

Abstract
Background
DNA copy number variants play an important part in the development of common birth defects such as oral clefts. Individual patients with multiple birth defects (including oral clefts) have been shown to carry small and large chromosomal deletions.
Methods
We investigated the role of polymorphic copy number deletions by comparing transmission rates of deletions from parents to offspring in case-parent trios of European ancestry ascertained through a cleft proband with trios ascertained through a normal offspring. DNA copy numbers in trios were called using the joint hidden Markov model in the freely available PennCNV software (www.openbioinformatics.org/penncnv). All statistical analyses were performed using Bioconductor tools (www.bioconductor.org) in the open source environment R.
Results
We identified a 67 kilo-base (kb) region in the gene MGAM on chromosome 7q34, and a 206 kb region overlapping genes ADAM3A and ADAM5 on chromosome 8p11, where deletions are more frequently transmitted to cleft offspring than control offspring.
Conclusions
These genes or nearby regulatory elements may be involved in the etiology of oral clefts.
Keywords: Oral clefts, DNA copy numbers, inherited deletions, case-parent trios, arrays
Introduction
Oral clefts, including cleft lip (CL), cleft palate (CP) and cleft lip and palate (CLP) are the most common craniofacial malformations in humans, and many different genes have been shown to influence risk (Marazita, 2012). Oral clefts can occur as a feature of many recognized Mendelian malformation syndromes (see Box 1 in Dixon et al., 2011). Distinguishing between “syndromic” and “non-syndromic” cases can be challenging, and some genes known to be causal for syndromic forms also show evidence of association with apparent “non-syndromic” forms (e.g. IRF6), perhaps due to ambiguity in diagnosis. For example, causal variants in IRF6 can reflect point mutations or small deletions, but the DiGeorge critical region on 22q11.21 includes the best documented group of microdeletions associated with cleft palate and other congenital malformations. Approximately 70% of all CL and CLP cases are classified as isolated (i.e. no other congenital anomaly) and non-syndromic, while roughly 50% of CP cases appear isolated and non-syndromic (Jones, 1988; Maarse et al., 2012). Genome-wide associations studies (GWAS) have identified at least a dozen confirmed genetic risk factors for isolated, non-syndromic oral clefts, where polymorphic markers have yielded evidence of association and/or linkage (Ludwig et al., 2012; Beaty et al., 2013). There have been few systematic studies of structural variants associated with oral clefts, however.
Transmitted and de novo chromosomal anomalies in severely affected cleft probands (usually those with multiple congenital anomalies) have identified chromosomal regions potentially harboring genes critical to normal craniofacial development, although it is difficult to generalize from these case reports to the broader group of isolated, non-syndromic cases. Detectable chromosomal abnormalities (including deletions) are not unusual in patients with oral clefts and another malformation, and estimated rates of microscopic and sub-microscopic anomalies range from 11 to 23% (Maarse et al., 2012). The rates of such chromosomal anomalies among infants with isolated oral clefts are much lower, however (about 1-2%, see Maarse et al., 2012).
The chromosomal regions found to be altered (deleted or duplicated) in severely affected oral cleft patients often encompass recognized candidate genes showing evidence of association or linkage in broader samples of isolated, non-syndromic cleft. Ounap et al. (2005) used fluorescence in situ hybridization (FISH) to identify a large duplication on chr2q13-22 in a single CP patient (with multiple anomalies), but additional cleft probands with duplications in this region were also found. Osoegawa et al. (2008) used array comparative hybridization (array-CGH) on 63 syndromic cleft patients and identified one CLP patient with a de novo 2.7 Mb deletion in the DiGeorge critical region of chr22q11.21. As technology has improved, smaller deletions can be identified from chip arrays covering the entire genome. Sahoo et al. (2011) used microarray chips to identify microdeletions in chr20p12.3 in three patients with CP (and other anomalies) that all included the candidate gene BMP2.
Osoegawa et al. (2008) also examined 104 non-syndromic oral cleft patients with array-CGH to identify two individuals with deletions: one CLP proband with a de novo 3.2 Mb deletion on chr6q25.1-25.2 and one CL proband with a 2.2 Mb deletion on 10q26.11-26.13 transmitted from the mother (who also had a CL). Tan et al. (2013) identified a de novo deletion spanning 2.3Mb of chr1q32 in a CLP proband with lip pits (fitting the clinical phenotype for Van der Woude syndrome, although there was no family history of oral clefts or lip pits). This microdeletion included the IRF6 gene, where multiple distinct point mutations and a few small deletions (ranging in size from a few base pairs up to 17kb) account for most inherited Van der Woude syndromes, the most common autosomal dominant Mendelian malformation syndrome including oral clefts (Salahshourifar et al., 2012). The deletion identified by Tan et al. (2013) started in a known copy number variant site and extended into a large intron of the neighboring gene STY14 - but IRF6 remained the gene predicted to be causal in this case.
Using genomic array data from trios of oral cleft probands and their parents (all examined and apparently isolated and non-syndromic) and control trios consisting of children and their parents not ascertained for a specific phenotype, we previously identified a 62 kb non-coding region on chromosome 7p14 where de novo deletions occurred more frequently among oral cleft probands than controls (Younkin et al., 2014). We used these genome-wide data to identify polymorphic copy number deletions across the entire genome. We compared rates of transmission of such copy number polymorphisms (CNPs) between a collection of 467 cleft case-parent trios and a control group of 391 child-parent trios of similar ethnicity. This systematic approach allowed tests for linkage and association between CNPs and risk of isolated, non-syndromic oral clefts.
Materials and Methods
Case-parent trios were ascertained through probands with an isolated, non-syndromic oral cleft as part of an international collaboration with subjects recruited from 13 different locations in the United States, Europe, Southeast and East Asia (Beaty et al., 2010). This project was included in the GENEVA consortium (Cornelis et al., 2010). For this study, oral clefts were categorized as either cleft lip (CL), cleft palate (CP) or cleft lip and palate (CLP). DNA samples were collected via whole blood, buccal brush/swab, saliva, mouthwash and dried blood spot samples, in varying proportions across recruitment sites. Parental interviews specifically asking about oral clefts (or other major birth defects) spanning at least three generations provided family history data on ∼75% of all probands of European ancestry from the consortium data used for the GWAS, and of these approximately two-thirds reported a negative family history. We assembled a control group of unaffected children and their parents by drawing from a family-based study of dental caries (Polk et al., 2008). Like the cleft group, DNA samples for the control trios were derived from whole blood, buccal brush/swab, saliva, and mouthwash. All samples were hybridized to the Illumina 610 quad array and typed at the Center for Inherited Disease Research (CIDR) at Johns Hopkins University (http://www.cidr.jhmi.edu). Because the control group included only subjects of European American ancestry, we only used cleft case-parent trios of European ancestry to minimize ethnic heterogeneity. These data are available from the Database of Genotypes and Phenotypes (dbGaP; www.ncbi.nlm.nih.gov/gap).
Data processing and quality control closely followed Younkin et al. (2014). DNA copy number deletions were inferred via PennCNV using the log R ratios (LRRs, standardized estimates of the probe intensities which quantify the total number of copies at a locus of interest) and B allele frequencies (BAFs, standardized estimates for the proportion of the B allele's contribution to the total probe intensity, assessing the genotype at each locus of interest) provided as part of genotyping array data. To avoid excessive false positive identifications due to poor data quality, we excluded all trios which contained DNA generated by whole genome amplification or any sample with a LRR median absolute deviation (MAD) above 0.30, a commonly recommended threshold (Wang et al., 2008; Scharpf et al., 2012; Younkin et al., 2014), along with trios with members flagged by CIDR's internal quality control pipeline, yielding 467 cleft trios composed of 1,384 subjects, and 391 control trios composed of 902 subjects (nuclear families with multiple offspring yielded multiple trios). All analyses except the CNV identification via PennCNV were carried out using the open source statistical environment R (http://cran.r-project.org) and Bioconductor tools (http://www.bioconductor.org; Gentleman et al., 2004).
The PennCNV algorithm employs a hidden Markov model (HMM) to jointly model the (hidden) copy number states in the trio members. Using the observed LRRs and BAFs in these samples, the most likely set of copy number states in the father, mother and offspring are jointly inferred via maximum likelihood methods. The output is returned as a simple numerical code: a normal DNA copy number (two alleles) in a sample is designated as a 3, a hemizygous deletion (one allele copy) is indicated as a 2, and a homozygous deletion (zero allele copies) is indicated as a 1. For example, trio-states of “322” and “232” (father/mother/offspring) indicate that a deletion was transmitted from one parent to the offspring, and “323” and “233” indicate that such a deletion was not transmitted. Generally, if nfmo represents the number of PennCNV trio copy number states, we calculated the number of observed deletion transmissions as
and the number of non-transmitted deletions as
This is equivalent to the counting methods used in the conventional allelic transmission disequilibrium test (TDT). In particular, trio-state “221” represents two transmitted deletions (one from each parent), “223” represents two non-transmitted deletions (from a heterozygous-by-heterozygous mating), and “222” represents one transmitted and one non-transmitted deletion from this same mating type. Also, “212” and “122” each represent one non-transmitted deletion (i.e. the proband has a hemizygous deletion, inherited from the parent with a homozygous deletion, but the deletion from the hemizygous parent was not inherited), while “211” and “121” each represent one transmitted deletion from the hemizygous parent.
Deletions were inferred separately for each trio, and each sample deletion was required to span at least 10 contiguous array probes. We used the GenomicRanges Bioconductor package (Lawrence et al., 2013) to delineate CNV components, defined as sets of probes where no change of copy number state occurred among any of the cleft or control trios, thereby defining homogeneous sets of CNV states. Contiguous sets of components were defined as regions. To count transmissions per region, we defined a deleted region as transmitted if any CNV component within the region was transmitted. Regions within 1 Mb of centromeres or telomeres, the HLA super locus (hg18 chr6:29,940,311-32,788,048), and regions recognized as being difficult to interrogate due to highly repetitive sequence content (Derrien et al., 2012) were removed (281 Mb total).
We defined polymorphic CNV regions as those with at least 25 potential transmissions (T + U ≥ 25) in both the cleft and control groups. Among these 467 case-parent and 391 control trios, we used a one-sided Fisher exact test to test the null hypothesis of no difference between groups to the alternative hypothesis of a higher transmission frequency among the cleft trios. Statistical significance for these largely unlinked regions was assessed using a Bonferroni correction. Additionally, we carried out association tests for the individual CNV components via one-sided Fisher exact tests. Since the components within regions (84 total) are contiguous and thus generate highly dependent p-values, we carried out permutation tests to correct for multiple comparisons and assess their statistical significance separately. We shuffled case and control status across all probands and recorded the minimum p-value across components. Using 100,000 permutations, we created an empirical distribution for the minimum p-value under the null hypothesis (in presence of dependency between components). Observed p-values were compared to this distribution to assess genome-wide significance.
Results
We compared the parent to offspring transmission rates of deletions identified by PennCNV (Wang et al., 2008) in parent-offspring trios with offspring ascertained through an isolated, non-syndromic oral cleft (case trios), and trios not ascertained through an affected proband (control trios) to identify polymorphic deletions associated with increased risk of oral clefts. All subjects used in this analysis are of European ancestry. Case trios were selected from an international consortium conducting a genome- wide association study (GWAS) of non-syndromic oral clefts (Beaty et al., 2010, 2011), while the control trios came from a US study where children and their parents were recruited without regard to phenotype from a GWAS searching for genes influencing dental caries (Polk et al., 2008).
Parental hemizygous deletions were inferred in 467 CL/P and 391 control trios, yielding 39 regions with CNPs (at least 25 parental deletions among both case and control trios). Of these regions, the majority (26) were within 1 Mb of genomic regions known to be difficult to interrogate (see Methods), likely constituting false positive identifications. Among the remaining 13 regions, we identified a 67 kb region of the gene MGAM on chromosome 7q34 supported by 13 array probes (Table 1) where the allele with the deleted DNA segment is more frequently transmitted from parent to offspring of cleft trios (πˆCleft = 0.64; 95% CI: 0.54 – 0.73) than in the control trios (πˆControl = 0.47; 95% CI: 0.38 – 0.55). Formally testing for a difference in transmission rates using a one-sided Fisher's exact test yielded a nominal p = 0.0044 (Table 2). Among the 69 affected probands in the case trios who inherited a DNA copy number deletion in this region from a parent, 17 had a cleft lip, 21 had a cleft palate, and 31 had a cleft lip and palate. A second region, spanning 206 kb encompassing the genes ADAM3A and ADAM5 on chromosome 8p11 (supported by 33 array probes) also showed an over-transmission of the deleted DNA segment (πˆCleft = 0.58 in the case trios versus πˆControl = 0.42 in the control trios, p = 0.0094; Tables 1 and 2). Among the 147 affected probands in the case trios who inherited a DNA copy number deletion in this region from a parent, 51 / 38 / 58 had CL / CP / CLP respectively.
Table 1.
Locations (hg18) and sizes of the two genomic regions on chromosomes 7q34 (upper row) and 8p11 (lower row) that showed the most significant over-transmission of the allele with the missing DNA segment from a hemizygous parent to an offspring, comparing trios with affected probands to control trios. These regions overlapped with the MGAM gene (chromosome 7), and ADAM3A / ADAM5 (chromosome 8), respectively.
| chr | start | end | size | genes |
|---|---|---|---|---|
| 7 | 141,380,317 | 141,447,476 | 67.2 kb | MGAM |
| 8 | 39,341,981 | 39,548,228 | 206.2 kb | ADAM3A, ADAM5 |
Table 2.
Transmission rates (π) comparing trios with affected probands (Cleft) to control trios (Control) in the two genomic regions on chromosomes 7q34 (upper row) and 8p11 (lower row). Transmission rates are based on counts of transmitted (T) and untransmitted (U) alleles, and compared using a one-sided Fisher's exact test. The statistical significance is reported as the nominal p-value (p).
| TCleft | UCleft | TControl | UControl | πˆCleft | πˆControl | p |
|---|---|---|---|---|---|---|
| 69 | 39 | 68 | 78 | 0.64 | 0.47 | 0.0044 |
| 147 | 108 | 32 | 45 | 0.58 | 0.42 | 0.0094 |
None of these loci remained significant after a strict Bonferroni correction for multiple comparisons (Figure 1). Nonetheless, assuming the 13 regions represent independent test statistics (supported by the fact that these regions are either on different chromosomes, or quite far apart), the observed p-values for these region are smaller than one would expect to see by chance alone (Figure 2A). It is noteworthy that the deletions at the ADAM3A / ADAM5 locus were not only transmitted more frequently in the cleft group, they were also more abundant among parents of the cleft probands (255 observable transmissions in 467 cleft trios; 55%) compared to control trios (77 observable transmissions in 391 control trios; 20%), possibly indicating sub-clinical oral clefting in some of the parents of cleft children (Table 1).
Figure 1.
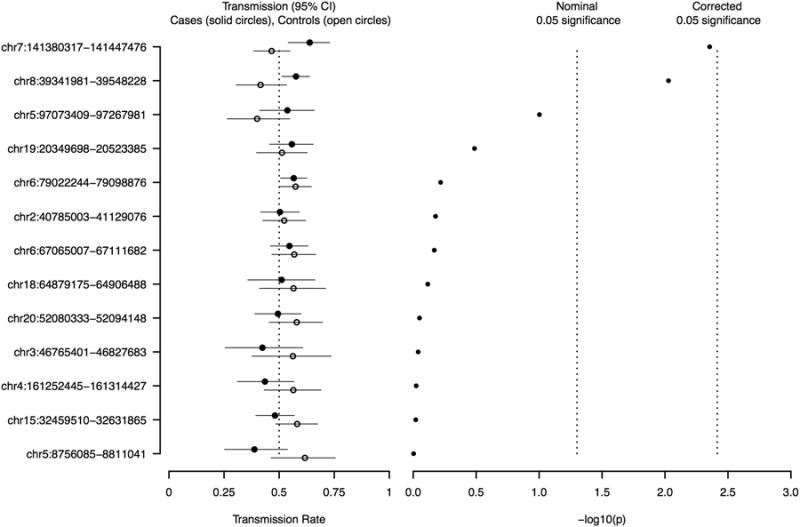
Left panel: Allele transmission rate estimates (cleft trios - solid circle; control trios - open circle) with unadjusted 95% confidence intervals, for the 13 copy number polymorphism regions identified. Expected transmission under independent assortment is 0.5. Right panel: The -log10 p-values derived from one-sided Fisher's exact tests for equality of transmission rates in cleft and control trios, versus the alternative of increased transmission rates in the cleft group. The vertical lines indicate the nominal significance (-log10(0.05) = 1.30), and the Bonferroni corrected p-values (-log10(0.05/13) = 2.41).
Figure 2.
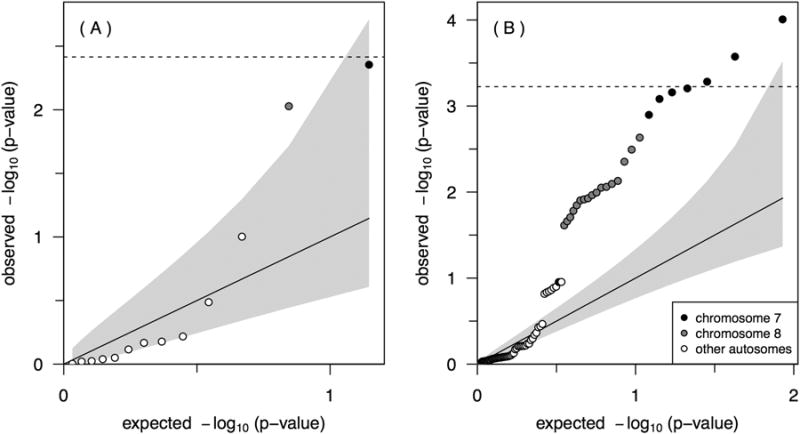
Quantile-quantile plots of expected versus observed −log10 p-values for the 13 CNP regions (A) and the corresponding 84 CNP components (B). The grey regions indicate confidence regions for the respective order statistics under an independence assumption, though the CNP components are highly correlated due to their proximity within regions.
These 13 regions tested above are comprised of 84 individual CNV components (see Methods) with multiple neighboring components per region, and thus, components cannot yield independent test statistics. Using permutation tests based on the empirical cumulative distribution function of the minimum p-value taking this correlation structure into account, significant components in the MGAM region on 7q34 were found (195 out of 100,000 permutation p-values were lower than the observed, p = 0.002), as well as in the chromosome 8p11 region overlapping ADAM3A and ADAM5 genes (4,023 out of 100,000 permutations, p = 0.040). The observed p-values for the CNP components in these two regions are also smaller than one would expect to see by chance alone (Figure 2B). For both regions the rates of transmitted and non-transmitted deletions were essentially the same in the control trios, but differed substantially from the case trios (Figure 3).
Figure 3.
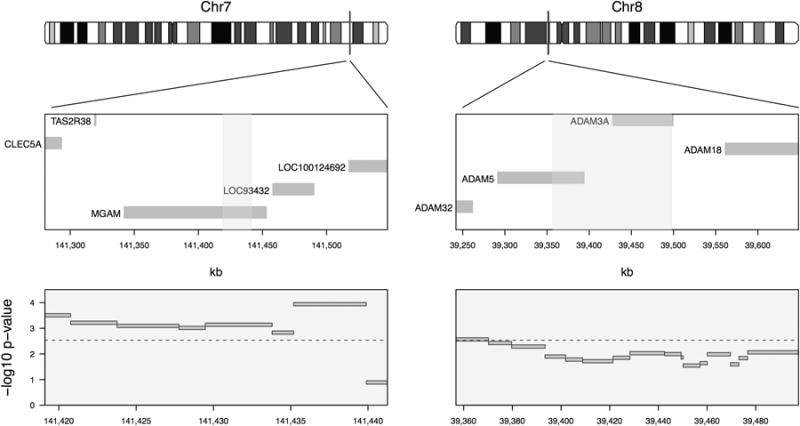
Location of DNA copy number deletions and test results for equal transmission rates of copy number components between case and control trios in the MGAM region on chromosome 7q34 (left) and the ADAM3A / ADAM5 region on chromosome 8p11 (right). The genomic locations of the components are indicated by vertical bars on the ideograms (top panels), and overlapped with nearby genes (middle panels). Test results are shown as nominal −log10(p) from Fisher's exact test for the respective CNV components, with the genome-wide significance level indicated by a dashed line (bottom panels). There is clear evidence of over-transmission of a deletion CNP in the MGAM region. The ADAM3A / ADAM5 region also contains a possible region of over-transmission.
Discussion
In this manuscript, we compared the parent to offspring transmission rates of deletions identified in European ancestry trios with offspring ascertained through an isolated, non-syndromic oral cleft, and trios not ascertained through an affected proband, to identify polymorphic deletions associated with increased risk of oral clefts. We identified a 67 kb region in gene MGAM on chromosome 7q34 and a 206 kb region spanning genes ADAM3A and ADAM5 on chromosome 8p11, which showed significant over-transmission of the deleted segments among the oral cleft trios compared to control trios. MGAM (maltase-glucoamylase) codes for a brush border membrane enzyme that plays a role in the final steps of digestion of starch to glucose in the small intestine. There is no obvious causal relationship between MGAM with oral clefts, although some nearby genes are better candidate genes (e.g. HOXA2 and EEC1). The association suggested in this analysis of transmitted deletions could represent 1) new biological effects for MGAM itself, 2) effects of a long-range enhancer influencing EEC1 or HOXA2, 3) the existence of long-range linkage disequilibrium between this region and an unobserved causal element, or 4) a spurious association. It is always possible that distant regulatory elements can control expression of a remote causal gene, and recently Uslu et al. (2014) showed a specific region, termed the medionasal regulatory domain, on the mouse homolog of chromosome 8q24 increased the risk of CLP in a transgenic mouse model. Therefore, deletions could easily contain a specific causal or regulatory element critical to normal craniofacial development. Similarly, ADAM3A and ADAM5 (both classified as pseudogenes) are about 1 Mb from FGFR1, a recognized candidate gene for oral clefts (Nie et al., 2006; Dode et al., 2007; Riley et al., 2007; Riley and Murray, 2007; Menezes et al., 2008; Stanier and Pauws, 2012; Wang et al., 2013). It is currently not clear what features in the chromosomal regions harboring MGAM and ADAM3A / ADAM5 may contribute to the observed phenotypes, and it is likely that many regulatory elements influence risk of oral clefts.
As described in the Methods section and also in Younkin et al. (2014), trios with at least one sample of poor data quality were excluded. Nonetheless, probe intensity signals used to identify regions of copy number changes are noisy, and we observed somewhat larger variability among control trios even after strict quality control (see Figure S4 in the supplementary material of Younkin et al., 2014). These artifacts typically result in slightly larger rates of CNV calls among controls (Scharpf et al., 2012; Younkin et al., 2014), but our one-sided statistical procedure for inferring transmission of alleles guards against spurious associations (and thus type I error inflation) since only a larger transmission of deletions was assumed to increase risk. Since we only assessed transmissions from parents with hemizygous deletions, in a principle transmission disequilibrium test (TDT) for case trios only could also be used. However, since there might be some inherent calling bias between “322” and “323” PennCNV states, we simply compared transmission rates in oral cleft and control trios. We note that the 95% confidence intervals for all estimated transmission rates among control trios covered the expected value of 0.5 under Mendel's law of independent assortment (Figure 1, open circles).
The findings reported here may help to narrow causal regions and identify how structural variants control risk to oral clefts. Genomic analysis based on arrays or sequencing will become routinely used in birth defects, as costs drop and our confidence in interpreting the importance of variants improves. As we translate genomic research into clinical practice, the yield of genetic testing will improve for patients with oral clefts. Better awareness of potential involvement of other body systems (e.g. neurodevelopmental outcomes), and more accurate recurrence risks are among the benefits of accurate diagnosis provided by genomic testing in cleft patients.
Acknowledgments
We thank the families who participated in the studies and gratefully acknowledge the invaluable assistance of clinical, field, and laboratory staff who contributed to this study, in particular the Center for Oral Health Research in Appalachia. The International Cleft Consortium involved many recruitment sites directed by separate investigators: Jeffrey C. Murray (University of Iowa), Rolf Terje Lie (University of Bergen), Allen Wilcox (NIEHS), Kare Christensen (University of Southern Denmark), Yah-Huei Wu-Chou (Chang Gang Memorial Hospital), Vincent Yeow (KK Women's & Children's Hosptial), Xiaoqian Ye (Wuhan University), Bing Shi (Sichaun University), and Samuel Chong (National University of Singapore).
We gratefully acknowledge the financial support provided by the National Institute of Health grants R03 DE021437 (SGY, IR), R01 DE016148 (MLM), R00 HG005015 (RBS) and Deutsche Forschungsgemeinschaft grant SCHW 1508/3-1 (HS). The consortium for GWAS genotyping and analysis was supported by the National Institute for Dental and Craniofacial Research through U01 DE018993 and U01 DE018903 (THB, MLM). Part of the original recruitment of Norwegian case-parent triads was supported by the Intramural Research Program of the National Institute of Health, National Institute of Environmental Health Sciences.
Funding: NIH R03 DE021437, R01 DE016148, R00 HG005015, Deutsche Forschungsgemeinschaft.
References
- Beaty TH, Murray JC, Marazita ML, Munger RG, Ruczinski I, Hetmanski JB, Liang KY, Wu T, Murray T, Fallin MD, Redett RA, Raymond G, Schwender H, Jin SC, Cooper ME, Dunnwald M, Mansilla MA, Leslie E, Bullard S, Lidral AC, Moreno LM, Menezes R, Vieira AR, Petrin A, Wilcox AJ, Lie RT, Jabs EW, Wu-Chou YH, Chen PK, Wang H, Ye X, Huang S, Yeow V, Chong SS, Jee SH, Shi B, Christensen K, Melbye M, Doheny KF, Pugh EW, Ling H, Castilla EE, Czeizel AE, Ma L, Field LL, Brody L, Pangilinan F, Mills JL, Molloy AM, Kirke PN, Scott JM, Scott JM, Arcos-Burgos M, Scott AF. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet. 2010;42(6):525–529. doi: 10.1038/ng.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty TH, Ruczinski I, Murray JC, Marazita ML, Munger RG, Hetmanski JB, Murray T, Redett RJ, Fallin MD, Liang KY, Wu T, Patel PJ, Jin SC, Zhang TX, Schwender H, Wu-Chou YH, Chen PK, Chong SS, Cheah F, Yeow V, Ye X, Wang H, Huang S, Jabs EW, Shi B, Wilcox AJ, Lie RT, Jee SH, Christensen K, Doheny KF, Pugh EW, Ling H, Scott AF. Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet Epidemiol. 2011;35(6):469–478. doi: 10.1002/gepi.20595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty TH, Taub MA, Scott AF, Murray JC, Marazita ML, Schwender H, Parker MM, Hetmanski JB, Balakrishnan P, Mansilla MA, Mangold E, Ludwig KU, Noethen MM, Rubini M, Elcioglu N, Ruczinski I. Confirming genes influencing risk to cleft lip with/without cleft palate in a case-parent trio study. Hum Genet. 2013;132(7):771–781. doi: 10.1007/s00439-013-1283-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis MC, Agrawal A, Cole JW, Hansel NN, Barnes KC, Beaty TH, Bennett SN, Bierut LJ, Boerwinkle E, Doheny KF, Feenstra B, Feingold E, Fornage M, Haiman CA, Harris EL, Hayes MG, Heit JA, Hu FB, Kang JH, Laurie CC, Ling H, Manolio TA, Marazita ML, Mathias RA, Mirel DB, Paschall J, Pasquale LR, Pugh EW, Rice JP, Udren J, van Dam RM, Wang X, Wiggs JL, Williams K, Yu K Consortium GENEVA. The Gene, Environment Association Studies consortium (GENEVA): maximizing the knowledge obtained from GWAS by collaboration across studies of multiple conditions. Genet Epidemiol. 2010;34(4):364–372. doi: 10.1002/gepi.20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Estell J, Marco Sola S, Knowles DG, Raineri E, Guig R, Ribeca P. Fast computation and applications of genome mappability. PLoS One. 2012;7(1):e30377. doi: 10.1371/journal.pone.0030377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12(3):167–178. doi: 10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dode C, Fouveaut C, Mortier G, Janssens S, Bertherat J, Mahoudeau J, Kottler ML, Chabrolle C, Gancel A, Franois I, Devriendt K, Wolczynski S, Pugeat M, Pineiro-Garcia A, Murat A, Bouchard P, Young J, Delpech M, Hardelin JP. Novel FGFR1 sequence variants in Kallmann syndrome, and genetic evidence that the FGFR1c isoform is required in olfactory bulb and palate morphogenesis. Hum Mutat. 2007;28(1):97–98. doi: 10.1002/humu.9470. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MC. Etiology of facial clefts: prospective evaluation of 428 patients. Cleft Palate J. 1988;25(1):16–20. [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9(8):e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig KU, Mangold E, Herms S, Nowak S, Reutter H, Paul A, Becker J, Herberz R, AlChawa T, Nasser E, Boehmer AC, Mattheisen M, Alblas MA, Barth S, Kluck N, Lauster C, Braumann B, Reich RH, Hemprich A, Poetzsch S, Blaumeiser B, Daratsianos N, Kreusch T, Murray JC, Marazita ML, Ruczinski I, Scott AF, Beaty TH, Kramer FJ, Wienker TF, Steegers-Theunissen RP, Rubini M, Mossey PA, Hoffmann P, Lange C, Cichon S, Propping P, Knapp M, Noethen MM. Genome- wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat Genet. 2012;44(9):968–971. doi: 10.1038/ng.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarse W, Rozendaal AM, Pajkrt E, Vermeij-Keers C, van der Molen ABM, van den Boogaard MJH. A systematic review of associated structural and chromosomal defects in oral clefts: when is prenatal genetic analysis indicated? J Med Genet. 2012;49(8):490–498. doi: 10.1136/jmedgenet-2012-101013. [DOI] [PubMed] [Google Scholar]
- Marazita ML. The evolution of human genetic studies of cleft lip and cleft palate. Annu Rev Genomics Hum Genet. 2012;13:263–283. doi: 10.1146/annurev-genom-090711-163729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes R, Letra A, Ruff J, Granjeiro JM, Vieira AR. Studies of genes in the FGF signaling pathway and oral clefts with or without dental anomalies. Am J Med Genet A. 2008;146A(12):1614–1617. doi: 10.1002/ajmg.a.32341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X, Luukko K, Kettunen P. FGF signalling in craniofacial development and developmental disorders. Oral Dis. 2006;12(2):102–111. doi: 10.1111/j.1601-0825.2005.01176.x. [DOI] [PubMed] [Google Scholar]
- Osoegawa K, Vessere GM, Utami KH, Mansilla MA, Johnson MK, Riley BM, L'Heureux J, Pfundt R, Staaf J, van der Vliet WA, Lidral AC, Schoenmakers EFPM, Borg A, Schutte BC, Lammer EJ, Murray JC, de Jong PJ. Identification of novel candidate genes associated with cleft lip and palate using array comparative genomic hybridisation. J Med Genet. 2008;45(2):81–86. doi: 10.1136/jmg.2007.052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ounap K, Ilus T, Laidre P, Uibo O, Tammur P, Bartsch O. A new case of 2q duplication supports either a locus for orofacial clefting between markers D2S1897 and D2S2023 or a locus for cleft palate only on chromosome 2q13-q21. Am J Med Genet A. 2005;137A(3):323–327. doi: 10.1002/ajmg.a.30890. [DOI] [PubMed] [Google Scholar]
- Polk DE, Weyant RJ, Crout RJ, McNeil DW, Tarter RE, Thomas JG, Marazita ML. Study protocol of the Center for Oral Health Research in Appalachia (COHRA) etiology study. BMC Oral Health. 2008;8:18. doi: 10.1186/1472-6831-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dod C, Mohammadi M, Marazita ML, Murray JC. Impaired FGF signaling contributes to cleft lip and palate. Proc Natl Acad Sci U S A. 2007;104(11):4512–4517. doi: 10.1073/pnas.0607956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BM, Murray JC. Sequence evaluation of FGF and FGFR gene conserved non-coding elements in non-syndromic cleft lip and palate cases. Am J Med Genet A. 2007;143A(24):3228–3234. doi: 10.1002/ajmg.a.31965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo T, Theisen A, Sanchez-Lara PA, Marble M, Schweitzer DN, Torchia BS, Lamb AN, Bejjani BA, Shaffer LG, Lacassie Y. Microdeletion 20p12.3 involving BMP2 contributes to syndromic forms of cleft palate. Am J Med Genet A. 2011;155A(7):1646–1653. doi: 10.1002/ajmg.a.34063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahshourifar I, Sulaiman WAW, Halim AS, Zilfalil BA. Mutation screening of IRF6 among families with non-syndromic oral clefts and identification of two novel variants: review of the literature. Eur J Med Genet. 2012;55(6-7):389–393. doi: 10.1016/j.ejmg.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Scharpf RB, Beaty TH, Schwender H, Younkin SG, Scott AF, Ruczinski I. Fast detection of de novo copy number variants from SNP arrays for case-parent trios. BMC Bioinformatics. 2012;13(1):330. doi: 10.1186/1471-2105-13-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanier P, Pauws E. Development of the lip and palate: FGF signalling. Front Oral Biol. 2012;16:71–80. doi: 10.1159/000337618. [DOI] [PubMed] [Google Scholar]
- Tan EC, Lim EC, Lee ST. De novo 2.3 Mb microdeletion of 1q32.2 involving the Van der Woude syndrome locus. Mol Cytogenet. 2013;6(1):31. doi: 10.1186/1755-8166-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uslu VV, Petretich M, Ruf S, Langenfeld K, Fonseca NA, Marioni JC, Spitz F. Long-range enhancers regulating Myc expression are required for normal facial morphogenesis. Nat Genet. 2014;46(7):753–758. doi: 10.1038/ng.2971. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhang T, Wu T, Hetmanski JB, Ruczinski I, Schwender H, Liang KY, Murray T, Fallin MD, Redett RJ, Raymond GV, Jin SC, Chou YHW, Chen PKT, Yeow V, Chong SS, Cheah FSH, Jee SH, Jabs EW, Scott AF, Beaty TH. The FGF and FGFR Gene Family and Risk of Cleft Lip With or Without Cleft Palate. Cleft Palate Craniofac J. 2013;50(1):96–103. doi: 10.1597/11-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Chen Z, Tadesse MG, Glessner J, Grant SFA, Hakonarson H, Bucan M, Li M. Modeling genetic inheritance of copy number variations. Nucleic Acids Res. 2008;36(21):e138. doi: 10.1093/nar/gkn641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younkin SG, Scharpf RB, Schwender H, Parker MM, Scott AF, Marazita ML, Beaty TH, Ruczinski I. A genome-wide study of de novo deletions identifies a candidate locus for non-syndromic isolated cleft lip/palate risk. BMC Genet. 2014;15:24. doi: 10.1186/1471-2156-15-24. [DOI] [PMC free article] [PubMed] [Google Scholar]