

Abstract
Niemann-Pick type C1 (NPC) disease is a lysosomal storage disease caused by mutations in the NPC1 gene, leading to an increase in unesterified cholesterol and several sphingolipids, and resulting in hepatic disease and progressive neurological disease. Whereas subcutaneous administration of the pharmaceutical excipient 2-hydroxypropyl-beta-cyclodextrin (HPβCD) ameliorated hepatic disease, doses sufficient to reduce neurological disease resulted in pulmonary toxicity. In contrast, direct administration of HPβCD into the cisterna magna of presymptomatic cats with NPC disease prevented the onset of cerebellar dysfunction for greater than a year and resulted in a reduction in Purkinje cell loss and near normal concentrations of cholesterol and sphingolipids. Moreover, administration of intracisternal HPβCD to NPC cats with ongoing cerebellar dysfunction slowed disease progression, increased survival time, and decreased the accumulation of brain gangliosides. An increase in hearing threshold was identified as a potential adverse effect. Together, these studies in the feline animal model have provided critical data on efficacy and safety of drug administration directly into the CNS that will be important for advancing HPβCD into clinical trials.
INTRODUCTION
Niemann-Pick type C (NPC) disease is a severe inherited disorder characterized by progressive cerebellar ataxia, dementia and early death due to neurological disease (1–3). More than 350 disease-causing mutations have been identified in the NPC1 gene and over 25 in the NPC2 gene. NPC1 and NPC2 proteins normally function in concert to facilitate egress of unesterified cholesterol and sphingolipids from the late endosomal/lysosomal compartment (2, 4, 5). Dysfunction of either protein results in lysosomal storage of unesterified cholesterol and multiple sphingolipids (6–10), along with impaired export of lipoprotein-derived cholesterol (11–15). Despite the identification of causative mutations and a partial understanding of the function of the NPC1 and NPC2 proteins, the disease pathogenesis is not well understood.
The juvenile form of NPC disease, which is the most common, presents with progressive learning disabilities and ataxia beginning at 6–15 years of age that is often preceded by hepatosplenomegaly. Vertical supranuclear gaze palsy, cataplexy, seizures, dysarthria, and dysphagia are also seen, with death commonly occurring in the first or second decade (2, 16). Neuropathological abnormalities include widespread neuronal cytoplasmic vacuolization, neuronal loss most severely affecting Purkinje cells, neuroaxonal dystrophy, gliosis, and inflammation (3, 7, 9, 17, 18). Lysosomal storage of unesterified cholesterol in neurons can be demonstrated by histochemical methods (8), whereas sphingolipid accumulation, particularly of gangliosides GM2 and GM3, can be demonstrated by both immunocytochemistry and biochemistry. Miglustat, a small imino sugar that partially inhibits glucosylceramide synthase and the synthesis of all glucosylceramide-based glycosphingolipids, delays the onset of clinical signs in animal models of NPC disease (19, 20). Whereas miglustat has been approved in Europe for the treatment of NPC disease since 2009, and subsequently in over 40 countries, its use for the treatment of NPC disease remains off-label in the USA (21–23). There are currently no FDA-approved therapies for NPC disease.
The cholesterol-lowering agents cholestyramine, lovastatin, and nicotinic acid, and a low cholesterol diet are ineffective in altering the neurological course of NPC disease (24, 25). However, in 2001, Camargo et al. evaluated the therapeutic effect of 2-hydroxypropyl-beta-cyclodextrin (HPβCD) in a mouse model of NPC disease (26). Structurally, HPβCD contains a hydrophilic exterior and a hydrophobic interior, allowing it to increase the solubility of poorly water-soluble compounds such as cholesterol. Notably, in vitro studies showed that millimolar concentrations of HPβCD efficiently and rapidly removed cholesterol from cultured cells (27–29). In vivo, intraperitoneal or subcutaneous administration of HPβCD to Npc1−/− mice decreased unesterified cholesterol storage in liver, and delayed onset of neurological disease, increased lifespan, increased Purkinje cell survival, and reduced cerebrocortical cholesterol and ganglioside accumulation (26, 30, 31).
Given that HPβCD does not readily cross the blood brain barrier (32), its apparent efficacy in the treatment of the neurological aspects of NPC disease is unexpected. To determine if direct intrathecal injection would be even more efficacious, we turned to a feline model of NPC disease. Feline NPC disease results from a single missense mutation in the NPC1 gene (p.C955S) that is evolutionarily conserved and found in a cysteine-rich region commonly mutated in patients (33). Disease progression in this naturally-occurring model recapitulates both the neuropathological and biochemical abnormalities observed in human patients, with the closest parallels to the juvenile form of NPC disease (9, 20, 34). In contrast to the murine model, the feline model is large enough to permit repeated administration of HPβCD either subcutaneously or intrathecally and repeated sampling of blood and cerebrospinal fluid (CSF) to evaluate mechanistic, pharmacologic, and toxicity issues. This model also allows for validation of biochemical markers of disease severity and therapeutic effects that are specific to CNS disease (20, 35–40).
In the present study, we show that administration of HPβCD into the subarachnoid space at the cisterna magna of affected cats completely resolved the clinical neurological signs of disease and Purkinje cell loss up to at least 24 weeks of age (the median age when untreated NPC cats die), providing critical data on efficacy and safety of drug administration directly into the CNS (41).
RESULTS
Untreated NPC cats developed CNS and hepatic disease and survived for a mean of 21 weeks
Thirty-nine untreated NPC cats were evaluated (Table 1). Untreated NPC cats showed increased serum hepatic alanine aminotransferase activity (ALT) and serum total cholesterol, decreased serum albumin and body weight, and progressive cerebellar ataxia and intention tremor compared to normal control cats (Table 2, Figures 1A–C, Figure S1, Video S1). Untreated NPC cats were euthanized when they were non-ambulatory and no longer able to remain in sternal recumbency without support, which occurred at a mean age of 20.7 ± 5 weeks (range 9–29 weeks). Liver histology and biochemistry showed severe and extensive vacuolization of hepatocyte and Kupffer cell cytoplasm accompanied by pronounced increases in unesterified cholesterol, sphingomyelin, bis(monoacylglycero)phosphate (BMP), glucosylceramide, lactosylceramide, globotriaosylceramide, free sphingosine (Figure 2), and GM3 ganglioside (Figure S2A). Examination of the brain revealed diffuse neuronal cytoplasmic vacuolization with intracellular cholesterol storage identified by filipin staining, abundant storage of gangliosides (GM2 and GM3) in several brain regions examined (neocortex, caudate nucleus, and cerebellum), and severe Purkinje cell loss (Figure 3, Figure S3). Biochemical analysis of cerebral gray matter in untreated NPC cats with end stage disease (median 25 weeks, range 21–29 weeks, n=9), revealed that GM2 and GM3 constituted 17.4 ± 0.6 % and 20.4 ± 1.5 % of the total gangliosides, respectively, compared to a normal proportion of 2.5 ± 0.6% for GM3 and 2.0 ± 0.5 % for GM2 (mean± SD) (Figure 4A). Other biochemical lipid abnormalities were a marked accumulation of lactosylceramide (Figure 4B) and glucosylceramide (Figure S2B), and a 3-fold increase of free sphingosine (Figure 4C). Studies in younger NPC cats (Figure 4D) showed that the increase in GM2 and GM3 gangliosides preceded the onset of neurological dysfunction. At 4 weeks of age, GM2 already constituted 9.5% of the total gangliosides, and increased to 14% at 11 weeks. The increase of GM3 started later (5.3% at 4 weeks) but reached a higher concentration with time. A similar developmental pattern has been described in NPC mouse models (42). Finally, no clinical evidence of pulmonary disease was evident in untreated NPC cats although lung histology showed foamy macrophages within alveolar spaces and septa (Figure 5A). For comparison, histological, biochemical and clinical data obtained in normal control cats are reported in Table 2 and Figures 2–5.
Table 1.
Route of administration and dose of HPβCD administered to 11 groups of cats.
| Genotype | Route of Administration | Dose | Dosing Interval | Animal Numbers | Survival time mean and SD, or longest (wks) | |
|---|---|---|---|---|---|---|
| Males | Females | |||||
|
| ||||||
| No mutant allele (normal control) | untreated | untreated | untreated | 26 | 24 | >76† |
| NPC1 | untreated | untreated | untreated | 22 | 17 | 20.7 ± 5.0 |
| NPC1 | SC | 1000 mg/kg & 25 mg/kg allopregnanolone | 7 days | 5 | 1 | 21.8 ± 6.5 |
| NPC1 | SC | 4000 mg/kg | 7 days | 0 | 2 | 31, 36‡ |
| NPC1 | SC | 8000 mg/kg | 7 days | 3 | 2 | 35.2 ± 12.4 |
| NPC1 | IC | 3.8 mg | 14 days | 1 | 2 | 49‡ |
| NPC1 | IC | 7.5 mg | 14 days | 1 | 2 | 62‡ |
| NPC1 | IC | 15 mg | 14 days | 2 | 1 | 66‡ |
| NPC1 | IC | 30 mg | 14 days | 1 | 5 | >76† |
| NPC1 | IC | 60 mg | 14 days | 2 | 1 | >76† |
| NPC1 | IC | 120 mg | 14 days | 2 | 7 | >76† |
| NPC1 | IC, SC | 120 mg, 1000 mg/kg | 14 days, 7 days | 5 | 3 | >76† |
| NPC1 | IC | 120 mg postsymptomatic* | 14 days | 4 | 4 | 43.5 ± 5.8 |
Treatment first began at 16 weeks of age
SC, subcutaneous; IC, intracisternal
No cats in this group died of NPC disease during the study period of 76 weeks
Longest survival time of individual cats that died due to signs of NPC disease. Remaining cats in this group were euthanized at ~24 weeks of age to collect histological data.
Table 2.
Serum ALT activity, and albumin and cholesterol concentrations in NPC cats.
| Untreated normal control | Untreated NPC | 1000 mg/kg SC | 4000 mg/kg SC | 8000 mg/kg SC | 30 mg IC | 120 mg IC | 120 mg IC & 1000 mg/kg SC | |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| ALT (U/L) | 62.1 ± 15.4 | 395.8 ± 169 *p=.0000002 |
128.1 ± 127.6 †p=.0003 |
37.5 | 77 ± 66.9 †p=.00001 |
295.3 ± 107 *p=.003 |
223.8 ± 87 *p=.003 †p=.003 |
135 ± 91 †p=.00004 |
| Albumin (g/dl) | 2.9 ± .3 | 2.6 ± .3* *p=.00035 |
2.8 ± .2 | 3.0 | 3.3 ± .38 *p=.002 †p=.000002 |
2.5 ± .01 *p=.00002 |
2.5 ± 0.2* *p=.00006 |
3.0 ± 0.3 †p=.008 |
| Cholesterol (mg/dl) | 120.3 ± 23 | 240.3 ± 72 *p=.000001 |
189.9 ± 48.5 *p=.004 †p=.04 |
131 | 166 ± 58 †p=.04 |
191 .2 ± 37.3 *p=.004 †p=.04 |
180.6 ± 22.4* *p=.0001 †p=.004 |
166 ± 34.6* *p=.007 *†p=.002 |
Mean ± SD for serum alanine aminotransferase (ALT), albumin, and total cholesterol of 24-wk-old cats. p-values are provided for groups significantly different from untreated normal control cats (*) or significantly different from untreated NPC cats (†). SD not provided for 4000 mg/kg group where n=2.
Fig 1. Age of onset and severity of ataxia in untreated NPC cats and NPC cats administered IC HPβCD.

(A) A dose-related effect on the age of onset of ataxia was noted in NPC cats receiving IC HPβCD compared to untreated NPC cats (†p<0.05). At 24 weeks of age, the severity of ataxia (B) and head tremor (C) was diminished in cats dosed presymptomatically; individual cats receiving 60 mg HPβCD or more showed no ataxia or head tremor at an age when untreated NPC cats were euthanized due to disease progression. No cats receiving 15 mg or greater HPβCD developed head tremor. At 76 weeks of age, only cats receiving 30 mg HPβCD or greater were alive, and these cats showed mild to moderate ataxia. Cats first administered HPβCD at 16 weeks of age (postsymptomatically), showed the same onset of ataxia as untreated cats (A), with individual cats showing less ataxia and head tremor at 24 weeks of age compared to untreated NPC cats (B, D). Ataxia was graded on a 0–4 scale with 0, none; +1, mild ataxia; +2, moderate ataxia resulting in falling when running; +3, severe ataxia resulting in falling when walking; +4 no longer able to stand. Head tremor graded on a 0–3 scale with 0, none; +1, mild; +2, moderate; +3, severe.
Fig 2. Liver histology and biochemistry in NPC cats administered SC or IC HPβCD.

(A) Untreated NPC cats showed severe vacuolization of hepatocyte and Kupffer cell cytoplasm that was ameliorated by 1000 mg/kg HPβCD but was unaffected by 120 mg IC HPβCD. All cats were 24 weeks of age; scale bar = 100 microns. (B) HPTLC of total lipids in 2 mg tissue samples. Migration in chloroform-methanol-H2O 65:25:4, anisaldehyde spray. The prominent storage of cholesterol and sphingomyelin was greatly reduced in all SC-treated cats but remained unchanged with IC administration. (C) HPTLC showing a similar effect of treatment on neutral glycosphingolipids from 5 mg tissue samples after saponification of total lipids. Migration as in B; stained with orcinol-H2SO4 spray. (D) Free sphingosine concentrations (measured by HPLC) that were increased ~50-fold in untreated NPC cats, decreased ~5 times after SC-HPβCD treatment. For the SC graph, both cats received 8000 mg/kg HPβCD. For the IC graph, data from cats receiving 15 mg, 30 mg, and 60 mg were included as there was no dose effect. Normal values: 37±15 pmol/mg protein (mean ± SD, n=7). Abbreviations: Untr’d: untreated; Norm: normal; SC8, SC4, SC1: subcutaneous, 8000, 4000 or 1000 mg/kg; IC120, IC60: intracisternal, 120 or 60 mg; Chol: unesterified cholesterol; BMP: bis(monoacylglycero)phosphate; Sph: sphingomyelin; GlcCer: glucosylceramide; Lac Cer: lactosylceramide; Gb3: globotriaosylceramide; HPTLC, high performance thin-layer chromatography; HPLC, high pressure liquid chromatography.
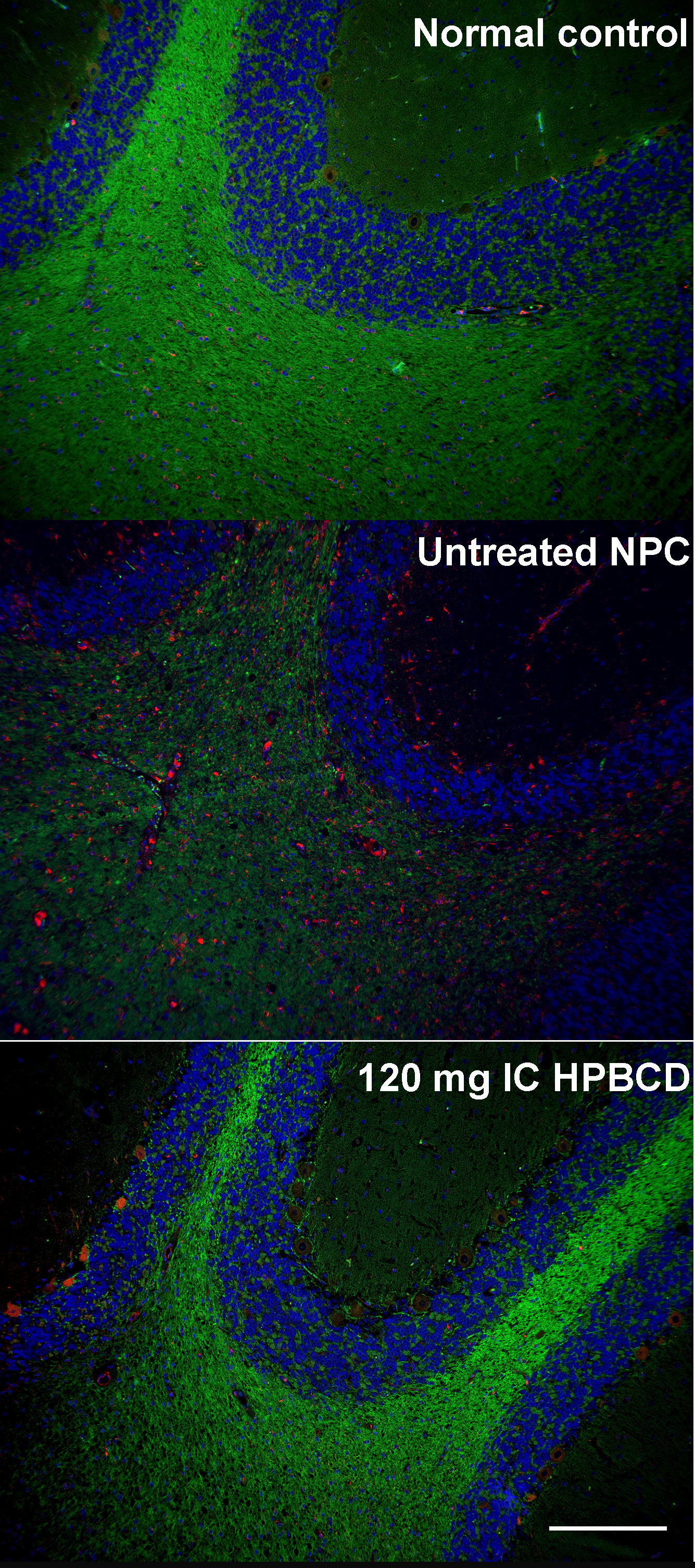
Fig 3. Calbindin and GM2 immunohistochemical staining and filipin staining of cerebellum from normal control, untreated NPC, and HPβCD-treated NPC cats.

Top row depicts immunohistochemical calbindin labeling of Purkinje cells (PCs), whose cell body and dendritic arbor are highlighted in brown. Middle row shows filipin labeling of unesterified cholesterol, accumulation of which is noted by white cytoplasmic staining. Bottom row demonstrates GM2 accumulation (dark brown punctae). Normal control column depicts a full complement of PCs lacking cholesterol and GM2 storage, typical of a normal cat at 25 wks of age. Untreated NPC column shows a large axonal spheroid (red arrow) in one of the remaining PCs within this 25-wk-old NPC cat as well as abundant storage of cholesterol and GM2 gangliosides present in all remaining PCs. Treatment of an NPC1 cat with 120 mg IC HPβCD (third column) resulted in substantial PC rescue as well as decreased cholesterol and GM2 ganglioside accumulation within PCs of this 29-wk-old cat. Abbreviations: MC, molecular cell layer; PC: Purkinje cell layer; GC: granular cell layer; scale bar = 50 microns.
Fig 4. Cerebral gray matter lipids in normal control, untreated NPC, and NPC cats administered IC HPβCD.

(A) Upper panel: HPTLC of total gangliosides (from 3 mg tissue samples), showing striking and selective reduction of GM3 and GM2 in NPC cats administered IC HPβCD; the gangliosides patterns remained essentially unchanged in NPC cats administered SC HPβCD (migration in chloroform-methanol-0.2% CaCl2 55:45:10, resorcinol-HCl spray). Lower panel: Quantitative data (24–26 week-old cats; GM3 and GM2 expressed as % of total gangliosides). IC treatment at 7.5 and 3.5 mg doses appeared less efficient at reducing GM2 and GM3 than 15 mg or higher doses. (B) Lactosylceramide concentrations (HPTLC from 10 mg tissue samples) were reduced in cats treated with IC HPβCD. (C) Free sphingosine concentrations (measured by HPLC, expressed as pmol/mg protein) were normalized in all IC treated cats. (D) HPTLC of total gangliosides (from 3 mg tissue samples) of untreated NPC cats at various ages (left), and developmental profiles of GM2 and GM3 storage (right). Normal proportions (% of total gangliosides) are 2.0 ± 0.5 % for GM2, and 2.5 ± 0.6% for GM3 (Mean ± SD, n=5). Abbreviations: Untr’d: untreated; Norm: normal; SC8, SC4, SC1: subcutaneous, 8000, 4000 or 1000 mg/kg; IC120, IC60: intracisternal, 120 or 60 mg; HPTLC, high performance thin-layer chromatography; HPLC, high pressure liquid chromatography.
Fig 5. Pulmonary histology from 6-month-old untreated NPC cats and NPC cats administered 8000 mg/kg SC HPβCD.
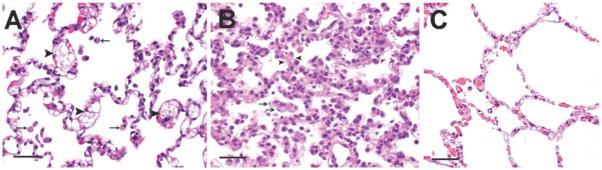
(A) In untreated NPC cats, the alveolar septa were expanded by foam cells (black arrowheads) as well as by macrophages containing larger irregular clear vacuoles. Alveolar spaces similarly contained foam cells (arrows). (B) Cats given 8000 mg/kg SC HPβCD had evidence of acute to subacute diffuse alveolar damage. Alveolar spaces contained abundant proteinaceous fluid with wispy strands of fibrin, foamy macrophages and neutrophils. The alveolar septa were lined by hypertrophied type II pneumocytes with multifocal hyaline membrane formation. The septa were congested and contained foamy macrophages (black arrows), neutrophils and lymphocytes. Arrowheads denote thickened alveolar septae. No evidence of alveolar damage was seen in the other treatment groups. (C) Lung from a normal control cat; scale bar = 100μm.
Subcutaneous HPβCD ameliorated hepatic disease and improved CNS disease only at doses with negative pulmonary consequences
Before assessing the potential effects of subcutaneous (SC) administration of HPβCD on NPC cats, we first determined serum and cerebrospinal fluid (CSF) concentrations in 24-week-old normal control cats 60 minutes after administration of a single dose of HPβCD. The achieved serum concentrations were 546 μg/ml, 2570 μg/ml, and 3485 μg/ml, after administration of 1000, 4000, and 8000 mg/kg, respectively. CSF concentrations of HPβCD were not detectable in cats receiving 1000 mg/kg, and were 19 μg/ml and 21.1 μg/ml in cats receiving 4000 mg/kg and 8000 mg/kg, respectively. A full pharmacokinetic study was not performed.
To test therapeutic efficacy in NPC cats, HPβCD was repeatedly administered SC at one of three doses (1000 mg/kg HPβCD with 25 mg/kg allopregnanolone, 4000 mg/kg HPβCD, or 8000 mg/kg HPβCD) every 7 days to NPC cats beginning at three weeks of age (Table 1). All three groups showed improvements in body weight (Figure S1), and in serum ALT, albumin and total cholesterol compared to untreated NPC cats (Table 2); since only two cats were evaluated in the 4000 mg/kg group, statistical analyses were not performed. Administration of 1000 mg/kg was sufficient to improve liver function (Table 2), ameliorate hepatic and Kupffer cell swelling (Figure 2A), and to decrease hepatic cholesterol, sphingomyelin, neutral glycolipids, free sphingosine (Figure 2B–D), and GM3 ganglioside storage (Figure S2A).
Cats that received 1000 mg/kg SC HPβCD had a similar onset of neurological dysfunction and mean survival time (21.8 ± 6.5 weeks; p=0.8) compared to untreated NPC cats. In contrast, two cats that received 4000 mg/kg showed modest amelioration of neurological disease and survived to 31 and 36 weeks of age, which was longer than any untreated cat. Mean survival time in cats receiving 8000 mg/kg (35.2 ± 12.4 weeks; p=.0003) was significantly greater than untreated NPC cats. However, the first two cats that received this dose were euthanized at 21 and 25 weeks of age due to the acute onset of severe dyspnea within 24 hours of the weekly HPβCD administration. These cats had diffuse alveolar damage characterized by thickening of the alveolar septa; hypertrophied type II pneumocytes with multifocal hyaline membrane formation, neutrophils and lymphocytes within alveolar septa; and proteinaceous fluid, fibrin, neutrophils, and foamy macrophages within alveolar spaces (Figure 5B). In contrast, pulmonary histology in NPC cats receiving 1000 mg/kg and 4000 mg/kg showed no alveolar damage, although foamy macrophages remained within the alveolar spaces as are present in untreated NPC cats (Figure 5A). Therefore, in the remaining three cats receiving 8000 mg/kg, HPβCD was discontinued at ~20 weeks of age, an age at which mild ataxia and head tremor were present but cats were otherwise clinically normal (Video S2). Remarkably, these three cats went on to survive more than twice as long as untreated cats (45 ± 8.5 weeks; p=6.4 E-07) without any further therapy, although they eventually developed neurological signs indistinguishable from untreated NPC cats. Notably, following the discontinuation of therapy, these cats survived an additional 21 weeks, which is the mean survival time of untreated NPC cats.
Evaluation of the brains of 24-week-old cats treated with 8000 mg/kg HPβCD showed a decrease in cerebrocortical filipin staining (Figure 6) and improved Purkinje cell survival (Figure 7). The biochemical study of gangliosides indicated a small reduction in the proportion of GM2 ganglioside in a 21-week-old cat treated with 8000 mg/kg (14%) and in another 31-week-old cat receiving 4000 mg/kg (15%). In a 37-week-old cat that had received 8000 mg/kg HPβCD until 24 weeks of age, the reduction was no longer present (19%) (Figure 4A); these data were confirmed by immunohistochemical staining (Figure S3). GM3 ganglioside followed the same trend (Figure 4A). Although gangliosides were no longer reduced, a decrease in cerebrocortical filipin staining was still observed in this 37-week-old cat (Figure 6). Finally, the lowest sphingosine concentration in the SC category (Figure 4C) was observed in the 21-week-old treated cat administered 8000 mg/kg HPβCD.
Fig 6. Filipin-staining of marginal gyrus of cerebral cortex.

Untreated 24-week-old NPC cats (B) showed storage of cholesterol (visualized as white cytoplasmic labeling within cells) in the marginal gyrus of the cerebral cortex; no cholesterol storage was observed in healthy control animals (A). Cats receiving 1000 mg/kg SC HPβCD showed no reduction in cholesterol staining (C). Cats receiving 8000 mg/kg SC HPβCD (D) showed substantially less cholesterol staining than did untreated NPC cats. Indeed, the example shown is from a 37-week-old cat that had received SC HPβCD until 24 weeks of age at which time it was discontinued. 24-week-old cats receiving 30 mg or greater IC HPβCD presymptomatically showed the greatest amelioration of the cholesterol storage defect (E, H). In contrast, treatment of NPC cats with 3.8 mg IC HPβCD (G) or 120 mg IC HPβCD administered post-symptomatically (F) revealed little effect on stored cholesterol in cats at 24 weeks of age, while both 30 mg and 120 mg HPβCD administered presymptomatically showed substantial reductions in cholesterol storage. Scale bar = 100 microns.
Fig 7. Calbindin immunofluorescence of cerebellar paravermis in 6-month-old cats.

Untreated NPC cats (B) showed a severe loss of Purkinje cells compared to normal control cats (A). Subcutaneous (SC) HPβCD administration at a dose of 8000 mg/kg resulted in increased Purkinje cell survival (D), whereas cats receiving 1000 mg/kg SC HPβCD were indistinguishable from untreated NPC cats (C). Cats treated with IC HPβCD at 30 mg or greater (H) resulted in substantial Purkinje cell survival. In contrast, 3.8 mg IC HPβCD (G) or 120 mg IC HPβCD given either presymptomatically (E) or postsymptomatically (120 mg IC late) (F) resulted in small but non-significant increases in Purkinje cell number compared to untreated NPC cats. Scale bar = 200 microns.
These studies show that SC administration of 1000 mg/kg was sufficient to improve body weight and hepatic disease but had no effect on neurological disease or survival time. In contrast, SC administration of 8000 mg/kg improved weight gain and hepatic disease, onset and severity of neurological dysfunction, cholesterol (and, marginally, ganglioside storage), and survival time. Unfortunately, pulmonary toxicity limited continued long-term peripheral administration of HPβCD at or above this concentration.
Intracisternal HPβCD resulted in neurologically normal cats at 24 weeks of age
To overcome the observed pulmonary toxicity, HPβCD was administered intrathecally at the cisterna magna (IC) to evaluate the efficacy and safety of drug delivered directly to the CNS. A dose of 120 mg, equivalent to an approximate dose of 4000 mg/kg brain weight, was initially evaluated. Following administration to seven 24-week-old normal control cats, CSF was sampled at 0.25 (n=2), 1 (n=2), 4 (n=3), and 24 hours (n=3) demonstrating a Cmax = 11645 μg/ml, t1/2 = 0.7 – 2.6 hr, and AUC = 28614 μg.h/ml (Table 3). High concentrations of HPβCD were also observed in cerebellum and cerebrum, up to 689 and 418 ug/ml, respectively, within 4 hours after IC administration.
Table 3.
Pharmacokinetics of HPβCD after intracisternal (IC) or intravenous (IV) administration in normal control cats.
| Analyte | Route of Administration | Dose | Matrix | C0 (μg/mL) | AUC0-∞ (μg·h/mL) | AUC0–24h (μg;·h/mL) | t½ (h) | CL/F (mL/h·kg) | Vdss (mL/kg) |
|---|---|---|---|---|---|---|---|---|---|
| HPβCD (Sigma) | IC | 120 mg | CSF | 11645 | 28614 | .7–2.6 | |||
| HPβCD (Kleptose) | IC | 30 mg | CSF | 15400 | 23300 | 23100 | 3.22 | 1.29 | 4.14 |
| HPβCD (Kleptose) | IV | 1000 mg/kg | CSF | 13.1 | NC | 346 | NC* | NC* | NC* |
| HPβCD (Kleptose) | IV | 1000 mg/kg | Plasma | 3760 | 4660 | 6770 | 1.11 | 215 | 336 |
Not calculated due to below detectable concentrations for some samples
NPC cats were treated with 120 mg IC HPβCD beginning at 3 weeks of age and repeated every 14 days thereafter. Remarkably, these cats were neurologically normal at 24 weeks of age and showed only mild ataxia at 76 weeks of age (Videos S3 and S4; Figures 1A–D). In the brain of 4 cats euthanized at 24 weeks of age, by filipin staining and immunostaining there was a marked reduction in storage of unesterified cholesterol and GM2 ganglioside (Figures 3 and 6; Figure S3), and Purkinje cell loss was completely abrogated (Figure 7). Purkinje cell numbers were significantly greater than those found in untreated NPC cats (p=.02). Indeed, on hematoxylin/eosin stained sections, treated cats were indistinguishable from normal animals (Figure 8A). Additionally, granular cell layer thickness was improved and cholesterol and ganglioside storage was decreased, astrogliosis of the cerebellar gray and white matter was reduced, and cerebellar white matter appeared normal (Figure 3, Figure S4). Biochemical study of the ganglioside patterns (Figure 4A) demonstrated drastically reduced storage of gangliosides GM2 (5.3%) and GM3 (4.6%), with unchanged concentrations of the major brain gangliosides. However, there was no improvement in serum albumin, and ALT and cholesterol concentrations were only slightly decreased when compared to untreated NPC cats (Table 2). Liver histology and lipid biochemistry were similar to the findings in untreated NPC cats (Figure 2).
Fig 8. Purkinje cell quantification and hearing threshold in NPC cats administered IC HPβCD.

(A) Untreated 24-week-old NPC cats showed significantly fewer Purkinje cells per unit length compared to age-matched normal control cats (*p<0.05). 24-week-old cats receiving 30 mg or greater IC HPβCD showed significantly greater Purkinje cell numbers compared to untreated NPC cats (†p<0.05). In contrast, 24-week-old cats treated postsymptomatically with 120 mg IC HPβCD at 16 weeks of age, showed significantly fewer Purkinje cells compared to normal control cats (*p<0.05). (B) Untreated 24-week-old NPC cats showed no significant difference in hearing threshold compared to normal control cats. In contrast, cats receiving 7.5 mg or 120 mg HPβCD developed significant increases in hearing threshold compared to untreated NPC cats (†<0.05). Individual cats receiving lower doses of HPβCD also showed elevated hearing thresholds although, as a group, these were not significant. (C) At 76 weeks of age, all NPC cats receiving 30 mg or greater HPβCD showed significant elevations in hearing threshold.
Additionally, one cohort of NPC cats was treated with a combination of both 120 mg IC and 1000 mg/kg SC HPβCD (which contained no allopregnanolone) (Table 1). These cats were neurologically normal at 24 weeks of age and either remained normal or showed only mild ataxia at 76 weeks of age (Figure 1A–D). However, they also maintained serum ALT activity and albumin concentration similar to that found in normal cats, accompanied by significant decreases in serum cholesterol when compared to untreated NPC cats (Table 2; p=.002). Biochemical analysis of liver showed a marked decrease of cholesterol, sphingomyelin, GM3 ganglioside, neutral glycolipids, and free sphingosine concentrations (Figure 2B, C). In the brain, similar results as described for IC treatment alone were observed, with strong decreases in filipin-stained cholesterol, GM2 and GM3 gangliosides (but not in the major gangliosides GM1, GD1a, GD1b, GT1b), lactosylceramide, sphingosine, and no loss of Purkinje cells (Figures 4, 8).
The above studies illustrate the ability of 120 mg IC HPβCD to ameliorate neurological disease, brain biochemical abnormalities, and Purkinje cell loss when treatment is initiated prior to the onset of neurological deficits. As human NPC patients are most often diagnosed after the onset of symptoms, we next determined the effects of instituting therapy when neurological dysfunction was already present. For this, a cohort of NPC cats was administered 120 mg HPβCD IC every 14 days beginning at 16 weeks of age, an age at which moderate ataxia (+2) and tremor (+2) exist. All eight NPC cats treated in this manner showed either no progression or slowed progression of clinical signs when evaluated at 24 weeks of age (Video S5; Figure 1B–C). Histological and biochemical evaluation of these cats showed an accumulation of cerebrocortical cholesterol, which was similar to that found in untreated NPC cats (Figure 6). The concentration of GM2 ganglioside (11.7 ± 0.4% of total gangliosides) was clearly less than at the age when treatment began, and not much higher than in 4-week old untreated cats (Figure 4A and D). Evaluation of Purkinje cells also suggested that loss of these cells was not as pronounced as that found in untreated cats (Figure 7), however quantification studies identified no difference between cats treated postsymptomatically and untreated NPC cats (Figure 8).
Intracisternal HPβCD improved neurological function and survival time in cats with NPC disease
To determine the effect of different doses of IC HPβCD on neurological signs of NPC disease in cats, cohorts were administered doses from 3.8 mg to 60 mg beginning at 3 weeks of age and doses were repeated every 14 days thereafter (Table 1). The age of onset of ataxia and the severity of neurological dysfunction at 24 weeks of age varied directly with the administered dose (Figure 1A–C). A dose of 3.8 mg HPβCD IC or greater resulted in a delay in the onset of ataxia and less severe clinical signs at 24 weeks of age when compared to untreated NPC cats. Doses of 15 mg or greater alleviated all head tremor. Doses of 30 mg or greater resulted in survival to at least 1.5 years of age and the development of only mild to moderate ataxia. Doses of 60 mg or greater resolved ataxia in cats at 24 weeks of age (the median age at which untreated cats are euthanized due to the inability to walk or maintain sternal recumbancy). No significant differences in outcome measures could be determined between cats dosed with 60 mg or 120 mg HPβCD, although, in this study, cats were not evaluated beyond 76 weeks of age.
To evaluate the histological and biochemical effects of dose, two or more cats in each group were sacrificed at 24 weeks of age to evaluate brain pathology and biochemistry (Figures 4, 6–8). Cerebrocortical neurons showed the greatest amount of filipin staining in cats receiving 3.8 mg, and the least staining in cats receiving 30 mg or greater (Figure 6). This trend held true for ganglioside accumulation as well, with those NPC cats receiving 30 mg or higher exhibiting less storage of GM2 ganglioside than those cats receiving less than 30 mg IC HPβCD (Figure S3). Although there was some Purkinje cell loss at lower doses, doses of 30 mg or greater resulted in Purkinje cell preservation (Figure 7 and 8). Biochemical studies showed that increasing doses of HPβCD were associated with decreases in cerebral GM2 ganglioside concentrations although this effect plateaued at 15 mg or greater (Fig 4A).
One or more cats in each IC-treated group were monitored long-term and euthanized only when they were non-ambulatory and no longer able to remain in sternal recumbency without support, or when they reached 76 weeks of age. Cats receiving 3.8 mg, 7.5 mg, or 15 mg survived to 49 weeks, 62 weeks, and 66 weeks of age, respectively. In contrast, all cats in treatment groups receiving 30 mg or greater survived the 76 week observation period (Figure 1D). Neurological dysfunction in cats receiving 30 mg or more progressed slowly and at 76 weeks of age these cats showed only mild or moderate ataxia with no evidence of tremor (Figure 1D). Male cats from the 120 mg IC group are currently over two years of age and are breeding and producing kittens.
Four cats that began treatment postsymptomatically, 120 mg IC HPβCD at 16 weeks of age, were observed beyond the 24 week observation period. These cats survived significantly longer than untreated NPC cats (43.5 ± 5.8 weeks of age; p=9.06 E-11). Three cats were euthanized due to inability to maintain sternal recumbency. One cat developed severe diarrhea and was euthanized at 42 weeks of age, although it remained able to walk at this age. The diarrhea was associated with an endemic corona virus in the animal colony, which had also required euthanasia of normal control cats.
The data suggested that a 30 mg dose or greater of IC HPβCD was sufficient to profoundly influence disease progression. Pharmacokinetics (PK) of this dose revealed Cmax= 15400 μg/ml, t1/2 = 3.22 hr, and AUC = 23100 μg.h/ml, which were not significantly different from PK data for the 120 mg dose (Table 3). The PK results suggested that the 30 mg IC dose was sufficient to achieve maximal exposure in the CSF of cats. Brain parenchymal concentrations of HPβCD at this dose were not determined.
Subcutaneous and intracisternal HPβCD administration caused injection-site inflammation and increased hearing threshold
We next assessed potential safety issues associated with both SC and IC HPβCD administration. A dose of 8000 mg/kg SC HPβCD resulted in dose-limiting pulmonary toxicity as described above. However, 1000 mg/kg, 4000 mg/kg, and 8000 mg/kg injected SC each produced increasing pain at the injection site with advancing age. Cats receiving both SC and IC HPβCD developed a progressively hunched posture and a short-strided gait in the thoracic limbs that began between 52 and 76 weeks of age. MRI of the shoulder of these cats revealed evidence of cellulitis, myositis, and arthritis limited to the shoulder joints (Figure S5). Subcutaneous administration of HPβCD was, therefore, discontinued in these cats after 76 weeks of age. This hunched posture was not seen in cats treated with IC HPβCD alone.
Although hearing thresholds of untreated NPC cats were not significantly different from unaffected cats at 24 weeks of age (Figure 8B), dose-dependent elevations in mean hearing threshold in cats receiving SC HPβCD were observed (1000 mg/kg = 87 dB SPL; 4000 mg/kg = 95 dB SPL; 8000 mg/kg = 112 dB SPL). In cats receiving IC HPβCD, a significant elevation in hearing threshold was seen in groups of cats receiving either 7.5 mg or 120 mg IC compared to untreated NPC cats (Figure 8B). Individual cats receiving 15 mg or greater HPβCD could be found with hearing threshold greater than or equal to 90 dB SPL, although a dose-dependent elevation in hearing threshold between group means did not reach significance. The elevation in hearing threshold was more pronounced in cats evaluated at 76 weeks of age where groups receiving 30 mg or greater IC HPβCD had significant elevation in hearing threshold with many having thresholds equal to or greater than 125 dB, which was the limit of sound generation in the equipment used (Figure 8C). These results in NPC cats confirm our previous results in normal cats (36), that both SC and IC HPβCD elevate hearing threshold and that continued administration resulted in more profound hearing loss.
DISCUSSION
Cyclodextrin therapy for NPC disease was first evaluated in 2001 when it was found that intraperitoneal HPβCD (500 mg/kg given 3 times/week) lowered liver unesterified cholesterol and delayed the onset of continuous extensor tremor of the limbs in Npc1−/− mice (26). In a subsequent study, a single dose of SC-administered allopregnanolone (25 mg/kg) dissolved in a 20% HPβCD solution (4000 mg/kg) increased Purkinje cell and granule cell survival, reduced brain ganglioside accumulation, and nearly doubled the lifespan of Npc1−/− mice (43). Although the substantial positive effects were attributed to the reconstitution of allopregnanolone in the Npc1−/− mouse brain (43), follow-up studies by two labs independently found that 4000 mg/kg SC HPβCD alone was sufficient to reduce storage of cholesterol and gangliosides, and to positively affect NPC-related neurological disease (30, 31, 44). Our data in the feline model support evidence of a dose related effect of SC HPβCD on NPC disease. 1000 mg/kg SC HPβCD, in the presence or absence of allopregnanolone, had substantial positive effects on hepatic disease in feline NPC disease, but no effect on neurological disease. In contrast to mice, 4000 mg/kg SC HPβCD resulted in only modest disease amelioration, whereas 8000 mg/kg SC HPβCD resulted in reduced NPC-associated neurological disease and increased survival time. However, 8000 mg/kg also resulted in life-threatening pulmonary toxicity that was not anticipated, as previous studies had not involved chronic administration of HPβCD at this dose (45–48). Histological evaluation confirmed the proliferation of cuboidal epithelial cells (type II pneumocytes) consistent with lung damage (49). It is possible that the pulmonary toxicity, and injection site inflammation, associated with HPβCD may have been due to differences in the degree of substitution or impurities present in the HPβCD formulation and not directly due to the cyclodextrin. The powdered form of HPβCD (HPβCD-H107; Sigma Aldrich, St Louis MO) was used for subcutaneous injections as high doses were necessary and this was the least expensive formulation available. In contrast, the cell-culture system tested form (HPβCD -C0926; Sigma Aldrich, St Louis MO) was used for all intracisternal injections. Our study cannot rule out whether the degree of substitution or impurities in different drug formulations were responsible for the pulmonary toxicity seen in cats.
Pulmonary disease is a rarely described finding in NPC patients, with greater severity found in NPC2 rather than NPC1 disease (2, 50–54). In untreated NPC cats, lung histology showed thickened septae and foamy macrophages within the alveolar spaces and septa, changes identical to those seen in the Npc1−/− mouse (55). Data from NPC mouse lung showed that 4000 mg/kg HPβCD did not reverse pulmonary disease (30, 56–58), nor did it decrease unesterified cholesterol concentrations or cholesterol synthesis, suggesting that macrophages in the lung did not have access to plasma HPβCD (56). It is interesting to note that treated cats have thus far shown no evidence of developing pneumonitis as seen in Npc1−/− mice, however, foamy macrophages continue to be evident in lungs from cats treated with SC HPβCD.
Although it is clear that administration of SC HPβCD positively affects CNS disease in both NPC mice and cats, it apparently does so without blood-brain barrier penetration (26, 32). Our current study shows that SC doses of 4000 and 8000 mg/kg resulted in very low CSF concentrations (~20 μg/ml; ~14.3 μM) measured 60 minutes following administration, and a brain to plasma ratio of 0.7 %, that is similar to what was found in mice (40), and approximates the serum/CSF ratio reported for human albumin (59, 60). To more fully evaluate the pharmacokinetics of this drug, we administered control cats a single dose of 1000 mg/kg HPβCD intravenously and determined CSF and plasma concentrations (Table 3). This IV dose resulted in a maximal serum concentration similar to what was found 60 minutes following SC administration of 8000 mg/kg HPβCD. The CSF concentration of 13.1 μg/ml was also similar, again indicating poor CNS penetration of HPβCD and a serum/CSF ratio of 0.3%. Therefore, one proposed explanation for the clinical efficacy seen in NPC cats receiving high SC doses is that, similar to serum albumin (40), HPβCD crosses the blood-brain barrier in small quantities through fluid-phase transcytosis and thus requires high serum concentrations of HPβCD to mitigate CNS disease. The evaluation of brain parenchymal concentrations of HPβCD following SC administration may further our understanding of whether serum HPβCD concentrations affect CNS disease directly or via indirect effects on brain endothelial cells as has previously been proposed (32, 61, 62).
Our results clearly demonstrate that direct administration of HPβCD into the CNS avoids the need for high serum concentrations and associated pulmonary toxicity. A dose-related effect on the age of onset of ataxia, severity of ataxia, brain cholesterol and ganglioside storage, and Purkinje cell survival was identified in cats treated prior to the onset of symptoms. Improvement in neurological function and Purkinje cell survival were always accompanied by concomitant decreases in cholesterol and ganglioside storage. Doses of 30 mg or greater resulted in the most profound amelioration of biochemical disease, and although significant differences in Purkinje cell numbers in cats administered 30, 60, or 120 mg IC HPβCD could not be identified, age of onset of ataxia and severity of ataxia suggest that 120 mg was the most efficacious. Based on these data, the maximally effective dose of IC HPβCD has not been determined. Planned studies include the administration of doses above 120 mg; the evaluation of neurological function in cats beyond 76 weeks of age; the evaluation of cerebellar pathology in cats at 76 weeks of age and beyond; and the effect of IC HPβCD administration on CNS tissue outside of the cerebellum.
Importantly, post-symptomatic therapy prolonged lifespan and slowed the progression of neurological dysfunction. However, no cat in this group survived beyond one year of age and no increase in Purkinje cell numbers were found when compared to 24-week-old untreated NPC cats. NPC cats euthanized at 16 weeks of age, when cats were first administered HPβCD, were not available for histological comparison. Post-symptomatically treated cats showed biochemical evidence of decreased ganglioside storage compared to age-matched untreated NPC cats, but the decrease was less profound than in cats treated presymptomatically. Indeed, immunohistochemistry showed less evidence of decreased gangliosides in the brains of post-symptomatically-treated cats. Similarly, little improvement in concentrations of cholesterol storage in the late endosomal/lysosomal compartment was identified by histochemical staining with filipin. These data suggest that presymptomatic therapy is most effective at slowing disease progression and that cholesterol and ganglioside storage may be more difficult to correct postsymptomatically. Biomarkers that are currently being validated to correlate with clinical improvement may shed light on whether decreases in cholesterol, sphingolipids, or some other metabolite best correlate with Purkinje cell survival and clinical improvement.
An elevated hearing threshold was identified as an adverse effect in cats administered either SC or IC HPβCD. Hearing loss is not a characteristic of feline NPC disease (34, 36), however, HPβCD administration resulted in a dose-dependent and duration-of-treatment-dependent increase in hearing threshold in both affected and control cats (36). Studies performed in mice confirmed that HPβCD-induced elevations in hearing threshold were accompanied by loss of outer hair cells within the cochlea, presumably due to changes in membrane composition and integrity and consequent effects on the outer hair cell motor protein prestin (63). Our studies in the cat indicate that chronic HPβCD at therapeutic doses is always accompanied by severe hearing loss. These findings have important implications for clinical use in human patients where deafness may be an expected outcome of therapy. Potential methods to mitigate HPβCD-mediated hearing loss including adjunct therapy, limiting access of HPβCD to the inner ear, using other cyclodextrins which may be less toxic, or managing hearing loss with either hearing aids or cochlear implants are being evaluated.
A complete understanding of how HPβCD reduces neuronal storage of cholesterol and sphingolipids and results in Purkinje cell survival remains to be determined. In vitro studies of HPβCD in cultured murine Npc1−/− neurons show a concentration-dependent effect on cholesterol storage (15, 64) with low doses (0.1 mM) releasing cholesterol from the late endosomal/lysosomal compartment to the endoplasmic reticulum; higher doses (5–10 mM) are toxic and deplete cholesterol from the plasma membrane. Intraventricular administration of HPβCD in the Npc1−/− mouse resulted in Purkinje cell survival and was accompanied by evidence of intracellular cholesterol mobilization from the lysosome to the metabolic pool of the cytosolic compartment (suppressed cholesterol synthesis, elevated cholesterol esters, suppressed SREBP2 target genes, and activated LXR-control genes) (65, 66). In the cat and mouse models, as well as in human patients, HPβCD administered directly into the CSF transiently increased both plasma and CSF 24(S)-hydroxycholesterol (24(S)-HC) concentrations (40). Given that conversion of unesterified cholesterol to 24(S)-HC is the principal route of excretion of cholesterol from the CNS, its elevation indicates that cholesterol flux through the lysosome to the cytosolic compartment was normalized following treatment with HPβCD. However, other studies also suggest that cyclodextrins may directly interact with sphingolipids or other membrane constituents as well as cholesterol (67). Genetic reductions of complex gangliosides in Npc1−/− mice have been shown to be accompanied by reduced intraneuronal cholesterol sequestration (68, 69). Finally, cyclodextrins may function through substrate-independent mechanisms such as modulation of autophagy (70) or stimulation of exocytosis (71). Indeed, dose-related clinical effects of HPβCD were accompanied by parallel changes in cholesterol, sphingolipids, and Purkinje cell survival in the feline model and we are currently evaluating effects on autophagy.
Direct injection of CSF with HPβCD ameliorates neuronal storage of cholesterol and gangliosides and improves Purkinje cell survival in both NPC mice (40, 64, 65) and cats, supporting its therapeutic potential in human patients. Here, we used direct injection into the cerebellomedullary cistern of our feline model, but intrathecal injections in humans are typically carried out by lumbar puncture or intraventricular injection. In the current Phase I trial, HPβCD (Kleptose® HPB, D.S. 0.4, average M.W. 1400) is being administered by monthly lumbar puncture. Therefore, to increase the translational potential of these studies, we are currently evaluating the safety and efficacy of lumbar injections in the feline model. Also, repeated cisternal administration of HPβCD in cats required general anesthesia using propofol every two weeks. Untreated NPC cats were also anesthetized every 14 days in order to acquire CSF for biomarker evaluation (40), and there was no evidence that disease progression was altered by anesthesia alone. In the current Phase I trial, only local anesthetic or sedation are being used. Finally, human patients present with learning disabilities, dementia, vertical supranuclear gaze palsy, seizures, dysarthria, and dysphagia in addition to cerebellar ataxia. Unfortunately, dementia and dysarthria are difficult to model in cats and monitoring seizures by electroencephalography is challenging in awake cats (71). As supranuclear gaze palsy and dysphagia were not observed in our feline model, we plan to verify regional cerebrocortical and brain stem effects of HPβCD using histological and biochemical methods. Thus far, we predict that intrathecal therapy with HPβCD will improve cerebellar ataxia in patients, and therefore could be used to enhance the overall clinical well-being of patients with NPC disease.
MATERIALS AND METHODS
Study design
This was a prospective study. Each treatment cohort (Table 1) was comprised of at least three cats in order to make statistical comparisons, except for the group of cats that received 4000 mg/kg HPβCD SC (see below). Clinical endpoints for study were defined prior to study onset and included 1) inability to walk or maintain sternal recumbancy, and 2) uncontrolled diarrhea resulting in dehydration. Cats were euthanized when these endpoints were reached. No outliers were defined and no cats enrolled were excluded from study. The objective of the study was to determine if direct administration of 2-hydroxypropyl-beta-cyclodextrin (HPβCD) to the intrathecal (IT) space at the cerebellomedullary cistern provides greater NPC disease amelioration than peripheral subcutaneous (SC) administration. First, cohorts of NPC cats were compared that received 1000 mg/kg, 4000 mg/kg, and 8000 mg/kg HPβCD SC every 7 days starting at three weeks of age. Cats were assigned to each cohort in a dose-escalation fashion with the first treated cohort receiving the lowest dose. Survival data in the two cats in the 4000 mg/kg cohort indicated insufficient effects at this dose, and, therefore, due to the limited number of cats which could be produced, no additional cats were evaluated in this cohort. Pulmonary toxicity limited the continued dosing of cats in the 8000 mg/kg group (see results). Several cohorts of affected NPC cats were evaluated using multiple IT administered doses. A dose of 120 mg IT, equivalent to an approximate dose of 4000 mg/kg brain weight, was initially evaluated. Following data collection from this group, five different groups of cats received 3.8, 7.5, 15, 30, or 60 mg HPβCD IT, respectively, every 14 days beginning at three weeks of age. Cats were assigned to each group by placing pieces of paper with the dose written on them into a container. When cats were born, the investigator drew a piece of paper from the container and assigned cats to each group based on the results. Finally, one group of NPC cats received a combination of 1000 mg/kg HPβCD SC every 7 days along with 120 mg HPβCD IT every 14 days starting at three weeks of age, and one group of NPC cats received 120 mg HPβCD IT every 14 days starting at 16 weeks of age. Age-matched cohorts of untreated NPC cats were available for comparison to treated cats up to the typical time of end-stage disease; treated versus untreated cohorts were determined by a coin flip method. Efficacy of therapy was evaluated by survival data, neurological examination performed weekly in all cats, body weight determined weekly in all cats, serum collected at various intervals for serum chemistry evaluation to determine the effect of therapy on liver enzymes, and microscopic examination of selected tissues. HPβCD treated cats were sacrificed at approximately 24 weeks of age to compare histologic and biochemical data to data from age-matched untreated cats. Additionally, a number of treated cats (indicated in the results section) were followed long term to assess the effect of HPβCD over time in cats.
Both male and female cats were evaluated in this study. Investigators were not blinded during the administration of HPβCD doses to cats, during the clinical evaluation of cats, or during the evaluation of tissue collected from cats except where specified.
Animals
Cats were raised in the National Referral Center for Animal Models of Human Genetic Disease of the School of Veterinary Medicine of the University of Pennsylvania (NIH OD P40-10939) under National Institutes of Health and USDA guidelines for the care and use of animals in research. The experimental protocol was approved by the University’s Institutional Animal Care and Use Committee. Peripheral blood leukocytes from all the cats were tested at one day of age for the NPC1 missense mutation using a polymerase chain reaction-based DNA test to identify affected as well as normal, control cats as previously described (20, 33, 34). Body weight was measured and physical and neurologic examinations were performed weekly from birth until death for all cats. The onset and progression, as well as the severity of signs of neurologic dysfunction were identified. Cerebellar ataxia was graded on a 0–4 scale (0, none; +1, mild ataxia; +2, moderate ataxia resulting in falling when running; +3, severe ataxia resulting in falling when walking; +4 no longer able to stand). Head tremor was graded on a 0–3 scale (0, none; +1, mild; +2, moderate; +3, severe). Brain stem auditory evoked response (BAER) testing was performed using previously described methods (36).
HPβCD formulations and treatment groups
The powdered form of HPβCD (HPβCD -H107; Sigma Aldrich, St Louis MO) was used in all subcutaneous administrations and the cell-culture tested form (HPβCD -C0926; Sigma Aldrich, St Louis MO) was used for all intracisternal administrations except where indicated. Kleptose® HPB (D.S. 0.4, average M.W. 1400) was also used in IC and IV PK experiments, and was provided by Janssen Research & Development (Beerse, Belgium). All HPβCD was administered in a 20% (weight/volume) solution dissolved in 0.9% saline (Hospira Inc, Lake Forest, IL) except when administered as a 3% solution dissolved in saline due to the small volume of administration (3.8 mg, 7.5 mg, and 15 mg). In cats receiving 1000 mg/kg SC alone, the 20% solution of HPβCD also contained 25 mg/kg allopregnanolone (provided by S. Mellon); when 1000 mg/kg SC HPβCD was administered in combination with IC therapy, allopregnanolone was not included in either dose. The inclusion of allopregnanolone in the cats receiving 1000 mg/kg SC HPβCD alone was due to these cats being treated soon after the publication of the Griffin et al study (43). Subsequent murine studies, however, have shown that cyclodextrin alone was sufficient to positively affect NPC-related neurological disease (30, 31, 44), and therefore, allopregnanolone was not administered to any other groups of cats. Dosing volumes for subcutaneous administration varied from 5 ml/kg to 40 ml/kg. Dosing volumes for intracisternal injections varied from 0.1 – 1 ml with 20% HPβCD formulations.
Thirteen cohorts of cats were evaluated (Table 1) including normal control cats and untreated NPC cats. Three groups received HPβCD subcutaneously (SC) every 7 days beginning at 3 weeks of age by tenting a region of skin over the neck and injecting the solution into the subcutaneous space. Six groups received HPβCD intrathecally at the cerebellomedullary cistern (IC) every 14 days beginning at 3 weeks of age. One group received a combination of 1000 mg/kg HPβCD SC every 7 days and 120 mg HPβCD IC every 14 days beginning at 3 weeks of age. Finally, one group received 120 mg HPβCD IC every 14 days beginning at 16 weeks of age. All intracisternal dosing and CSF collected was performed in cats anesthetized with propofol (up to 6 mg/kg intravenously; Abbott Laboratories, Chicago, IL).
Blood and cerebrospinal fluid collection
Approximately 3 ml of blood was collected from either the cephalic or jugular vein at 8, 16 and 24 weeks of age to perform a complete blood count and serum chemistry analysis. Approximately 1 ml of cerebrospinal fluid (CSF) was collected from the cerebellomedullary cistern at the time of each dosing, and every two weeks from untreated NPC cats. Remaining serum and CSF were frozen at −80°C for biomarker studies
Histologic analysis
For pathological evaluations, cats were sacrificed at ~24 weeks of age, an age at which untreated NPC cats can no longer remain sternal and are euthanized. A number of treated, affected cats were also observed for a longer period of time to assess the efficacy of HPβCD; these cats were euthanized when they were no longer able to remain sternal or when they reached 76 weeks of age. Euthanasia was performed using an overdose of intravenous barbiturate. Immediately prior to euthanasia, cats were given an intravenous dose of 200 U heparin to prevent clotting during the tissue harvest. After sacrifice, animals were perfused through the left ventricle with 750 mL of 0.9% cold saline and samples of brain, liver, and lung were acquired and flash frozen. After perfusion, tissue samples were collected, sectioned, and dropped-fixed in 4% paraformaldehyde for 48 hours. Transverse sections of brain at the level of the caudate nucleus and cerebellar nuclei tissue were placed in cold 0.1 M phosphate buffer and shipped overnight to the Walkley laboratory for processing. These sections were dissected into selected blocks, cut on a vibratome (35 μm) and processed to visualize cholesterol storage (via filipin labeling) and ganglioside storage (via immunohistochemistry or immunofluorescence) using methods previously described (19, 73). The remainder of the brain as well as tissue from other organs were paraffin-embedded, sectioned at 5 μm and stained with hematoxylin/eosin (HE). For immunohistochemistry sections on charged slides were deparaffinized through xylenes and graded ethanols and antigen retrieval was performed in a microwave using Antigen Retrieval Citra Solution (BioGenex, Fremont, CA). Purkinje cells were identified using rabbit anti-calbindin (Swant, Switzerland) diluted 1:3,000 and granule cells (used as a reference) were identified using mouse anti-NeuN (Millipore #MAB377, Billerica, MA) diluted 1:500 in Antibody Diluent Reagent Solution (Invitrogen, Grand Island, NY). After a 12 hour incubation at 4°C, unbound primary antibodies were washed off and the sections incubated for 30 minutes at 37°C with Alexafluor 568 and 488-conjugated secondary antibodies (Invitrogen, Grand Island, NY). Two fields from each specimen were imaged at 2.5x on a Leitz DM RBE fluorescence microscope equipped with a QImaging Retiga-2000DC CCD camera (Surrey BC, Canada) controlled by iVision-Mac image acquisition and analysis software (Biovision Technologies, Exton, PA). Images were analyzed using iVision-Mac. Calbindin-positive Purkinje cells were counted for each specimen and a line was then drawn along the Purkinje cell layer to determine a total length in pixels. The total number of labeled cells per specimen divided by the total length generated a labeling index for the specimen. The myelin basic protein (MBP) antibody used was a mouse anti MBP from Abcam (Cambridge, MA #ab24567). The LAMP1 antibody used was a rabbit polyclonal from Abcam (Cambridge, MA #ab24170)
Biochemical analysis of lipids
Lipid studies on dissected cerebral cortex, cerebellum, and liver were conducted on frozen tissues following exactly the same procedures as described in a recent study on NPC cats treated with miglustat (20). Part of the control biological material (untreated NPC cats and normal cats) could thus be shared in both studies. The methodology has been described earlier in more detail (20, 30, 74, 75). Isolation of gangliosides from total lipid extracts was performed by reverse-phase chromatography on Bond-Elute C18 100 mg columns. Silica gel high performance thin-layer plates were from Merck (Darmstadt); densitometry of the plates at 580 nm was performed using a Camag TLC II scanner with CATS software. In the present study, individual ganglioside concentrations were not expressed in nmol/g wet weight as in previous publications, but as percentage of total gangliosides. This mode of expression was elected because it was independent of potential dehydration of the tissue samples during storage, and thus reduced methodological variation when comparing the effect of different doses of HPβCD.
Bioanalytical and Pharmacokinetic (PK) analyses of HPβCD
Plasma and CSF HPβCD were quantified using a RP-UPLC-MS/MS method (72). PK of the plasma and CSF concentrations of HPβCD was performed to determine the concentration at zero time (C0 = Cmax) by the extrapolation of drug concentration back to the time of dosing the area under the plasma concentration versus time curve (AUC0-∞ and AUC0–24h), terminal half-life (t½), plasma clearance (CL) and the apparent volume of distribution at steady state (Vdss) for IV (CSF only) and IT dose groups using the WinNonlin Version 5.3 (Pharsight) program.
Statistics
Mean values and standard deviations were calculated to describe the findings. The unpaired 2-tailed t-test was used to compare data from treated cats to both wild type and normal cats. Values were considered statistically significant at the p< 0.05 level. P-values are provided where significant differences exist.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Accessible Summary.
Cyclodextrin prevents brain degeneration
Niemann-Pick type C1 (NPC) disease is a severe hereditary nervous system disorder associated with the storage of cholesterol and other lipids inside nervous tissue. Injection of the pharmaceutical excipient cyclodextrin into the spinal fluid of cats with naturally occurring NPC disease prevented lipids from accumulating and prevented nervous system disease from developing. The only side effect found was a loss of hearing acuity associated with therapy. Together, these studies in the cat model have provided critical data on efficacy and safety of cyclodextrin administration directly into the spinal fluid that will be important for advancing this drug into clinical trials.
Acknowledgments
Data reported in the manuscript are archived with the FDA in the active IND as well as in the corresponding author’s laboratory at the University of Pennsylvania. The authors would like to thank X. Jiang for developing the very sensitive 2D-LC-IF-MS/MS analytical method that enabled quantification of HPβCD in plasma and CSF samples (72). We would also like to thank John Dietschy and Synthia Mellon for critical conversations on experimental design and Leslie King for her excellent assistance in writing this manuscript.
Funding:
This work was supported by the following grants: NIH R01-NS073661 (CHV), P40-02512 (CHV), Ara Parseghian Medical Research Foundation (CHV), Dana’s Angels Research Trust (CHV, DSO, SUW), Race for Adam (CHV), National Niemann Pick Disease Foundation (CHV), Support Of Accelerated Research for Niemann Pick Type C disease (SOAR-NPC; P30 HD071593) (CHV, DSO SUW), R01 NS081985 (DSO), P30 DK020579 (DSO); the German Research Foundation (DFG-STE 1069/2-1) (VMS); R01 NS053677 (SUW).
Footnotes
Author contributions: Each author contributed directly to the planning, collection, and analyses of data in this manuscript. Specfically, CH Vite and members of his lab (JH Bagel, GP Swain, M Prociuk, TU Sikora, VM Stein, P O’Donnell, T Ruane, A Crooks, S Li, and E Mauldin) were responsible for experimental planning and data analyses; caring for cats and administering cyclodextrin; planning and collecting all clinical and electrodiagnostic data; collecting, processing, and evaluating histological samples from cats; and for writing the manuscript. S. Stellar, M. de Meulder, and M.L. Kao were responsible for planning and analyses of all the pharmacokinetic data obtained for the cyclodextrin study. DS Ory, C Davidson, and S Walkley were involved in experimental planning and data analyses, and performed the filipin and ganglioside staining of cat tissue. M. Vanier performed all biochemical tissue lipid analyses.
Competing Interests:
Vite and Ory have received honoraria from Actelion and Biomarin.
Vanier has received travel expenses, consulting fees and presentation honoraria from Actelion Pharmaceuticals, Ltd.
Patents: Ory – Disease specific biomarkers for Niemann-Pick C disease (US Patent 8,497,122
Walkley – Methods for therapeutic use of glucosylceramide synthesis inhibitors and composition thereof (US Patent 6,683,076)
References
- 1.Pentchev PG, Comly ME, Kruth HS, Vanier MT, Wenger DA, Patel S, Brady RO. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc Natl Acad Sci U S A. 1985;82:8247–8251. doi: 10.1073/pnas.82.23.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea E, Neufeld EB, Blanchette-Mackie EJ, Pentchev PG. In: Metabolic and Molecular Bases of Inherited Disease. Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, editors. McGraw-Hill; NY: 2001. [Google Scholar]
- 4.Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, Infante RE. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci U S A. 2008;105:15287–15292. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walkley SU. Pyramidal neurons with ectopic dendrites in storage diseases exhibit increased GM2 ganglioside immunoreactivity. Neuroscience. 1995;68:1027–1035. doi: 10.1016/0306-4522(95)00208-z. [DOI] [PubMed] [Google Scholar]
- 7.Vanier MT, Millat G. Niemann-Pick disease type C. Clin Genet. 2003;64:269–281. doi: 10.1034/j.1399-0004.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- 8.Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol. 2001;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- 9.Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta. 2004;1685:48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 10.Peake KB, Vance JE. Defective cholesterol trafficking in Niemann-Pick C-deficient cells. FEBS letters. 2010;584:2731–2739. doi: 10.1016/j.febslet.2010.04.047. [DOI] [PubMed] [Google Scholar]
- 11.Wojtanik KM, Liscum L. The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J Biol Chem. 2003;278:14850–14856. doi: 10.1074/jbc.M300488200. [DOI] [PubMed] [Google Scholar]
- 12.Liscum L, Ruggiero RM, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is defective in Niemann-Pick type C fibroblasts. J Cell Biol. 1989;108:1625–1636. doi: 10.1083/jcb.108.5.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vance JE, Peake KB. Function of the Niemann-Pick type C proteins and their bypass by cyclodextrin. Curr Opin Lipidol. 2011;22:204–209. doi: 10.1097/MOL.0b013e3283453e69. [DOI] [PubMed] [Google Scholar]
- 14.Ory DS. Niemann-Pick type C: a disorder of cellular cholesterol trafficking. Biochim Biophys Acta. 2000;1529:331–339. doi: 10.1016/s1388-1981(00)00158-x. [DOI] [PubMed] [Google Scholar]
- 15.Vance JE, Karten B. Niemann-Pick C Disease and Mobilization of Lysosomal Cholesterol by Cyclodextrin. J Lipid Res. 2014;55:1609–1621. doi: 10.1194/jlr.R047837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patterson MC, Mengel E, Wijburg FA, Muller A, Schwierin B, Drevon H, Vanier MT, Pineda M. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J Rare Dis. 2013;8:12. doi: 10.1186/1750-1172-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braak H, Braak E, Goebel HH. Isocortical pathology in type C Niemann-Pick disease. A combined Golgi-pigmentoarchitectonic study. J Neuropathol Exp Neurol. 1983;42:671–687. doi: 10.1097/00005072-198311000-00007. [DOI] [PubMed] [Google Scholar]
- 18.Elleder M, Jirasek A, Smid F, Ledvinova J, Besley GT. Niemann-Pick disease type C. Study on the nature of the cerebral storage process. Acta Neuropathol. 1985;66:325–336. doi: 10.1007/BF00690966. [DOI] [PubMed] [Google Scholar]
- 19.Zervas M, Somers KL, Thrall MA, Walkley SU. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr Biol. 2001;11:1283–1287. doi: 10.1016/s0960-9822(01)00396-7. [DOI] [PubMed] [Google Scholar]
- 20.Stein VM, Crooks A, Ding W, Prociuk M, O’Donnell P, Bryan C, Sikora T, Dingemanse J, Vanier MT, Walkley SU, Vite CH. Miglustat improves purkinje cell survival and alters microglial phenotype in feline Niemann-Pick disease type C. J Neuropathol Exp Neurol. 2012;71:434–448. doi: 10.1097/NEN.0b013e31825414a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wraith JE, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, Giorgino R, Patterson MC. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Mol Genet Metab. 2010;99:351–357. doi: 10.1016/j.ymgme.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 22.Walterfang M, Chien YH, Imrie J, Rushton D, Schubiger D, Patterson MC. Dysphagia as a risk factor for mortality in Niemann-Pick disease type C: systematic literature review and evidence from studies with miglustat. Orphanet journal of rare diseases. 2012;7:76. doi: 10.1186/1750-1172-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patterson MC, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, Giorgino R, Wraith JE. Long-term miglustat therapy in children with Niemann-Pick disease type C. J Child Neurol. 2010;25:300–305. doi: 10.1177/0883073809344222. [DOI] [PubMed] [Google Scholar]
- 24.Patterson MC, Di Bisceglie AM, Higgins JJ, Abel RB, Schiffmann R, Parker CC, Argoff CE, Grewal RP, Yu K, Pentchev PG, et al. The effect of cholesterol-lowering agents on hepatic and plasma cholesterol in Niemann-Pick disease type C. Neurology. 1993;43:61–64. doi: 10.1212/wnl.43.1_part_1.61. [DOI] [PubMed] [Google Scholar]
- 25.Somers KL, Brown DE, Fulton R, Schultheiss PC, Hamar D, Smith MO, Allison R, Connally HE, Just C, Mitchell TW, Wenger DA, Thrall MA. Effects of dietary cholesterol restriction in a feline model of Niemann-Pick type C disease. J Inherit Metab Dis. 2001;24:427–436. doi: 10.1023/a:1010588112003. [DOI] [PubMed] [Google Scholar]
- 26.Camargo F, Erickson RP, Garver WS, Hossain GS, Carbone PN, Heidenreich RA, Blanchard J. Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci. 2001;70:131–142. doi: 10.1016/s0024-3205(01)01384-4. [DOI] [PubMed] [Google Scholar]
- 27.Kilsdonk EP, Yancey PG, Stoudt GW, Bangerter FW, Johnson WJ, Phillips MC, Rothblat GH. Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem. 1995;270:17250–17256. doi: 10.1074/jbc.270.29.17250. [DOI] [PubMed] [Google Scholar]
- 28.Atger VM, de la Llera Moya M, Stoudt GW, Rodrigueza WV, Phillips MC, Rothblat GH. Cyclodextrins as catalysts for the removal of cholesterol from macrophage foam cells. J Clin Invest. 1997;99:773–780. doi: 10.1172/JCI119223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu SM, Cogny A, Kockx M, Dean RT, Gaus K, Jessup W, Kritharides L. Cyclodextrins differentially mobilize free and esterified cholesterol from primary human foam cell macrophages. J Lipid Res. 2003;44:1156–1166. doi: 10.1194/jlr.M200464-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, Ory DS, Vanier MT, Walkley SU. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One. 2009;4:e6951. doi: 10.1371/journal.pone.0006951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu B, Turley SD, Burns DK, Miller AM, Repa JJ, Dietschy JM. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse. Proc Natl Acad Sci U S A. 2009;106:2377–2382. doi: 10.1073/pnas.0810895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pontikis CC, Davidson CD, Walkley SU, Platt FM, Begley DJ. Cyclodextrin alleviates neuronal storage of cholesterol in Niemann-Pick C disease without evidence of detectable blood-brain barrier permeability. J Inherit Metab Dis. 2013;36:491–498. doi: 10.1007/s10545-012-9583-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Somers KL, Royals MA, Carstea ED, Rafi MA, Wenger DA, Thrall MA. Mutation analysis of feline Niemann-Pick C1 disease. Mol Genet Metab. 2003;79:99–103. doi: 10.1016/s1096-7192(03)00074-x. [DOI] [PubMed] [Google Scholar]
- 34.Vite CH, Ding W, Bryan C, O’Donnell P, Cullen K, Aleman D, Haskins ME, Van Winkle T. Clinical, electrophysiological, and serum biochemical measures of progressive neurological and hepatic dysfunction in feline Niemann-Pick type C disease. Pediatr Res. 2008;64:544–549. doi: 10.1203/PDR.0b013e318184d2ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porter FD, Scherrer DE, Lanier MH, Langmade SJ, Molugu V, Gale SE, Olzeski D, Sidhu R, Dietzen DJ, Fu R, Wassif CA, Yanjanin NM, Marso SP, House J, Vite C, Schaffer JE, Ory DS. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci Transl Med. 2010;2:56ra81. doi: 10.1126/scitranslmed.3001417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ward S, O’Donnell P, Fernandez S, Vite CH. 2-hydroxypropyl-beta-cyclodextrin raises hearing threshold in normal cats and in cats with Niemann-Pick type C disease. Pediatr Res. 2010;68:52–56. doi: 10.1203/PDR.0b013e3181df4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan M, Sidhu R, Fujiwara H, Tortelli B, Zhang J, Davidson C, Walkley SU, Bagel JH, Vite C, Yanjanin NM, Porter FD, Schaffer JE, Ory DS. Identification of Niemann-Pick C1 disease biomarkers through sphingolipid profiling. J Lipid Res. 2013;54:2800–2814. doi: 10.1194/jlr.M040618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brinkmalm G, Portelius E, Ohrfelt A, Mattsson N, Persson R, Gustavsson MK, Vite CH, Gobom J, Mansson JE, Nilsson J, Halim A, Larson G, Ruetschi U, Zetterberg H, Blennow K, Brinkmalm A. An online nano-LC-ESI-FTICR-MS method for comprehensive characterization of endogenous fragments from amyloid beta and amyloid precursor protein in human and cat cerebrospinal fluid. J Mass Spectrom. 47:591–603. doi: 10.1002/jms.2987. [DOI] [PubMed] [Google Scholar]
- 39.Mattsson N, Olsson M, Gustavsson MK, Kosicek M, Malnar M, Mansson JE, Blomqvist M, Gobom J, Andreasson U, Brinkmalm G, Vite C, Hecimovic S, Hastings C, Blennow K, Zetterberg H, Portelius E. Amyloid-beta metabolism in Niemann-Pick C disease models and patients. Metab Brain Dis. 27:573–585. doi: 10.1007/s11011-012-9332-8. [DOI] [PubMed] [Google Scholar]
- 40.Tortelli B, Fujiwara H, Bagel JH, Zhang J, Sidhu R, Jiang X, Yanjanin N, Shankar RK, Carillo-Carasco N, Heiss J, Ottinger EA, Porter FD, Schaffer JE, Vite CH, Ory DS. Cholesterol homeostatic responses provide biomarkers for monitoring treatment for the neurodegenerative disease Niemann-Pick C1 (NPC1) Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu331. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ottinger EA, Kao ML, Carrillo-Carrasco N, et al. Collaborative development of 2-hydroxypropyl-beta-cyclodextrin for the treatment of Niemann-Pick type C1 disease. Curr Topics Med Chem. 2014;14:1–10. doi: 10.2174/1568026613666131127160118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maue RA, Burgess RW, Wang B, Wooley CM, Seburn KL, Vanier MT, Rogers MA, Chang CC, Chang TY, Harris BT, Graber DJ, Penatti CA, Porter DM, Szwergold BS, Henderson LP, Totenhagen JW, Trouard TP, Borbon IA, Erickson RP. A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum Mol Genet. 2012;21:730–750. doi: 10.1093/hmg/ddr505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat Med. 2004;10:704–711. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- 44.Liu B, Li H, Repa JJ, Turley SD, Dietschy JM. Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J Lipid Res. 2008;49:663–669. doi: 10.1194/jlr.M700525-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Pitha J, Gerloczy A, Olivi A. Parenteral hydroxypropyl cyclodextrins: intravenous and intracerebral administration of lipophiles. Journal of pharmaceutical sciences. 1994;83:833–837. doi: 10.1002/jps.2600830615. [DOI] [PubMed] [Google Scholar]
- 46.Irie T, Uekama K. Pharmaceutical applications of cyclodextrins. III. Toxicological issues and safety evaluation. Journal of pharmaceutical sciences. 1997;86:147–162. doi: 10.1021/js960213f. [DOI] [PubMed] [Google Scholar]
- 47.Rajewski RA, Stella VJ. Pharmaceutical applications of cyclodextrins. 2. In vivo drug delivery. Journal of pharmaceutical sciences. 1996;85:1142–1169. doi: 10.1021/js960075u. [DOI] [PubMed] [Google Scholar]
- 48.Gould S, Scott RC. 2-Hydroxypropyl-beta-cyclodextrin (HP-beta-CD): a toxicology review. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association. 2005;43:1451–1459. doi: 10.1016/j.fct.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Zhao C-z, Fang X-c, Wang D, Tang F-d, Wang X-d. Involvement of type II pneumocytes in the pathogenesis of chronic obstructive pulmonary disease. Respir Med. 2010;104:1391–1395. doi: 10.1016/j.rmed.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 50.Meiner V, Shpitzen S, Mandel H, Klar A, Ben-Neriah Z, Zlotogora J, Sagi M, Lossos A, Bargal R, Sury V, Carmi R, Leitersdorf E, Zeigler M. Clinical-biochemical correlation in molecularly characterized patients with Niemann-Pick type C. Genet Med. 2001;3:343–348. doi: 10.1097/00125817-200109000-00003. [DOI] [PubMed] [Google Scholar]
- 51.Millat G, Chikh K, Naureckiene S, Sleat DE, Fensom AH, Higaki K, Elleder M, Lobel P, Vanier MT. Niemann-Pick disease type C: spectrum of HE1 mutations and genotype/phenotype correlations in the NPC2 group. Am J Hum Genet. 2001;69:1013–1021. doi: 10.1086/324068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pin I, Pradines S, Pincemaille O, Frappat P, Brambilla E, Vanier MT, Bost M. A fatal respiratory form of type C Niemann-Pick disease. Arch Fr Pediatr. 1990;47:373–375. [PubMed] [Google Scholar]
- 53.Gonzalez de Dios J, Fernandez Tejada E, Diaz Fernandez MC, Ortega Paez E, Hernandez Gonzalez J, de la Vega Bueno A, Hierro Llanillo L, Larrauri Martinez J, Jara Vega P. The current state of Niemann-Pick disease: evaluation of six cases. An Esp Pediatr. 1990;32:143–148. [PubMed] [Google Scholar]
- 54.Schofer O, Mischo B, Puschel W, Harzer K, Vanier MT. Early-lethal pulmonary form of Niemann-Pick type C disease belonging to a second, rare genetic complementation group. Eur J Pediatr. 1998;157:45–49. doi: 10.1007/s004310050764. [DOI] [PubMed] [Google Scholar]
- 55.Roszell BR, Tao JQ, Yu KJ, Gao L, Huang S, Ning Y, Feinstein SI, Vite CH, Bates SR. Pulmonary abnormalities in animal models due to Niemann-Pick type C1 (NPC1) or C2 (NPC2) disease. PLoS One. 2013;8:e67084. doi: 10.1371/journal.pone.0067084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu B, Ramirez CM, Miller AM, Repa JJ, Turley SD, Dietschy JM. Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J Lipid Res. 2010;51:933–944. doi: 10.1194/jlr.M000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramirez CM, Liu B, Taylor AM, Repa JJ, Burns DK, Weinberg AG, Turley SD, Dietschy JM. Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann-Pick type C1 mouse and markedly prolongs life. Pediatr Res. 2010;68:309–315. doi: 10.1203/PDR.0b013e3181ee4dd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muralidhar A, Borbon IA, Esharif DM, Ke W, Manacheril R, Daines M, Erickson RP. Pulmonary function and pathology in hydroxypropyl-beta-cyclodextin-treated and untreated Npc1(−)/(−) mice. Mol Genet Metab. 2011;103:142–147. doi: 10.1016/j.ymgme.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seyfert S, Faulstich A, Marx P. What determines the CSF concentrations of albumin and plasma-derived IgG? Journal of the neurological sciences. 2004;219:31–33. doi: 10.1016/j.jns.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 60.Cutler RW, Deuel RK, Barlow CF. Albumin exchange between plasma and cerebrospinal fluid. Arch Neurol. 1967;17:261–270. doi: 10.1001/archneur.1967.00470270039006. [DOI] [PubMed] [Google Scholar]
- 61.Begley DJ. Brain superhighways. Sci Transl Med. 2012;4:147fs129. doi: 10.1126/scitranslmed.3004611. [DOI] [PubMed] [Google Scholar]
- 62.Taylor AM, Liu B, Mari Y, Liu B, Repa JJ. Cyclodextrin mediates rapid changes in lipid balance in Npc1−/− mice without carrying cholesterol through the bloodstream. J Lipid Res. 2012;53:2331–2342. doi: 10.1194/jlr.M028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crumling MA, Liu L, Thomas PV, Benson J, Kanicki A, Kabara L, Halsey K, Dolan D, Duncan RK. Hearing loss and hair cell death in mice given the cholesterol-chelating agent hydroxypropyl-beta-cyclodextrin. PLoS One. 2012;7:e53280. doi: 10.1371/journal.pone.0053280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peake KB, Vance JE. Normalization of cholesterol homeostasis by 2-hydroxypropyl-beta-cyclodextrin in neurons and glia from Niemann-Pick C1 (NPC1)-deficient mice. J Biol Chem. 2012;287:9290–9298. doi: 10.1074/jbc.M111.326405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aqul A, Liu B, Ramirez CM, Pieper AA, Estill SJ, Burns DK, Repa JJ, Turley SD, Dietschy JM. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J Neurosci. 31:9404–9413. doi: 10.1523/JNEUROSCI.1317-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramirez CM, Liu B, Aqul A, Taylor AM, Repa JJ, Turley SD, Dietschy JM. Quantitative role of LAL, NPC2, and NPC1 in lysosomal cholesterol processing defined by genetic and pharmacological manipulations. J Lipid Res. 52:688–698. doi: 10.1194/jlr.M013789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Monnaert V, Tilloy S, Bricout H, Fenart L, Cecchelli R, Monflier E. Behavior of alpha-, beta-, and gamma-cyclodextrins and their derivatives on an in vitro model of blood-brain barrier. The Journal of pharmacology and experimental therapeutics. 2004;310:745–751. doi: 10.1124/jpet.104.067512. [DOI] [PubMed] [Google Scholar]
- 68.Gondre-Lewis MC, McGlynn R, Walkley SU. Cholesterol accumulation in NPC1-deficient neurons is ganglioside dependent. Curr Biol. 2003;13:1324–1329. doi: 10.1016/s0960-9822(03)00531-1. [DOI] [PubMed] [Google Scholar]
- 69.Zhou S, Davidson C, McGlynn R, Stephney G, Dobrenis K, Vanier MT, Walkley SU. Endosomal/lysosomal processing of gangliosides affects neuronal cholesterol sequestration in Niemann-Pick disease type C. Am J Pathol. 2011;179:890–902. doi: 10.1016/j.ajpath.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Song W, Wang F, Lotfi P, Sardiello M, Segatori L. 2-Hydroxypropyl-beta-cyclodextrin promotes transcription factor EB-mediated activation of autophagy: implications for therapy. J Biol Chem. 2014;289:10211–10222. doi: 10.1074/jbc.M113.506246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen FW, Li C, Ioannou YA. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS One. 2010;5:e15054. doi: 10.1371/journal.pone.0015054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jiang H, Sidhu R, Fujiwara H, De Meulder M, de Vries R, Gong Y, Kao M, Porter FD, Yanjanin NM, Carillo-Carasco N, Xu X, Ottinger E, Woolery M, Ory DS, Jiang X. Development and validation of sensitive LC-MS/MS assays for quantification of HP-beta-CD in human plasma and CSF. J Lipid Res. 2014;55:1537–1548. doi: 10.1194/jlr.D050278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Micsenyi MC, Dobrenis K, Stephney G, Pickel J, Vanier MT, Slaugenhaupt SA, Walkley SU. Neuropathology of the Mcoln1(−/−) knockout mouse model of mucolipidosis type IV. J Neuropathol Exp Neurol. 2009;68:125–135. doi: 10.1097/NEN.0b013e3181942cf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fujita N, Suzuki K, Vanier MT, Popko B, Maeda N, Klein A, Henseler M, Sandhoff K, Nakayasu H. Targeted disruption of the mouse sphingolipid activator protein gene: a complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum Mol Genet. 1996;5:711–725. doi: 10.1093/hmg/5.6.711. [DOI] [PubMed] [Google Scholar]
- 75.Sleat DE, Wiseman JA, El-Banna M, Price SM, Verot L, Shen MM, Tint GS, Vanier MT, Walkley SU, Lobel P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci U S A. 2004;101:5886–5891. doi: 10.1073/pnas.0308456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.