


Abstract
Automatically extracting protein names from the literature and linking these names to the associated entries in sequence databases is becoming increasingly important for annotating biological databases. NLProt is a novel system that combines dictionary- and rule-based filtering with several support vector machines (SVMs) to tag protein names in PubMed abstracts. When considering partially tagged names as errors, NLProt still reached a precision of 75% at a recall of 76%. By many criteria our system outperformed other tagging methods significantly; in particular, it proved very reliable even for novel names. Names encountered particularly frequently in Drosophila, such as white, wing and bizarre, constitute an obvious limitation of NLProt. Our method is available both as an Internet server and as a program for download (http://cubic.bioc.columbia.edu/services/NLProt/). Input can be PubMed/MEDLINE identifiers, authors, titles and journals, as well as collections of abstracts, or entire papers.
INTRODUCTION
Importance of protein name extraction. The amount of biological information that is produced and stored daily in the form of scientific articles continues to grow exponentially (1). Automatic text analysis mines this wealth of information quickly. One crucial task of automatic text mining is the extraction of named entities such as protein and gene names from natural language. This seemingly simple task is complicated by the largely missing nomenclature rules and by names for proteins and genes that resemble chemical compounds (Caeridin/Cantharidin), cell cultures (CD4+-cells/CD4 protein) and even non-scientific English words (white, wing, bizarre). To make things even more challenging for text miners, protein and gene names often resemble each other (myc-c gene/myc-c protein). Previously developed methods were reported to have reached F-measures (harmonic average between accuracy and coverage) (Equation 1) ∼70% (2–6). However, very few of these methods were tested on large, curated databases, and very few successfully identify novel names. We developed a novel method because one of our research problems required the extraction of protein sequences from over 3000 PubMed abstracts. We found only one publicly available method to solve this task and that one failed for our purposes. We imagine that our novel method may help others as much as it helped us to automatically create databases of e.g. protein–protein interactions, gene-regulation patterns, protein function and protein–disease associations.
Encoding textual information for machine-learning. A consecutive stretch of words such as a protein name can be represented as a vector in which each component codes for certain words of the name. Given a pre-defined word list with five words (A B C D E) and the hypothetical protein name ‘B C’, we can represent this name through a position-unspecific vector by simply counting the occurrences of each word in the name (one B and one C) {0,1,1,0,0}. We can also include information of position and order by reserving the first five slots (because the word list contains five words) for the first word in the name (B), and generally the nth five slots for the nth word. For our example, this position-specific vector is {0,1,0,0,0, 0,0,1,0,0}. The context of a named entity, i.e. the words surrounding the name, can be coded similarly. We encoded contextual information in position-specific vectors and used these to train the support vector machine (SVM) (7) building blocks of our system. Finally, we linked the differently trained SVM building blocks to reach maximal performance (Methods and Results).
Related work. Three basic approaches tag protein names, namely, rule-based (8–11), dictionary-based (3,4,12,13) and machine-learning methods (5,6,14–16). Fukuda et al. (9) suggested that even extremely simple rules can yield F∼96% when tailored to a certain subject, such as 30 SH3-domain-related articles. One rule-based system is Yapex (10), with an estimated F = 67%. Krauthammer et al. (3) developed a dictionary-based system that—after translating the input text into nucleotide sequences—uses BLAST (a fast sequence alignment method) (17) to find names in a database of protein names. The authors considered partial matches as correct (CD4 instead of CD4 kinase) and reported an F = 75% on a set of two review articles. Tsuruoka and Tsujii (4) used dynamic programming instead of BLAST. After additionally filtering high-scoring examples through a simple Bayesian classifier, they reported F = 70%. Hanisch et al. (13) used a semi-automatically generated dictionary in combination with a linear algorithm and reported an F = 93% without specifying the dataset used for this estimate. One problem with dictionary-based systems is the limitation to names that are present in the dictionary. Collier et al. (6) and Morgan et al. (5) reported levels of F = 73% and F = 75%, respectively, by using Hidden Markov Models. The only publicly available method currently is GAPSCORE (16), which uses a statistical model for gene and protein names by taking their appearance, morphology and context into account; it is reported to reach F = 58%.
METHODS AND RESULTS
We describe the details of the design and performance of NLProt elsewhere (S. Mika and B. Rost, manuscript submitted). Here, we summarize those aspects that might help users to optimally use our system.
Dictionaries. A dictionary with protein names can help in linking each name to an associated database identifier. UniProt and their pillars SWISS-PROT (a database of protein sequences) and TrEMBL (translation of the EMBL nucleotide database) (18,19) collect over one million protein sequences along with the most commonly used names of these proteins (DE field), and the names of related genes (GN field). We generated a list of all SWISS-PROT + TrEMBL protein and gene names and refer to this dictionary as the ‘protein dictionary’. A second, built-in dictionary is our ‘common dictionary’, which contains non-protein names and is based on the online-version of the Merriam-Webster (MW) dictionary (http://www.m-w.com). We expanded the common dictionary through medical terms (http://cancerweb.ncl.ac.uk/omd/), species names (http://us.expasy.org/cgi-bin/speclist), tissue types (http://us.expasy.org/cgi-bin/lists?tisslist.txt) and minerals/formulas (http://un2sg4.unige.ch/athena/mineral/min_lists.html). We used this common dictionary for pre-filtering text. We also derived a list of 130 typical endings (last 4 letters) for chemical names (http://www.speclab.com). We used this list by removing all words with identical endings (*hyde and *enyl).
Algorithm. The NLProt algorithm starts by slicing the input text into all possible samples of 9–13 consecutive tokens. We consider words, numbers and punctuation characters except for hyphens (-) and slashes (/) as tokens as long as they are separated from each other by spaces. A sample is characterized by holding 1–5 centre tokens (A–E in Figure 1) plus the preceding 4 tokens (1–4 in Figure 1) and the following 4 tokens (5–8 in Figure 1). The tokens 1–8 are together referred to as the ‘environment’. Note that the environment always contains 8 tokens, whereas the number of tokens in the centre of a sample can vary from 1 to 5. Each sample is then passed through a pre-filtering procedure (a complete list of filtering rules is on our website). The remaining samples are passed through three differently specialized SVMs that were trained on text represented as vectors (Introduction). SVM1 was trained on the central words, SVM2 on the environments and SVM3 on the overlap between the two. Each SVM independently produces a score. A fourth score results from searching for the central words in our protein name dictionary (= 0 if not found). The last SVM in our system (SVM4) is trained to find a ‘good compromise’ between these four initial scores. We used an off-the-shelf package to implement the SVMs [‘svm-light’ (20); http://www.joachims.org].
Figure 1.

Schema for text parsing. Two examples of text from a MEDLINE abstract; we considered the one on the top as ‘true positive’ and the one at the bottom as ‘true negative’ (no protein name). Letters and numbers in brackets label words. A text sample will only count as ‘true positive’ if the centre contains a complete protein name (no fragment). We divided samples (continuous words) into two parts: the environment (before the name, words 1–4, and after the name, words 5–8) and the centre (words A–E). Note that the environment always contains 8 tokens (words), whereas we varied the size of the centre from 1 to 5.
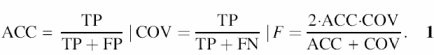
Performance measures. The commonly used measure for the performance of name-taggers is the harmonic mean of accuracy and coverage, called the F-measure (Equation 1). Given the number of true positives (TP), false positives (FP) and false negatives (FN), the values for accuracy, coverage and F-measure are defined through Equation 1:
Sustained high performance for many datasets. We tested our method on different corpora that were not tagged by us (data available from our website). Our program reached an F-measure of 75% on the Yapex corpus, which was tagged by Franzen et al. (10) for developing their protein name-tagger (Yapex). The Yapex tagger only reached an F-measure of 67% on the same corpus. For 10 000 sentences from the BioCreative competition (http://www.mitre.org/public/biocreative/), NLProt reached an F-score of 74%.
NLProt without dictionary and without redundancy in the datasets reached 60%. The necessity of using unbiased, redundancy-free datasets for evaluating predictors has become common sense in the field of protein and DNA sequence analysis. The problem was solved by finding measures for sequence-similarity, such as percentage sequence-identity (PIDE), BLAST E-values (17,21) and the HSSP (homology derived structure of proteins) value (22,23). Here, any protein name showing up in the training set, as well as in the test set, would mislead any evaluation of the results of a machine-learning-based name-tagger. However, this problem has never been addressed in any of the papers on named entity extraction. We reduced bias in the datasets using three rules. First, we ignored names identical between the testing and training sets (counted neither as true nor as false). Second, we ignored multiple occurrences of the same name if correctly identified (note that the same name may still contribute many times to our false-negative or false-positive count). Third, in order to test the success of our machine-learning component, we removed all names in the test set from our protein dictionary. Our system reached F = 60% on the Yapex corpus applying these bias-reduction rules. Note that, by definition, all dictionary-based methods would yield F = 0% on this test! This lower limit of F = 60% was striking given that it applied for situations in which all the names correctly identified were neither in the dictionary nor in the training set; i.e. the system had learned to correctly discover names it had never encountered. The only two papers that report results on data entirely unknown to the system are a Hidden Markov Model (HMM)-based tagger reaching F = 33% (5) and the system combining dictionaries with BLAST searches reaching F = 4% for unknown samples (3).
INPUT, OUTPUT AND OPTIONS
Input. Our server accepts practically any natural language text in ASCII (American Standard Code for Information Interchange) format, i.e. simple text without formatting. For example, the user can cut-and-paste from any word document or web browser into the input field of our server (Figure 2). NLProt will perform at its best only if the user feeds it with full sentences from typical scientific publications (text has to be in English). This is because the building blocks of NLProt were trained within the context of protein names in PubMed abstracts. The online version of NLProt accepts at most 50 000 characters as input text (this corresponds to 2–3 papers or 20–40 abstracts). However, there are no restrictions on the input size when using the command-line version of NLProt. Our server can also be queried with a list of PubMed/MEDLINE identifiers. In such a case, it will retrieve the corresponding abstracts from PubMed/MEDLINE and identify the protein names/sequences in these. A third input option is to use our PubMed/MEDLINE search form from the NLProt website. The program will then present a list with the search results so that users can pick from this list those abstracts in which they want to tag protein names.
Figure 2.

Screen-shot of NLProt submission webpage. In the upper part, users choose output and processing options (output formats and databases to search). The middle part shows a field for ASCII text-input (natural language text or PubMed identifiers). As an alternative to these two types of input, the user may submit requests directly through a PubMed search form (lower part). NLProt needs to know which part of the input form to use (radio buttons ‘input type’).
Options and output. By default, the program shows only the most likely database identifier (from SWISS-PROT or TrEMBL) for each name. In this mode, NLProt additionally scans each name found for surrounding words indicating a certain organism and gives only the protein identifier that fits both the protein and the species. However, users can opt to display alternative, sub-optimal solutions, and they can also specify a database (currently SWISS-PROT, TrEMBL or both). A third option is the output format [either plain text or HTML (Hyper Text Markup Language)-formatted files]. The output file contains the tagged input text and a list of all names identified by NLProt. If plain text is chosen, the 〈n〉 tag indicates protein names and the 〈/n〉 tag terminates a given name. In HTML-formatted output, names are indicated in red. For both output formats, a list is generated which shows all the protein names found sorted by their position in the text. This list contains information about the final score (SVM4) of each name identified, the corresponding organism, its exact text position and the database identifier(s) that could be found in association with this name. Finally, users can opt to receive a second file that contains all sequences in FASTA format.
Download NLProt to run on your machine. NLProt is available as a command-line tool for Windows (DOS) and LINUX machines. The programs are downloadable from our website as zip files and simply need to be unpacked and installed on a local machine by following simple steps, which are described in the included README.txt files. Note that NLProt is rather fast: tagging one PubMed abstract required ∼1–2 s, on average, when applying the system to 380 000 PubMed abstracts.
CONCLUSIONS
NLProt constitutes very novel territory for the research activities in our group. The features of the web server are heavily biased by what we would have liked to do with such a method when we looked for it. Our experience with this web server will determine how future versions will look.
Acknowledgments
ACKNOWLEDGEMENTS
Thanks to Jinfeng Liu and Megan Restuccia (Columbia) for computer assistance. Thanks to Amos Bairoch (SIB, Geneva), Rolf Apweiler (EBI, Hinxton), Phil Bourne (San Diego University) and their crews for maintaining excellent databases and to all experimentalists who enabled this tool by making their data publicly available. This work was supported by the grants R01-GM63029-01 and R01-GM64633-01 from the National Institute of Health (NIH) and 1-R01-LM07329-01 from the National Library of Medicine (NLM).
REFERENCES
- 1.Wheeler D.L., Church,D.M., Edgar,R., Federhen,S., Helmberg,W., Madden,T.L., Pontius,J.U., Schuler,G.D., Schriml,L.M., Sequeira,E. et al. (2004) Database resources of the National Center for Biotechnology Information: update. Nucleic Acids Res., 32, D35–D40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hou W. and Chen,H. (2003) Enhancing performance of protein name recognizers using collocation. ACL-03 Workshop on Natural Language Processing in Biomedicine, Sapparo, Japan, pp. 25–32. [Google Scholar]
- 3.Krauthammer M., Rzhetsky,A., Morozov,P. and Friedman,C. (2000) Using BLAST for identifying gene and protein names in journal articles. Gene, 259, 245–252. [DOI] [PubMed] [Google Scholar]
- 4.Tsuruoka Y. and Tsujii,J. (2003) Boosting precision and recall of dictionary-based protein name recognition. ACL-03 Workshop on Natural Language Processing in Biomedicine, Sapparo, Japan, pp. 41–48. [Google Scholar]
- 5.Morgan A., Hirschman,L., Yeh,A. and Colosimo,M. (2003) Gene name extraction using FlyBase resources. ACL-03 Workshop on Natural Language Processing in Biomedicine, Sapparo, Japan, pp. 1–8. [Google Scholar]
- 6.Collier N., Nobata,C. and Tsujii,J. (2000) Extracting the names of genes and gene products with a Hidden Markov Model. Proc. COLING 2000, 201–207. [Google Scholar]
- 7.Cortes C. and Vapnik,V. (1995) Support Vector Networks. Mach. Learn., 20, 273–297. [Google Scholar]
- 8.Proux D., Rechenmann,F., Julliard,L., Pillet,V.V. and Jacq,B. (1998) Detecting gene symbols and names in biological texts: A first step toward pertinent information extraction. Genome Inform. Ser. Workshop Genome Inform., 9, 72–80. [PubMed] [Google Scholar]
- 9.Fukuda K., Tsunoda,T., Tamura,A. and Takagi,T. (1998) Toward information extraction: identifying protein names from biological papers. Pac. Symp. Biocomput., 707–718. [PubMed] [Google Scholar]
- 10.Franzen K., Eriksson,G., Olsson,F., Asker,L., Liden,P. and Cöster,J. (2002) Protein names and how to find them. Int. J. Med. Inf., 67, 49–61. [DOI] [PubMed] [Google Scholar]
- 11.Narayanaswamy M., Ravikumar,K.E. and Vijay-Shanker,K. (2003) A biological named entity recognizer. Pac. Symp. Biocomput., 427–438. [DOI] [PubMed] [Google Scholar]
- 12.Rindflesch T.C., Bean,C.A. and Sneiderman,C.A. (2000) Argument identification for arterial branching predications asserted in cardiac catheterization reports. Proc. AMIA Symp., 704–708. [PMC free article] [PubMed] [Google Scholar]
- 13.Hanisch D., Fluck,J., Mevissen,H. and Zimmer,R. (2003) Playing biology's name game: identifying protein names in scientific text. Pac. Symp. Biocomput., 403–414. [PubMed] [Google Scholar]
- 14.Tanabe L. and Wilbur,W.J. (2002) Tagging gene and protein names in biomedical text. Bioinformatics, 18, 1124–1132. [DOI] [PubMed] [Google Scholar]
- 15.Weinstein J.N., Scherf,U., Lee,J.K., Nishizuka,S., Gwadry,F., Bussey,A.K., Kim,S., Smith,L.H., Tanabe,L., Richman,S. et al. (2002) The bioinformatics of microarray gene expression profiling. Cytometry, 47, 46–49. [DOI] [PubMed] [Google Scholar]
- 16.Chang J., Schutze,H. and Altman,R. (2004) GAPSCORE: finding gene and protein names one word at a time. Bioinformatics, 20, 216–225. [DOI] [PubMed] [Google Scholar]
- 17.Altschul S., Madden,T., Shaffer,A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D. (1997) Gapped Blast and PSI-Blast: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bairoch A. and Apweiler,R. (2000) The Swiss-Prot protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res., 28, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apweiler R., Bairoch,A., Wu,C.H., Barker,W.C., Boeckmann,B., Ferro,S., Gasteiger,E., Huang,H., Lopez,R., Magrane,M. et al. (2004) UniProt: the Universal Protein knowledgebase. Nucleic Acids Res., 32, D115–D119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joachims T. (1999) Making large-scale Support Vector Machine learning practical. In Schoelkopf,B., Burges,C.J.C. and Smola,A.J. (eds), Advances in Kernel Methods—Support Vector Learning. MIT-Press, Cambridge MA, pp. 169–184. [Google Scholar]
- 21.Altschul S. and Gish,W. (1996) Local alignment statistics. Methods Enzymol., 266, 460–480. [DOI] [PubMed] [Google Scholar]
- 22.Sander C. and Schneider,R. (1991) Database of homology-derived structures and the structural meaning of sequence alignments. Proteins, 9, 56–68. [DOI] [PubMed] [Google Scholar]
- 23.Mika S. and Rost,B. (2003) UniqueProt: creating representative protein sequence sets. Nucleic Acids Res., 31, 3789–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]