

Abstract
Autophagy, or “self eating,” is an adaptive process that helps cells cope with metabolic, toxic, and even infectious stressors. While the adaptive capability of autophagy is generally beneficial, autophagy can also facilitate enhanced nutrient utilization and improved growth characteristics in cancer cells. Moreover, autophagy can promote greater cellular robustness in the context of therapeutic intervention. This has proven to be the case in advanced prostate cancer, where preclinical data largely supports that autophagy facilitates both disease progression and therapeutic resistance. Notably, androgen deprivation therapy, taxane-based chemotherapy, targeted kinase inhibition, and nutrient restriction all induce significant cellular distress. Autophagy is subsequently up-regulated through core metabolic regulatory signaling cascades (i.e. AMPK, PI3K, and mTOR), and more favorable growth and nutrient conditions are established. Current research also demonstrates that when the autophagic machinery is inhibited, greater cell killing and tumor responsiveness can be obtained. In this review, we will cover current prostate cancer treatments associated with alterations in autophagy; data supporting autophagic modulation with added emphasis on alterations occurring within prostate cancer models; and finally, research supporting adjuvant autophagic modulation with current prostate cancer treatment paradigms.
Introduction
In health and disease, the biomolecular adaptions to “stress” are necessarily dynamic. Complex and integrated biological processes such as inflammation, immunity, and nutrient depletion require coordinated regulation of cellular machinery and efficient utilization of available nutrients. Autophagy, or “self-eating” is an essential and highly conserved homeostatic process that can do just this. While “self-eating” sounds detrimental, research shows that a basal level of autophagy is critical for normal development and homeostasis. [2-4] Autophagy maintains cellular homeostasis by metabolizing cytoplasmic protein waste through the lysosomal system. Furthermore, research has also shown that autophagy can suppress tumor development. [5, 6] But autophagy is multi-faceted, and it may not always be protective. In established tumors, autophagy can be utilized to meet the heightened nutrient demand found in proliferative cancer cells. [6] Moreover, autophagy can be used by cancer cells as a survival strategy during therapeutic intervention (e.g., radiation, chemotherapy, and hormone therapy). [8] Understanding the context-dependent role of autophagy in cancer development, and specifically treatment resistance, has the potential to improve current treatment paradigms, including advanced prostate cancer. [9-11] Current treatment paradigms for prostate cancer have demonstrated limited and transient efficacy. The adaptive capabilities of autophagy potentially contribute to this, and concurrent autophagy manipulation is under therapeutic investigation. This review will cover current prostate cancer treatments associated with alterations in autophagy; data supporting autophagic modulation with added emphasis on alterations occurring within prostate cancer models; and finally, research supporting adjuvant autophagic modulation with current prostate cancer treatment paradigms.
Autophagy and Autophagic Modulation
Autophagy
Autophagy (also known as macroautophagy) is a highly conserved evolutionary process that is involved in a number of cellular homeostatic processes which regulate cytoplasmic biomass, organellar abundance, and organellar distribution, and which remove harmful protein aggregates and intracellular toxins. [3, 12, 13] Autophagy also serves important immunological roles and is capable of removing intracellular pathogens and influencing both innate and adaptive immunity. [13-16]
Perhaps not surprisingly, given autophagy's diverse metabolic and immunological involvement, autophagic dysregulation has been linked to tumorigenesis. Mice that are heterozygous for the autophagy gene, Beclin 1, have been shown to develop multiple spontaneous malignancies. [17] Furthermore, the monoalleliac deletion of this same gene has also been found in several human cancers. [3] Autophagy has also been found to be upregulated at the immunological synapse during dendritic cell CD4+ T-cell contact – influencing T-cell activation and likely anti-tumor immunity as well (Note, there is tremendous plasticity in this system and the consequences of too little or too much autophagy in regards to immune system function requires more research). [13, 14] Nevertheless, while it is generally accepted that autophagy can act as a protective mechanism, in established tumors, autophagy may actually facilitate carcinogenesis. [18, 19] Studies have shown autophagy to be up-regulated in the internal environment of tumors, areas that are characteristically low in nutrient and oxygen levels. [5, 6, 20] Additionally, study models where essential autophagy genes (e.g., Beclin 1, Atg5, and Atg7) have been knocked out are much more sensitive to metabolic stress. [18] Moreover, the up-regulation of key autophagy markers in tumor tissues has been linked to poorer therapeutic outcomes. [11]
This apparent context-dependent role of autophagy – defined by the stage of disease – is complicated by the significant cross-talk between the autophagic and apoptotic molecular pathways. [3, 21, 22] Both pathways share important molecular intermediates (e.g., the inhibitory interaction between bcl-2 and beclin 1 and the interaction between LC3B and Fas), and these intermediates appear to be largely counter-regulatory. [1, 21] In other words, the autophagic and apoptotic pathways can be considered as competitive determinants of cell fate. Nevertheless, if the homeostatic balance between autophagy and apoptosis is lost, either pathway can potentially drive cellular demise. Studies have shown that cell lines over-expressing beclin 1, a key autophagy-regulated protein, result in hyper-active autophagy and ultimately complete self-digestion. [23-25] Cell death that is mediated through autophagy, or autophagy-associated cell death (Type II Programed Cell Death), is characteristically caspase-independent and histologically distinct from apoptosis (Type I Programmed Cell Death). [23] Therefore, autophagy may continue to facilitate cell death even in cases of apoptotic dysfunction. [23, 26] Apoptotic dysfunction is not uncommon in cancer pathogenesis; thus autophagy up-regulation, even in the case of established tumors, may have nuanced implications in terms of cell fate. [27] In summary, the contextual meaning of autophagic activity is complex, depending not only the stage of disease, but on the apoptotic status as well.[22]
Autophagic Modulation
Given the dynamic role of autophagy in tumor pathogenesis, and that most cancer treatments induce autophagy, adjuvant autophagy inhibition has become an area of research interest. The results have been encouraging; studies have shown that the pharmacological inhibition of autophagy can enhance the cell killing effect of cancer therapeutics in preclinical models. [28-30] Based upon the success of preclinical reports demonstrating therapeutic efficacy, concurrent autophagy inhibition is now also being explored in a number of clinical trials for patients with refractory malignancies. [31] (See Table of ongoing clinical trials) Several autophagy inhibitors have been explored in both preclinical and clinical models, and these inhibitors are typically classified by the stage in which they inhibit autolysomal formation (i.e. the mature autophagosome complex). [32] In prostate cancer research specifically, laboratory inhibitors of autophagy include 3 methyladenine (3-MA), and siRNAs targeting specific autophagy related genes (Atgs). (The use of siRNAs has largely been relegated to preclinical studies as a confirmatory tool of autophagy inhibition.) Alternatively, more clinically applicable inhibitors of autophagy include chloroquine, its derivative, hydroxychloroquine, and bafilomycin A1. [32] Chloroquine and hydroxychloroquine – lysosomotropic amines traditionally used to treat malaria and rheumatoid arthritis – have been the most widely studied. [12, 33] Both of these agents are inexpensive and have well known pharmacokinetic profiles, facilitating their use in clinical trials.
Table 1. Ongoing Clinical Trials (Clinicaltrials.gov).
| Phase: | Interventions: | Condition: | Status: | Study: |
|---|---|---|---|---|
| II | Drug: hydroxychloroquine | Prostate Cancer | Active, not recruiting | Hydroxychloroquine in Treating Patients With Rising PSA Levels After Local Therapy for Prostate Cancer |
| I | Drug: Akt inhibitor MK2206|Drug: hydroxychloroquine|Other: pharmacological study|Other: laboratory biomarker analysis | Recurrent Prostate Cancer|Recurrent Renal Cell Cancer|Stage III Prostate Cancer|Stage III Renal Cell Cancer|Stage IV Prostate Cancer|Stage IV Renal Cell Cancer|Unspecified Adult Solid Tumor, Protocol Specific | Recruiting | Akt Inhibitor MK2206 and Hydroxychloroquine in Treating Patients With Advanced Solid Tumors or Prostate or Kidney Cancer |
| II | Drug: docetaxel|Drug: hydroxychloroquine | Prostate Cancer | Terminated; Has Results | Docetaxel and Hydroxychloroquine in Treating Patients With Metastatic Prostate Cancer |
| II | Drug: Abiraterone|Drug: ABT-263|Drug: Hydroxychloroquine | Prostate Cancer | Recruiting | Phase II Study of ABT-263/Abiraterone or ABT-263/Abiraterone/Hydroxychloroquine in Prostrate Cancer |
| II | Drug: metformin hydrochloride | Prostate Cancer | Active, not recruiting | Metformin Hydrochloride as First-Line Therapy in Treating Patients With Locally Advanced or Metastatic Prostate Cancer |
| II | Drug: Placebo and Castration|Drug: Metformin and Castration | Prostate Cancer | Recruiting | Castration Compared to Castration Plus Metformin as First Line Treatment for Patients With Advanced Prostate Cancer |
| I/II | Drug: Metformin | Prostate Cancer | Active, not recruiting | Metformin in Castration-Resistant Prostate Cancer |
| II | Drug: METFORMIN|Drug: Placebo|Drug: TAXOTERE® | Prostatic Neoplasms | Recruiting | Metformin-Docetaxel Association in Metastatic Hormone-refractory Prostate Cancer |
| II | Drug: Metformin | Metastatic Prostate Cancer | Not yet recruiting | Impact of the Addition of Metformin to Abiraterone in Metastatic Prostate Cancer Patients |
| I/II | Drug: everolimus|Drug: gefitinib | Brain and Central Nervous System Tumors|Prostate Cancer | Completed | Everolimus and Gefitinib in Treating Patients With Progressive Glioblastoma Multiforme or Progressive Metastatic Prostate Cancer |
| II | Drug: Pasireotide|Drug: Everolimus|Other: Laboratory biomarker analysis | Castrate Resistant Prostate Cancer|Chemotherapy Naive Prostate Cancer|Prostate Cancer | Recruiting | Pasireotide (SOM230) With or Without Everolimus in Treating Patients With Hormone Resistant, Chemotherapy Naive Prostate Cancer |
| I/II | Drug: RAD001|Drug: Docetaxel | Metastatic, Androgen Independent Prostate Cancer|Prostate Cancer | Completed | The Use of RAD001 With Docetaxel in the Treatment of Metastatic, Androgen Independent Prostate Cancer |
| I | Drug: vorinostat|Drug: temsirolimus|Other: laboratory biomarker analysis|Procedure: positron emission tomography/computed tomography | Prostate Cancer|Adenocarcinoma of the Prostate|Hormone-resistant Prostate Cancer|Recurrent Prostate Cancer|Stage IV Prostate Cancer | Recruiting | Temsirolimus and Vorinostat in Treating Patients With Metastatic Prostate Cancer |
| I/II | Biological: cixutumumab|Other: diagnostic laboratory biomarker analysis|Drug: temsirolimus | Adenocarcinoma of the Prostate|Hormone-resistant Prostate Cancer|Recurrent Prostate Cancer|Stage IV Prostate Cancer | Active, not recruiting | Cixutumumab and Temsirolimus in Treating Patients With Metastatic Prostate Cancer |
| II | Drug: Temsirolimus|Drug: Diphenhydramine | Prostate Cancer | Terminated; Has Results | Impact of Temsirolimus Therapy on Circulating Tumor Cell Biology In Men With Castration Resistant Metastatic Prostate Cancer |
| I/II | Drug: RAD001, Docetaxel, Bevacizumab | Prostate Cancer | Recruiting | Safety Study & Effectiveness of Docetaxel With RAD001 and Bevacizumab in Men With Advanced Prostate Cancer |
| I | Other: Everolimus, lupron, bicalutamide, and radiation | Prostate Cancer | Recruiting | Study of Everolimus Added to Combined Hormonal and Radiation Therapy for High Risk Prostate Cancer |
| II | Drug: carboplatin|Drug: RAD 001|Drug: prednisone|Other: laboratory biomarker analysis|Other: pharmacological study | Prostate Cancer | Active, not recruiting | Carboplatin, Everolimus, and Prednisone in Treating Patients With Metastatic Prostate Cancer That Progressed After Docetaxel |
| II | Drug: torisel | Prostate Cancer | Active, not recruiting | Single Agent Temsirolimus (Torisel®) in Chemotherapy-naïve Castration-Resistant Prostate Cancer Patients |
| II | Drug: everolimus | Prostate Cancer | Active, not recruiting | Everolimus as First-Line Therapy in Treating Patients With Prostate Cancer |
| II | Drug: everolimus|Procedure: conventional surgery | Prostate Cancer | Active, not recruiting | Everolimus in Treating Patients With Newly Diagnosed Localized Prostate Cancer |
| I/II | Drug: temsirolimus|Biological: bevacizumab|Genetic: polymorphism analysis|Other: laboratory biomarker analysis | Prostate Cancer | Active, not recruiting | Temsirolimus and Bevacizumab in Hormone-Resistant Metastatic Prostate Cancer That Did Not Respond to Chemotherapy |
| II | Drug: RAD001 | Prostate Cancer | Recruiting | Phase II Study of RAD001 in a Neoadjuvant Setting in Men With Intermediate or High Risk Prostate Cancer |
| I | Drug: bicalutamide|Drug: everolimus|Drug: leuprolideacetate|Radiation: external beam radiation therapy | Prostate Cancer | Recruiting | Everolimus, Bicalutamide, and Leuprolide Acetate in Treating Patients Undergoing Radiation Therapy For High-Risk Locally Advanced Prostate Cancer |
| II | Drug: bicalutamide|Drug: Everolimus | Prostate Cancer | Active, not recruiting | Bicalutamide With or Without Everolimus in Treating Patients With Recurrent or Metastatic Prostate Cancer |
| II | Drug: RAD001 | Hormone Refractory Prostate Cancer | Completed | RAD001 in Patients With Metastatic, Hormone-Refractory Prostate Cancer |
| I/II | Drug: Docetaxel|Drug: Temsirolimus | Prostatic Neoplasms | Recruiting | CESAR Study in Prostate Cancer With Temsirolimus Added to Standard Docetaxel Therapy (CEPTAS) |
| II | Drug: temsirolimus|Procedure: conventional surgery|Procedure: neoadjuvant therapy | Prostate Cancer | Completed | Neoadjuvant CCI-779 Followed By Radical Prostatectomy in Treating Patients With Newly Diagnosed Prostate Cancer Who Have a High Risk of Relapse |
| I/II | Drug: sirolimus|Other: immunohistochemistry staining method|Procedure: neoadjuvant therapy | Prostate Cancer | Active, not recruiting | Sirolimus Before Surgery in Treating Patients With Advanced Localized Prostate Cancer |
| I | Drug: Everolimus (RAD001) | Prostate Cancer Patients With Detectable PSA Following Prostatectomy | Recruiting | Phase 1 Trial of the Mammalian Target of Rapamycin (mTOR) Inhibitor Everolimus Plus Radiation Therapy (RT) for Salvage Treatment of Biochemical Recurrence in Prostate Cancer Patients Following Prostatectomy |
| II | Drug: temsirolimus | Prostate Cancer | Completed | CCI-779 in Treating Patients With Prostate Cancer |
| II | Drug: RAD001 | Metastatic Hormone Refractory Prostate Cancer | Completed | Molecular, Genetic, and Genomic Assessments From Patients Treated With RAD001 |
| I | Biological: DEC-205-NY-ESO-1 fusion protein vaccine|Drug: sirolimus|Other: laboratory biomarker analysis | Patients With NY-ESO-1 Expressing Solid Tumors | Recruiting | Vaccine Therapy With or Without Sirolimus in Treating Patients With NY-ESO-1 Expressing Solid Tumors |
| I | Drug: temsirolimus|Drug: vinorelbineditartrate | Patients With Unresectable or Metastatic Solid Tumors | Active, not recruiting | Temsirolimus and Vinorelbine Ditartrate in Treating Patients With Unresectable or Metastatic Solid Tumors |
| I/II | Drug: temsirolimus | Prostatic Neoplasms | Completed | An Open Label Exploratory Study in Newly Diagnosed Prostate Cancer Patients |
| I | Drug: AZD2014 | Prostate Cancer | Not yet recruiting | Investigating the Effects of AZD2014 Therapy Given Prior to Radical Prostatectomy in Men With High Risk Prostate Cancer |
| II | Drug: ridaforolimus | Prostate Cancer | Completed | AP23573 in Patients With Taxane-Resistant Androgen-Independent Prostate Cancer (AIPC)(8669-017)(COMPLETED) |
| II | Drug: ridaforolimus (MK8669)|Drug: Comparator: Placebo|Drug: open-label ridaforolimus (MK8669) | Prostate Cancer | Completed | Bicalutamide and Ridaforolimus in Men With Prostate Cancer (MK-8669-002) |
| II | Drug: Pantoprazole | Prostate Cancer | Recruiting | Pantoprazole and Docetaxel for Men With Metastatic Castration-Resistant Prostate Cancer |
| I | Drug: ADI-PEG 20 | Solid Tumors|Prostate Cancer | Recruiting | Ph 1 Trial of ADI-PEG 20 Plus Docetaxel in Solid Tumors With Emphasis on Prostate Cancer and Non-Small Cell Lung Cancer |
Other notable autophagy inhibitors being investigated in prostate cancer research include metformin and desmethylclomipramine. Metformin, an oral biguanide that is commonly prescribed to treat non-insulin-dependent diabetes, has also been shown to inhibit autophagy. [32] Nevertheless, the exact mechanism of autophagic inhibition is still being explored. What is known is that metformin activates AMPK, a potent inhibitor of mTOR. [34] But mechanistically, this would suggest that metformin is an autophagy stimulator. This apparent contradiction can be explained by metformin's proposed effect on beclin 1, a core autophagy regulator. A recent study has shown that metformin can actually sequester beclin 1, thus inhibiting autophagic activity despite the inhibition of mTOR. [35] Beyond metformin's role in autophagy inhibition, metformin has also shown to have anti-proliferative effects in a variety of cancer cell lines (e.g., prostate, ovarian, breast, colorectal, and endometrial carcinoma models). [36-42] Moreover, several studies suggest that patients taking metformin for diabetes mellitus have decreased prostate cancer incidence and disease severity as well. [43, 44] Thus, metformin possesses several characteristics that make it not only a more readily translatable autophagy inhibitor, but also characteristics that strongly suggest better efficacy in prostate cancer specifically. Lastly, desmethylclomipramine, a tricyclic antidepressant, has also been shown to inhibit autophagy. [45] Like hydroxychloroquine and metformin, its widespread use improves its value as a clinically viable autophagy inhibitor. Desmethylclomipramine is believed to decrease autophagic flux by inhibiting the fusion of the autophagosome and the lysosome, but more work is needed to fully understand its mechanism of action. [46]
While we believe that many, if not most, prostate cancer treatments induce a cytoprotective variety of autophagy, several prostate cancer treatments may actually induce toxic, or unsustainably elevated, autophagic activity (e.g., Rad001, zoledronic acid, Atorvastatin, YM155, (-)-gossypol). [47] Mechanistically, these cases of cytotoxic autophagy have been attributed to the sustained activity of critical autophagy promoters, namely Atgs and Beclin 1. [47] Regardless, a direct link between up-regulated autophagy promoters and cell death has remained ellusive. A recent publication by Shen et. al. screened 1400 compounds for increased autophagic flux but found no evidence for autophagic death. Alternatively, some have proposed that autophagy may precede cell death but is not necessarily causal. For example, Giampietri et. al. demonstrated enhanced TNF-alpha-dependent apoptosis in PC3 cells (prostate cancer cells without functioning androgen receptor) when autophagy was induced. [48] Given the controversy currently surrounding cyotoxic autophagy, the term autophagy-associated cell death is commonly used. And while many other autophagy inducers are being investigated, much more research is required to improve our understanding of autophagy-associated cell death and whether or not there is a clinical role for adjuvant autophagy induction in prostate cancer.
Treatments and Autophagy
ADT and Androgen Receptor Inhibitors
Androgen ablation is a common therapeutic intervention for advanced prostate cancer, producing widespread death of the prostate epithelia and dramatic, albeit transient, clinical improvements. [49] Unfortunately, advanced prostate cancer being treated with ADT ultimately progresses to a lethal phenotype known as castration-resistant prostate cancer. [50] Research conducted in our lab and elsewhere indicates that ADT stimulates autophagic activity – facilitating the development of the castration resistant phenotype. [46] Prior research conducted by Li et. al. and Jiang et. al. has shown that androgens play an influential role in autophagy and that the androgen receptor is a key mediator. [51, 52] The androgen receptor negatively controls autophagic activity, and the targeted inhibition of this receptor by both Bennett et. al. and our lab has demonstrated that such inhibition positively influences autophagy. (See Figure 2) Bennett et. al., determined that LNCaP cells treated with bicalutamide, a non-steroidal androgen receptor antagonist, induced autophagy in the absence of any additional stressors. [2] Recently, research conducted by our lab group further substantiates these findings, and we were able to show that the targeted inhibition of the androgen receptor with enzalutamide produced significant autophagic flux in in vitro and in vivo prostate cancer models. [46] Taken together, these data suggest that ADT – and perhaps more importantly androgen receptor inhibition – can directly promote the cellular adaptions that help overcome the metabolic stress of hormone withdrawal.
Figure 2.
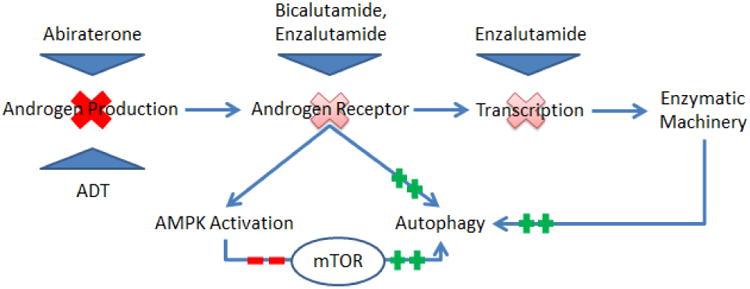
Depicts therapeutic interventions that either decrease total androgen or impede androgen receptor activity. The associated alterations in androgen receptor activity and down-stream synthetic capability influence autophagy, most notably through mTOR inhibition.
The exact molecular events that link androgen receptor inhibition with autophagy are still somewhat fragmented, but mTOR appears to play a critical role. Xuet. al. demonstrated that androgen treatment directly affected mTOR activity and that the androgen receptor promoted mTOR mRNA synthesis. This finding has been reinforced by a number of other studies that have shown androgen deprivation to specifically decrease downstream mTOR metabolites. [53] In our lab, androgen deprivation and prolonged androgen receptor blockade with enzalutamide were coupled with AMPK activation and ultimately mTOR suppression via phospho-Raptor. [46] Similarly, others have shown that AMPK directly regulates mTOR activity in response to imbalances in the core energy metabolites, AMP and ATP. [51, 54] A less direct effect of androgen receptor inhibition is hypoxia. Studies have shown that androgen withdrawal impedes angiogenesis, and that the resultant nutrient deprivation and hypoxia further stimulates autophagy. [55] While these data substantiate the androgen receptor's influence on autophagic flux, the androgen receptor alone cannot completely explain autophagy regulation. For example, PC3 lines lacking androgen receptor can also up-regulate autophagy during ADT. [51] The question is, how? Potential explanations include alterations in proto-oncogenes like PTEN, variations in tumor suppressors like p53, and the aberrant expression of downstream androgen receptor targets.
Given that androgens and the androgen receptor are valuable targets in prostate cancer treatment, and that their therapeutic benefit is in some ways limited by autophagy, studies are currently underway to determine if autophagy modulation could improve ADT. To date, the data supports that concurrent androgen receptor and autophagy inhibition synergistically promote cell death. [56] Additionally, the effects of this synergistic inhibition can occur in a dose- and time- dependent manner. [50] Bennett et. al. demonstrated that inhibiting autophagy with 3-MA enhanced cell death in cells treated with bicalutamide 1.5-fold. In another study by Colquhoun et. al., LNCaP cells treated with bicalutamide and metformin demonstrated a significant reduction in colony formation rates. This also held true for PC3 lines, albeit much higher doses of bicalutamide and metformin were required. [39] In our lab, clomipramine and metformin significantly improved the cyto-toxicity of enzalutamide in vitro. Moreover, these effects translated to in-vivo mouse models, where enzalutamide and clomipramine in combination decreased tumor size by 91 percent. Alternatively, when enzalutamide and metformin were used, tumor size decreased by 78 percent. [46] There are several ongoing clinical trials evaluating adjuvant autophagy inhibition in the context of ADT and androgen receptor blockade. (See Table of ongoing clinical trials) In summary, the androgen receptor plays important, but apparently non-essential roles in autophagy induction. More research is needed to understand the nuanced roles that the androgen receptor plays throughout the progression of prostate cancer.
Taxanes
Microtubules are cytoskeleton structures that rapidly fluctuate through polymerization states to meet a multitude of cellular needs (e.g., mitosis, protein synthesis, and sub-cellular organization). [57] These various processes are in high demand in rapidly dividing cells and consequently, targeting microtubules has been popular in cancer research. In prostate cancer, docetaxel (a microtubule stabilizing agent or taxol) is a current standard of care in men with symptomatic castration-resistance prostate cancer, providing a survival benefit of approximately 2 months. [58] Exploring explanations for this limited therapeutic efficacy has been exceedingly difficult, especially in regards to autophagy. Earlier studies show a variable degree of autophagic activity during taxane-based therapies, with some studies showing that taxanes induced autophagy and others showing that they inhibit autophagy. The discrepancies in these findings can in part be explained by the fact that different concentrations of taxanes and different cell lines were used in this earlier research. Furthermore, a recent study in breast cancer cells suggests that the autophagic response to taxanes is context dependent, with the duration of treatment, and the intensity of environmental factors contributing to the degree of autophagic activity. [59] Aggregately, these data once again highlight the contextual versatility of autophagy.
Despite the variability in the available data, there are mechanistic links between taxanes and autophagy that have been described. Theoretically, taxanes should inhibit autophagy, given that they target microtubules – the core constituents of autophagosome formation and trafficking. But this paradigm has significant limitations. Namely, evidence shows that autophagy can be up-regulated in response to taxanes. Some have proposed that taxanes only target specific elements of the microtubule structure (e.g., the mitotic spindle), leaving enough remaining function to stimulate the core events of autophagy. [60] It has also been proposed that structurally damaged microtubules can induce autophagy through Raf-1 mediated signaling cascades. [61] Perhaps the most well delineated explanations for how taxanes can induce autophagy are described by Notte et. al. [59] In this study they identified two key pathways; the first involved the inhibition of mTOR and the second involved the activation of the c-Jun N-terminal kinase (JNK). (JNK phosphorylates Bcl-2 and Bcl-XL, and disrupts the binding of beclin 1, freeing it to activate autophagy.[59]) Interestingly, while these pathways shared similar functions, they were differentially expressed based upon treatment duration and environmental stress. [59]
While more research is required to understand exactly how taxanes influence autophagic activity, autophagy seems to be an important factor in taxol resistance, specifically in prostate cancer cells. In a recent study by Bennett et. al., LNCaP-AI cells were approximately 2.5 times more resistant to docetaxel than LNCaP cells grown in androgen-replete media. (LNCaP-AI cells are prostate cancer cells that are chronically maintained in androgen-depleted conditions.) Moreover, the authors also demonstrated that the restoration of androgens to the LNCaP-AI cells was capable of restoring docetaxel sensitivity. Autophagy was believed to be responsible for this resistance, and when autophagy was suppressed with 3-MA, the cytotoxic effect of bicalutamide and docetaxel increased two-fold. [2] In summary, the autophagic response in taxanes is context dependent, with the data generally supporting a pro-survival function that can be impeded with adjuvant autophagy inhibition.
Kinase Inhibitors
Tyrosine kinases are essential mediators of signal transduction, and any aberrant activity of this family of kinases can potentially result in abnormalities in cellular proliferation, differentiation, apoptosis, and metabolism. [6, 13] Src Family Kinases (SFKs), a non-receptor tyrosine kinase, is perhaps the most widely characterized member of this family, and it has been shown to play important roles in tumor progression in several different cancers (e.g., glioblastoma, colon, lung, breast and prostate cancers). [62] In prostate cancer specifically, studies in our lab and elsewhere have implicated SFK activation with disease invasiveness and metastatic potential – specifically to bone. [63-65]Also, SFK activity has shown oncogenic effects in both androgen-dependent and independent prostate cancers, suggesting that SFK activity may also be involved in the progression of prostate cancer to castration resistance. [63, 66] The pathogenesis of SFK in prostate cancer has largely been attributed to its ability to activate the androgen receptor independent of androgen stimulation. Studies have shown that SFK activity can be regulated by a number of growth factors during androgen deprivation (e.g. epidermal growth factor [EGF], interleukin-6 [IL-6], interleukin-8 [IL-8], and neurokines [gastrin-releasing peptide]) and these factors activate important proliferative pathways – notably Ras/Raf/Erk and PI3K/Akt. (See Figure 3) [6, 63, 66-69]
Figure 3.
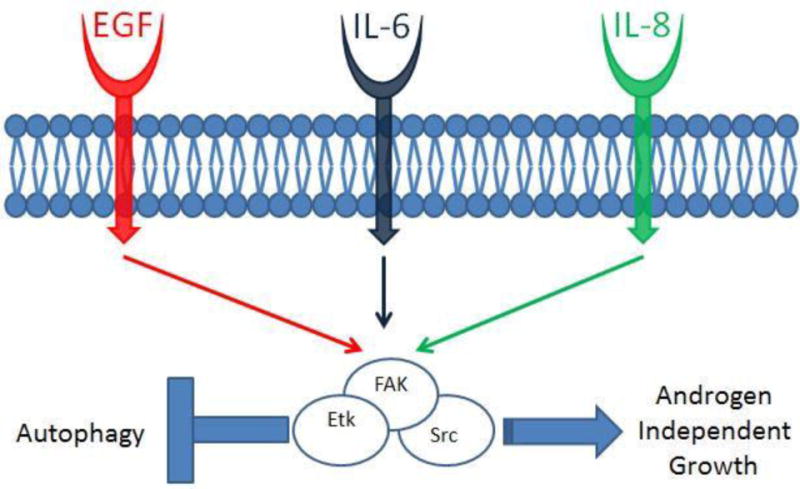
Depicts activation of Src kinase complex by epidermal growth factor (EGF), and cytokines Interleukin-6 (IL-6) and Interleukin-8 (IL-8). The Src kinase complex is composed of Src, Etk (A Btk tyrosine kinase family kinase) and FAK (Focal Adhesion Kinase) and undergoes cross-activation upon stimulation. The Src kinase complex negatively regulates autophagy and also promotes androgen independent growth.
Modified from [63]
Given the role of SFKs in prostate cancer pathogenesis, and that prominent molecular elements of its activity are known, SFKs inhibitors were introduced into clinical research. Dasatinib and saracatinib are two SFK inhibitors that have been investigated in prostate cancer. Saracatinib has proven that it can decrease tumor migration and metastasis in prostate cancer cell lines. [65] However, SFK inhibitors have been decidedly less successful at inducing significant levels of apoptosis in these cell lines, and once again, autophagy is one likely reason. [66, 70] Our research group has demonstrated that when SFK is inhibited, the PI3K/Akt/mTOR/S6K signaling cascade is impeded, resulting in the up-regulation of autophagy. [6, 63, 65, 66] Additionally, SFKs are believed to regulate glucose transport (e.g., GLUT1 and GLUT4), and once glucose transport is compromised, the cells will be energetically stressed. This also will further promote autophagy through AMPK activation. [6, 63] So, while various models directly implicate aberrant SFK activity in prostate cancer pathogenesis, and targeted inhibition of SFK has proven somewhat useful, autophagy provides the mechanistic escape that limits this drug class' therapeutic potential.
Studies exploring autophagy inhibition with concurrent SFK inhibition support that autophagy is associated with this drug class' limited efficacy. When our lab group inhibited autophagy by three independent approaches – using chloroquine, 3-MA, and siRNA directed Atg7 knockdown (an essential autophagy constituent) – each of these approaches enhanced PC3 cell death by means of increased apoptotic activity. Subsequently, we also demonstrated that chloroquine could enhance the therapeutic efficacy of saracatinib in PC3 xenographic mouse models, showing that after 14 days of tumor growth the addition of chloroquine to saracatinib inhibited tumor growth by 64%, versus 26% inhibition in tumors treated with saracatinib alone. [65] In summary, SFKs are influential components of the cellular proliferative and adaptive machinery, and the inhibition of these receptors can affect important characteristics of prostate cancer progression. However, the therapeutic impact of these agents is limited by the paradoxical molecular overlap that supports both treatment effect and treatment resistance. Fortunately, the data seem to suggest that adjunct autophagy inhibition undermines this resistance.
Arginine deiminase
Amino acid deprivation as a cancer treatment modality has long been established in cancer research. [71] A variety of amino acids (e.g., glutamine, methionine, leucine, and arginine) have, at one point, been selectively eliminated to challenge the metabolic needs of a growing cancer. [71] In prostate cancer research specifically, there is growing evidence to suggest that prostate cancer cells are especially susceptible to arginine restriction. In an analysis performed at our institution by Kim et. al., not one of the 88 prostate cancer specimens analyzed expressed arginosuccinate synthetase – suggesting significant limitations in de novo arginine synthesis. [71] Therefore, these prostate cancers should be exquisitely sensitive to any arginine depletion, and indeed when CWR22RV1 cells (a castrate-resistant prostate cancer cell line lacking arginosuccinate synthetase) were treated with arginine deiminase (ADI) – an enzyme that degrades arginine – this accelerated cell death. [71] These results were also replicated in CWR22RV1 xenographs in nude mice. [71] When this killing effect was further analyzed, AMPK was notably activated and the AKT/mTOR/S6K pathway was found to be deactivated. (See Figure 4) This suggests that not only was autophagy up-regulated, but that greater therapeutic efficacy could be achieved with autophagy inhibition.
Figure 4.
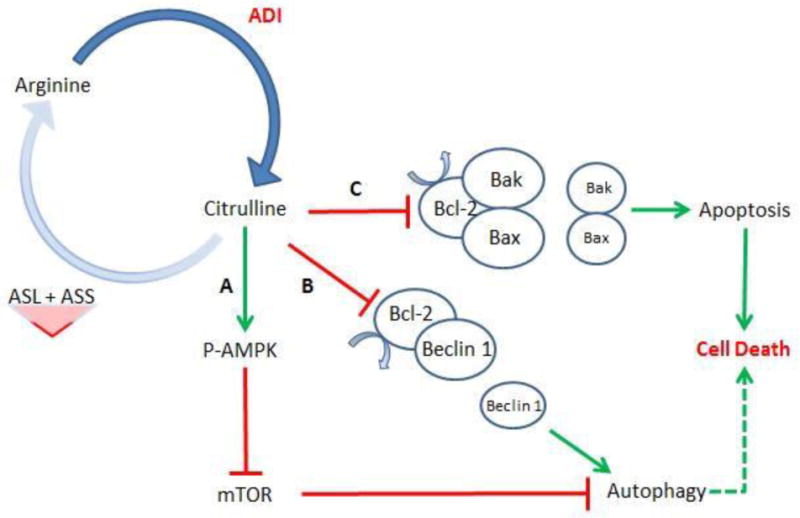
Arginine deiminase (ADI) hydrolyzes arginine into citrulline. To replenish arginine, cells utilize a two-step synthetic pathway involving argininosuccinate synthetase (ASS) and argininosuccinate lyase (ASL). Prostate cancers deficient in either ASL or ASS will not be able to synthesize the necessary amount of arginine de novo. At this point, arginine deficiency can activate several different cellular metabolic pathways. Pathway A, AMPK is activated and mTOR is subsequently inhibited. This will promote autophagy. Pathway B, Bcl-2 is inhibited, freeing Beclin 1 to stimulate autophagy. Pathway C, Bcl-2 is once again inhibited, but in this case freeing Bak/Bax to activate apoptosis. Note: A longer duration of treatment ultimately promotes cell death – mediated by either apoptosis or pro-longed autophagy.
To address this possibility, Kim et. al., treated prostate cancer lines with ADI and chloroquine. They found that chloroquine accelerated the apoptosis induced by ADI. It is worth noting that they also found that chloroquine itself had little effect on cellular viability – supporting therapeutic synergy with ADI. The pro-survival function of autophagy was confirmed with the siRNA knockdown of beclin 1, which effectively inhibited autophagy and promoted apoptosis. However, the synergistic effects of ADI and autophagy inhibition were limited to CWR22RV1 cells. In LNCaP cells that express functional arginosuccinate synthetase, combined ADI and chloroquine produced little effect. Collectively these data suggest that autophagy could be a valuable target in prostate cancers that are found to lack arginosuccinate synthetase. Phase I/II clinical trials in hepatoma and melanoma cell lines have already demonstrated some success, but clinical trials are needed in appropriately selected prostate cancer patients to fully determine the therapeutic efficacy of ADI with concurrent autophagy inhibition. [71]
Conclusion
Our understanding of prostate progression continues to evolve. The dynamic interplay of autophagy in prostate cancer progression has been a molecularly intriguing and potentially modifiable factor in prostate cancer pathogenesis. Fundamentally, the degree of autophagic activity is important; too little or too much autophagy portends either beneficial or deleterious consequences depending on the cellular context. [3] Here we have shown that the advances in prostate cancer treatments, specifically advances in targeted androgen synthesis and androgen receptor inhibition, microtubule inhibition, cell-signaling inhibition, and finally amino acid deprivation, stimulate the adaptive response of autophagy in prostate cancer models. Furthermore, autophagy can be modulated in these models to improve the therapeutic efficacy of the aforementioned therapeutic classes. To date, a number of human trials investigating autophagy in cancer have been undertaken, with several in later phases demonstrating therapeutic efficacy. [72] Prostate cancer specific clinical trials are ongoing, and hopefully these will further substantiate what has been observed pre-clinically. Moving forward, refinement of autophagy modulation is needed. Currently available autophagy modulators are relatively non-specific and cytotoxicity in non-cancerous tissues is still a concern. [19] This includes uncertain influences of autophagic manipulation upon immunological activity. Moreover, given the context-dependent role autophagy plays, the correct selection of the appropriate patient and the correct directional modification of autophagy are essential. [3] There is still a paucity of autopaphagy specific biomarkers, and the development of a greater set of biomarkers will certainly help better characterize prostate cancers that are more autophagy dependent and better inform treatment paradigms utilizing autophagic modulation.
Figure 1.

Starvation and growth-factor deprivation stimulates cell signaling cascades (the key intermediates identified above) that ultimately result in the initiation of autophagy. The autophagosome matures and is trafficked to lysosomes where it fuses and creates the autolysosome. The autolysosome digests cellular biomass and eliminates toxins, facilitating a host of favorable nutrient and growth conditions.
Box 1. Morphological and Molecular Considerations.
The exact morphological and molecular steps of autophagy have been extensively covered in a number of reviews; a comprehensive review can be found in Choi et. al., “Autophagy in Human Health and Disease.” [1] Regardless, the essential steps of autophagy are as follows:
Cytoplasmic material is engulfed by an isolation membrane called a phagophore, producing a double-membrane structure known as the autophagosome. [1]
The autopagosome is then trafficked to lysosomes where the contents are destroyed by lysosomal enzymes. [1]
These steps – to include prominent molecular intermediates – are highlighted in Figure 1. It is the flux through this system – the active cycling of cytoplasmic material through the lysosomal system – that truly determines autophagic activity. The critical role of autophagic flux and other methodological aspects are well covered in Klionsky et. al, “Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy.” [7]
Key Points.
Autophagy is a dynamic metabolic process that facilitates nutrient utilization and toxin removal.
The degree of autophagic activity is important; too little or too much autophagic activity has very different consequences depending upon the disease state.
Several autophagic modulators are under active investigation; notably, hydroxychloroquine is already being used in numerous clinical trials.
Pre-clinical prostate cancer models significantly up-regulate autophagy in response to ADT, taxane-based chemotherapy, targeted inhibition of kinases, and amino-acid restriction.
Adjuvant autophagy inhibition in pre-clinical prostate cancer models improves cell killing and tumor responsiveness to treatment.
Review Criteria.
A systematic literature review was conducted primarily via electronic database searches of PubMed/Medline. Searches were conducted with the following combinations and iteration of the following terms: autophagy, prostate cancer, castration-resistant prostate cancer, androgen deprivation therapy, abiraterone, enzalutamide, taxanes, docetaxel, kinase inhibitors, arginine deiminase, chloroquine, hydroxychloroquine, metformin, desmethylclomipramine, autophagy inhibitors, and autophagy modulators. All selected papers were full-text and printed in English. Preference was for papers located in core clinical journals and/or reviews. The majority of selected papers were between 2005 and 2014. Reference lists were read and informed on-going reference searching and selection.
Acknowledgments
Disclosure of potential conflicts of interests: Dr. Christopher Evans has a research grant from Medivation/Astellas and is a consultant/advisory board member, clinical trial investigator and shareholder of Medivation.
Grant support: The work is supported by funding from the following sources: the DOD, the SU2C/AACR/PCF Prostate Cancer Dream Team award, and by the National Center for Advancing Translational Sciences at the National Institutes of Health through grant number UL1 TR000002 and the linked TL1 TR000133 award.
Biographies
Jason M. Farrow, B.S.: Jason Farrow holds a Bachelor's Degree in Science and is a 4th year medical student at the University of California, Davis. He is a National Institutes of Health funded T32 scholar and will be receiving a Masters in Advanced Studies. Jason has strong interests in clinical research and hopes to pursue an academic career in urology.
Joy Yang, Ph.D.: Dr. Yang holds a Ph.D. in Microbiology and received postdoctoral training in molecular biology at the University of California, Davis. She has a long history of urological research at the University of California, Davis, and has made signicant contributions as both a co-investigator and co-author on numerous basic science publications. Her research focus is on delineating molecular mechanisms for targeted advanced prostate cancer therapies – utilizing her expertise in in vivo modeling.
Christopher P. Evans, M.D.: Dr. Evans is Professor and Chair for the Department of Urology at the University of California, Davis; he is also a Co-Leader of the Cancer Center Prostate Program. Dr. Evans completed urology residency at UCSF and was a Urological Oncology Fellow at the M.D. Anderson Cancer Center. Dr. Evans has published over 120 peer-reviewed papers encompassing both clinical and urological research fields. His research expertise is on target therapies for castration resistant prostate cancer using kinase inhibitors, androgen receptor sigaling inhibitors, and autophagy modulators.
References
- 1.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(19):1845–6. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 2.Bennett HL, et al. Does androgen-ablation therapy (AAT) associated autophagy have a pro-survival effect in LNCaP human prostate cancer cells? BJU Int. 2013;111(4):672–82. doi: 10.1111/j.1464-410X.2012.11409.x. [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–9. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 5.Degenhardt K, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leone RD, Amaravadi RK. Autophagy: a targetable linchpin of cancer cell metabolism. Trends Endocrinol Metab. 2013;24(4):209–17. doi: 10.1016/j.tem.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen N, Karantza V. Autophagy as a therapeutic target in cancer. Cancer Biol Ther. 2011;11(2):157–68. doi: 10.4161/cbt.11.2.14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chodak GW, Warren KS. Watchful waiting for prostate cancer: a review article. Prostate Cancer Prostatic Dis. 2006;9(1):25–9. doi: 10.1038/sj.pcan.4500857. [DOI] [PubMed] [Google Scholar]
- 10.Patel AR, Klein EA. Risk factors for prostate cancer. Nat Clin Pract Urol. 2009;6(2):87–95. doi: 10.1038/ncpuro1290. [DOI] [PubMed] [Google Scholar]
- 11.Cheong H, et al. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol. 2012;30(7):671–8. doi: 10.1038/nbt.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deretic V. Autophagosome and phagosome. Methods Mol Biol. 2008;445:1–10. doi: 10.1007/978-1-59745-157-4_1. [DOI] [PubMed] [Google Scholar]
- 13.Townsend KN, et al. Autophagy inhibition in cancer therapy: metabolic considerations for antitumor immunity. Immunol Rev. 2012;249(1):176–94. doi: 10.1111/j.1600-065X.2012.01141.x. [DOI] [PubMed] [Google Scholar]
- 14.Deretic V. Autophagy: an emerging immunological paradigm. J Immunol. 2012;189(1):15–20. doi: 10.4049/jimmunol.1102108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, et al. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood. 2012;119(12):2895–905. doi: 10.1182/blood-2011-08-372383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qu X, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7(12):961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–10. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lum JJ, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120(2):237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 21.Yonekawa T, Thorburn A. Autophagy and cell death. Essays Biochem. 2013;55:105–17. doi: 10.1042/bse0550105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marino G, et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115(10):2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehrpour M, et al. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20(7):748–62. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 25.Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Kondo Y, et al. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(9):726–34. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 27.Yang ZJ, et al. Autophagy modulation for cancer therapy. Cancer Biol Ther. 2011;11(2):169–76. doi: 10.4161/cbt.11.2.14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carew JS, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110(1):313–22. doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katayama M, et al. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007;14(3):548–58. doi: 10.1038/sj.cdd.4402030. [DOI] [PubMed] [Google Scholar]
- 30.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15(17):5308–16. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amaravadi RK, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17(4):654–66. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang ZJ, et al. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10(9):1533–41. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sotelo J, Briceno E, Lopez-Gonzalez MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006;144(5):337–43. doi: 10.7326/0003-4819-144-5-200603070-00008. [DOI] [PubMed] [Google Scholar]
- 34.Vakana E, Altman JK, Platanias LC. Targeting AMPK in the treatment of malignancies. J Cell Biochem. 2012;113(2):404–9. doi: 10.1002/jcb.23369. [DOI] [PubMed] [Google Scholar]
- 35.Ben Sahra I, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010;70(6):2465–75. doi: 10.1158/0008-5472.CAN-09-2782. [DOI] [PubMed] [Google Scholar]
- 36.Ben Sahra I, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27(25):3576–86. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 37.Cantrell LA, et al. Metformin is a potent inhibitor of endometrial cancer cell proliferation--implications for a novel treatment strategy. Gynecol Oncol. 2010;116(1):92–8. doi: 10.1016/j.ygyno.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cazzaniga M, et al. Is it time to test metformin in breast cancer clinical trials? Cancer Epidemiol Biomarkers Prev. 2009;18(3):701–5. doi: 10.1158/1055-9965.EPI-08-0871. [DOI] [PubMed] [Google Scholar]
- 39.Colquhoun AJ, et al. Metformin enhances the antiproliferative and apoptotic effect of bicalutamide in prostate cancer. Prostate Cancer Prostatic Dis. 2012;15(4):346–52. doi: 10.1038/pcan.2012.16. [DOI] [PubMed] [Google Scholar]
- 40.Zakikhani M, et al. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66(21):10269–73. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 41.Shank JJ, et al. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol Oncol. 2012;127(2):390–7. doi: 10.1016/j.ygyno.2012.07.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, et al. Effects of metformin on CD133+ colorectal cancer cells in diabetic patients. PLoS One. 2013;8(11):e81264. doi: 10.1371/journal.pone.0081264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Evans JM, et al. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spratt DE, et al. Metformin and prostate cancer: reduced development of castration-resistant disease and prostate cancer mortality. Eur Urol. 2013;63(4):709–16. doi: 10.1016/j.eururo.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rossi M, et al. Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. J Cell Sci. 2009;122(Pt 18):3330–9. doi: 10.1242/jcs.048181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen HG, et al. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene. 2014;0 doi: 10.1038/onc.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kung Hsing-Jien, PhD, C C, MS, Nguyen Hao G, MD, PhD, Yang Joy C, PhD, Evans Christopher P, MD, Bold Richard J, MD, Chuang Frank., MD, PhD . Autophagy and Prostate Cancer Therapeutics. In: Tindall DJ, editor. Prostate Cancer. Springer; New York: 2013. pp. 497–518. Protein Reviews. [Google Scholar]
- 48.Giampietri C, et al. Autophagy modulators sensitize prostate epithelial cancer cell lines to TNF-alpha-dependent apoptosis. Apoptosis. 2012;17(11):1210–22. doi: 10.1007/s10495-012-0752-z. [DOI] [PubMed] [Google Scholar]
- 49.Bennett HL, et al. Androgens modulate autophagy and cell death via regulation of the endoplasmic reticulum chaperone glucose-regulated protein 78/BiP in prostate cancer cells. Cell Death Dis. 2010;1:e72. doi: 10.1038/cddis.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaini RR, Hu CA. Synergistic killing effect of chloroquine and androgen deprivation in LNCaP cells. Biochem Biophys Res Commun. 2012;425(2):150–6. doi: 10.1016/j.bbrc.2012.07.054. [DOI] [PubMed] [Google Scholar]
- 51.Li M, et al. Autophagy protects LNCaP cells under androgen deprivation conditions. Autophagy. 2008;4(1):54–60. doi: 10.4161/auto.5209. [DOI] [PubMed] [Google Scholar]
- 52.Jiang Q, et al. Targeting androgen receptor leads to suppression of prostate cancer via induction of autophagy. J Urol. 2012;188(4):1361–8. doi: 10.1016/j.juro.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 53.Xu Y, et al. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66(15):7783–92. doi: 10.1158/0008-5472.CAN-05-4472. [DOI] [PubMed] [Google Scholar]
- 54.Chhipa RR, Wu Y, Ip C. AMPK-mediated autophagy is a survival mechanism in androgen-dependent prostate cancer cells subjected to androgen deprivation and hypoxia. Cell Signal. 2011;23(9):1466–72. doi: 10.1016/j.cellsig.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lozy F, Karantza V. Autophagy and cancer cell metabolism. Semin Cell Dev Biol. 2012;23(4):395–401. doi: 10.1016/j.semcdb.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ziparo E, et al. Autophagy in prostate cancer and androgen suppression therapy. Int J Mol Sci. 2013;14(6):12090–106. doi: 10.3390/ijms140612090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.CHapter 2 overview of cellular physiology in medical physiology. 2012 [Google Scholar]
- 58.Tannock IF, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 59.Notte A, et al. Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast cancer cells: role of autophagy and JNK activation. Cell Death Dis. 2013;4:e638. doi: 10.1038/cddis.2013.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Long BH, Fairchild CR. Paclitaxel inhibits progression of mitotic cells to G1 phase by interference with spindle formation without affecting other microtubule functions during anaphase and telephase. Cancer Res. 1994;54(16):4355–61. [PubMed] [Google Scholar]
- 61.Eum KH, Lee M. Crosstalk between autophagy and apoptosis in the regulation of paclitaxel-induced cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem. 2011;348(1-2):61–8. doi: 10.1007/s11010-010-0638-8. [DOI] [PubMed] [Google Scholar]
- 62.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4(6):470–80. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 63.Kung HJ. Targeting tyrosine kinases and autophagy in prostate cancer. Horm Cancer. 2011;2(1):38–46. doi: 10.1007/s12672-010-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee YC, et al. Src family kinase/abl inhibitor dasatinib suppresses proliferation and enhances differentiation of osteoblasts. Oncogene. 2010;29(22):3196–207. doi: 10.1038/onc.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu Z, et al. Autophagy Blockade Sensitizes Prostate Cancer Cells towards Src Family Kinase Inhibitors. Genes Cancer. 2010;1(1):40–9. doi: 10.1177/1947601909358324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lamoureux F, et al. Blocked autophagy using lysosomotropic agents sensitizes resistant prostate tumor cells to the novel Akt inhibitor AZD5363. Clin Cancer Res. 2013;19(4):833–44. doi: 10.1158/1078-0432.CCR-12-3114. [DOI] [PubMed] [Google Scholar]
- 67.DaSilva J, et al. The neuroendocrine-derived peptide parathyroid hormone-related protein promotes prostate cancer cell growth by stabilizing the androgen receptor. Cancer Res. 2009;69(18):7402–11. doi: 10.1158/0008-5472.CAN-08-4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee LF, et al. Neuropeptide-induced androgen independence in prostate cancer cells: roles of nonreceptor tyrosine kinases Etk/Bmx, Src, and focal adhesion kinase. Mol Cell Biol. 2001;21(24):8385–97. doi: 10.1128/MCB.21.24.8385-8397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee LF, et al. Interleukin-8 confers androgen-independent growth and migration of LNCaP: differential effects of tyrosine kinases Src and FAK. Oncogene. 2004;23(12):2197–205. doi: 10.1038/sj.onc.1207344. [DOI] [PubMed] [Google Scholar]
- 70.Yang JC, et al. Aberrant activation of androgen receptor in a new neuropeptide-autocrine model of androgen-insensitive prostate cancer. Cancer Res. 2009;69(1):151–60. doi: 10.1158/0008-5472.CAN-08-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim RH, Bold RJ, Kung HJ. ADI, autophagy and apoptosis: metabolic stress as a therapeutic option for prostate cancer. Autophagy. 2009;5(4):567–8. doi: 10.4161/auto.5.4.8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carew JS, Kelly KR, Nawrocki ST. Autophagy as a target for cancer therapy: new developments. Cancer Manag Res. 2012;4:357–65. doi: 10.2147/CMAR.S26133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Savaraj N, et al. Arginine deprivation, autophagy, apoptosis (AAA) for the treatment of melanoma. Curr Mol Med. 2010;10(4):405–12. doi: 10.2174/156652410791316995. [DOI] [PMC free article] [PubMed] [Google Scholar]