Bilayers Adsorbed to Micro Glass Beads. A New Vesicular Model System Studied by 2H-Nuclear Magnetic Resonance. Biophys. J. 1990, 58, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linseisen F. M.; Hetzer M.; Brumm T.; Bayerl T. M. Differences in the Physical Properties of Lipid Monolayers and Bilayers on a Spherical Solid Support. Biophys. J. 1997, 72, 1659–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mornet S.; Lambert O.; Duguet E.; Brisson A. The Formation of Supported Lipid Bilayers on Silica Nanoparticles Revealed by Cryoelectron Microscopy. Nano Lett. 2005, 5, 281–285. [DOI] [PubMed] [Google Scholar]
- Roggers R. A.; Lin V. S. Y.; Trewyn B. G. Chemically Reducible Lipid Bilayer Coated Mesoporous Silica Nanoparticles Demonstrating Controlled Release and HeLa and Normal Mouse Liver Cell Biocompatibility and Cellular Internalization. Mol. Pharmaceutics 2012, 9, 2770–2777. [DOI] [PubMed] [Google Scholar]
- Dengler E. C.; Liu J.; Kerwin A.; Torres S.; Olcott C. M.; Bowman B. N.; Armijo L.; Gentry K.; Wilkerson J.; Wallace J.; et al. Mesoporous Silica-Supported Lipid Bilayers (Protocells) for DNA Cargo Delivery to the Spinal Cord. J. Controlled Release 2013, 168, 209–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommerwerk A.; Brüßler J.; Schäfer J.; Baginski L.; Bandulik M.; Bakowsky U. Lipid Coated Chitosan Microparticles as Protein Carriers. Pulm. Pharmacol. Ther. 2011, 8, 1978–1984. [Google Scholar]
- Ji T.; Zhao Y.; Ding Y.; Nie G. Using Functional Nanomaterials To Target and Regulate the Tumor Microenvironment: Diagnostic and Therapeutic Applications. Adv. Mater. 2013, 25, 3508–3525. [DOI] [PubMed] [Google Scholar]
- Shipley L. A.; Brown T. J.; Cornpropst J. D.; Hamilton M.; Daniels W. D.; Culp H. W. Metabolism and Disposition of Gemcitabine, and Oncolytic Deoxycytidine Analog, in Mice, Rats, and Dogs. Drug Metab. Dispos. 1992, 20, 849–855. [PubMed] [Google Scholar]
- Shu C. J.; Campbell D. O.; Lee J. T.; Tran A. Q.; Wengrod J. C.; Witte O. N.; Phelps M. E.; Satyamurthy N.; Czernin J.; Radu C. G. Novel PET Probes Specific for Deoxycytidine Kinase. J. Nucl. Med. 2010, 51, 1092–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M. J.; Goldstein D.; Hamm J.; Figer A.; Hecht J. R.; Gallinger S.; Au H. J.; Murawa P.; Walde D.; Wolff R. A.; et al. Erlotinib Plus Gemcitabine Compared with Gemcitabine Alone in Patients with Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Cai T.-y.; Zhu H.; Yang L.-q.; Jiang H.; Dong X.-w.; Hu Y.-z.; Lin N.-m.; He Q.-j.; Yang B. Synergistic Antitumor Activity of Gemcitabine and ABT-737 in Vitro and in Vivo through Disrupting the Interaction of USP9X and Mcl-1. Mol. Cancer Ther. 2011, 10, 1264–1275. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Ji Z.; Xia T.; Meng H.; Low-Kam C.; Liu R.; Pokhrel S.; Lin S.; Wang X.; Liao Y.-P.; et al. Use of Metal Oxide Nanoparticle Band Gap To Develop a Predictive Paradigm for Oxidative Stress and Acute Pulmonary Inflammation. ACS Nano 2012, 6, 4349–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaCosta M. V.; Doughan S.; Han Y.; Krull U. J. Lanthanide Upconversion Nanoparticles and Applications in Bioassays and Bioimaging: A Review. Anal. Chim. Acta 2014, 832, 1–33. [DOI] [PubMed] [Google Scholar]
- Sugahara K. N.; Teesalu T.; Karmali P. P.; Kotamraju V. R.; Agemy L.; Greenwald D. R.; Ruoslahti E. Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science 2010, 328, 1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Maltzahn G.; Park J.-H.; Lin K. Y.; Singh N.; Schwppe C.; Mesters R.; Berdel W. E.; Ruoslahti E.; Sailor M. J.; Bhatia S. N. Nanoparticles That Communicate in Vivo To Amplify Tumour Targeting. Nat. Mater. 2011, 10, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H.; Xing G.; Sun B.; Zhao F.; Lei H.; Li W.; Song Y.; Chen Z.; Yuan H.; Wang X.; et al. Potent Angiogenesis Inhibition by the Particulate Form of Fullerene Derivatives. ACS Nano 2010, 4, 2773–2783. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.