

Abstract
Omics technologies enable unbiased investigation of biological systems through massively parallel sequence acquisition or molecular measurements, bringing the life sciences into the era of Big Data. A central challenge posed by such omics datasets is how to transform these data into biological knowledge, for example, how to use these data to answer questions such as: Which functional pathways are involved in cell differentiation? Which genes should we target to stop cancer? Network analysis is a powerful and general approach to solve this problem consisting of two fundamental stages, network reconstruction, and network interrogation. Here we provide an overview of network analysis including a step-by-step guide on how to perform and use this approach to investigate a biological question. In this guide, we also include the software packages that we and others employ for each of the steps of a network analysis workflow.
Keywords: network reconstruction, network interrogation, systems biology, big data, data integration, inter-omics network, transkingdom network
Introduction
In saying that we understand a biological process, we usually mean that we are able to predict future events and manipulate the process into a desired direction. Thus, biological inquiry could be viewed as an attempt to understand how a biological system transits from one state to another. Such transitions underlie a wide range of biological phenomena from cell differentiation to recovery from disease. In attempting to understand these transitions, a simple and frequently used approach is to compare two states of a system (eg, before and after stimulus, with and without mutation, or healthy and diseased). Although more sophisticated approaches with time-series data, dose-effect data, or three or more sample groups can be also used, here we discuss analysis of data from a two-class study design. Furthermore, most of the methods that we describe can, with slight modifications, be used for other study designs. Today, omics technologies enable unbiased investigation of biological systems through massively parallel sequence acquisition or molecular measurements, bringing the life sciences into the era of Big Data. A central challenge posed by such omics datasets is how to navigate through the haystack of measurements (eg, differential expression between two states) to identify the needles comprised of the critical causal factors.
Network analysis is a powerful and general approach to this problem, in which the biological system is modeled as a network whose nodes represent dynamical units (eg, genes, proteins, metabolites, etc) and edges stand for links between them. Network analysis consists of two fundamental stages: network reconstruction and network interrogation. For omics molecular measurements such as gene expression, a particular type of network analysis called covariation network analysis has become a dominant approach. In such networks, a node represents the expression of the gene being measured, and an edge indicates that the expressions of two genes are correlated. Multiple groups including ours have been successfully using such methods to gain a systems-level understanding of biological processes and to reveal mechanisms of different diseases.1–3 Several recent discoveries ranging from genes that drive progression of different cancers4,5 to microbes and microbial genes that cause a human illness6 became possible because of the predictive power of network analysis. In particular, such insights would be very difficult to achieve if analysis is limited to finding differentially expressed genes and follow-up data mining of those genes. Due to the rapid pace of evolution of techniques and omics technologies, the practical application of network analysis has usually required a dedicated computational biologist. This requirement has limited the extent to which the larger biological sciences community has benefited from network analysis. Here we provide an overview of covariation network reconstruction and interrogation, including a step-by-step guide on how to perform and use network analysis to investigate a biological question (Fig. 1). In this guide, we include the software packages that we employ (and specific pointers to the methods or software used by other groups) for each of the steps of a model network analysis workflow. Although in this guide we mostly focus on covariation networks, the analysis steps related to network interrogation are applicable to other types of networks such as semantic networks or molecular interaction networks.
Figure 1.

Workflow of network analysis. (A) Network analysis starts from data obtained from high-throughput experiments such as microarray experiments detecting expression of genes in samples. (B) Differentially expressed genes are found between two states of a system (eg, normal vs disease). (C) Correlations of DEGs based on their expression values are calculated to detect regulatory relationship among them. (D) Significant correlations suggest connections between differentially expressed genes (DEGs) and are used to generate a network of DEGs. (E) Network interrogation is performed to detect modules, key regulators, and functional pathways that are important for state transitions. (F) Based on the findings from network interrogation, new hypotheses are generated, which can be tested in newly designed experiments. Data from new experiments could also be subject to further analysis.
In general, the types of omics measurements that are amenable to network analysis include microarrays, next-generation sequencing (for genotyping, transcriptome profiling, or microbiome analysis), and mass spectrometry-based proteomics and metabolomics data. While network analysis is usually and most straightforwardly applied to one type of omics data at a time (ie, to a homogeneous dataset), integrative networks are becoming more popular under the premise that the resulting networks more comprehensively describe the underlying biology.7,8 Each type of omics measurement technology has a specific procedure for transforming the raw data (eg, DNA sequences, mass spectrum peaks, spot fluorescence intensity for microarrays) to a consensus abundance or frequency measure for each feature. These methods are reviewed elsewhere9–12 and are beyond the scope of this article. In this guide, we use gene expression data to illustrate the process of network reconstruction and interrogation.
Network reconstruction
The first stage of network analysis is network reconstruction, which is the data-driven discovery or inference of the entities/nodes (transcripts, proteins, genes, metabolites, or microbes) and relationships or edges between these entities that together constitute the biological network. Here, we describe the steps involved in network reconstruction starting from entity abundance or frequency data.
Normalization (data preprocessing)
Customarily, abundance data are normalized in order to correct for sample-to-sample variation in the overall distribution of abundance values (or more generally, to normalize specific quantities that depend on the distribution). Measurements of gene expression levels (as well as other types of omics data) can be affected by a variety of non-biological factors including unequal amount of starting RNA, different extents of labeling, or different efficiencies of detection between samples. Before normalization, data are often log-transformed in order to stabilize variances when measurements span orders of magnitude. Frequently used normalization schemes include median normalization, quantile normalization, LOWESS normalization13 for RNA microarray data, reads per kilobase per million mapped reads (RPKM),14 and trimmed mean of M-values15 for RNA-seq data. In practice, we use normalization procedures available in the software package BRB ArrayTools16 for normalization of microarray data (Table 1). In addition, most normalization procedures are available as software packages in the Bioconductor toolkit.17 Systematic evaluations of transcriptome normalization methods have been reported for both microarrays18 and RNA-seq19; however, evaluations using large numbers of sample groups are needed in order to determine which normalization method is most appropriate for covariance network inference. Selection of an appropriate normalization method is clearly important, given that selection of a suboptimal normalization scheme can lead to overestimation of gene–gene correlation coefficients.18 Beyond transcriptome profiling, different omics data types may benefit from different types of normalization. For example, new methods have been proposed for normalization of metabolomics20 and microbiome21 data. Although there is no consensus about the best methods for many types of data, in the experience of the authors,4,22–26 simple methods such as quantile, LOWESS, or even median normalization perform reasonably well for class comparison and correlation if there are no major biases in the data such as batch effects.
Table 1.
Tools for network reconstruction and interrogation.
| STEP | METHOD -(STATISTICS / MATHEMATICS) | TOOL | LINK | REF |
|---|---|---|---|---|
| Network reconstruction | ||||
| Normalization | Quantile, lowess | BRB Array tools | http://linus.nci.nih.gov/BRB-ArrayTools.html | 16 |
| Quantile, lowess, etc. Relevant mixture model framework |
Package ‘affy* in Bioconductor | http://www.bioconductor.org/packages/release/bioc/html/affy.html | 106 | |
| R package ‘phyloseq’ | http://joey711.github.io/phyloseq/ | 107 | ||
| Finding DEGs | t-test | BRB Array tools IDEG6 | http://linus.nci.nih.gov/BRB-ArrayTools.html | 16 |
| Different test statistics, choice with Bonferroni correction | http://telethon.bio.unipd.it/bioinfo/IDEG6_form/ | 108 | ||
| SVM | SIRENE | http://cbio.ensmp.fr/sirene/ | 109 | |
| Semi-supervised learning; Logistic regression | SEREND | http://www.cs.cmu.edu/~jemst/Ecoli/ | 109 | |
| Likelihood of mutual information | CLR | http://omictools.com/clr-s2342.html | 111 | |
| Mutual information | ARACNE | http://wiki.c2b2.columbia.edu/workbench/index.php/ | 38 | |
| Mutual Information | MIDER | ARACNe | ||
| Itemset mining | DISTILLER | http://www.iim.csic.es/~gingproc/mider.html | 112 | |
| Bayesian hierarchical clustering; conditional | LeMoNe | request from authors | 113 | |
| http://bioinformatics.psb.ugent.be/software/ | 114 | |||
| Entropy Context Likelihood of Relatedness | Inferelator | details/LeMoNe http://bonneaulab.bio.nyu.edu/networks.html | 115 | |
| Remove indirect links | Partial correlation | Corpcor | http://cran.r-project.org/web/packages/corpcor/index.html | 116 |
| Local partial correlation | Local partial correlation | http://compbio.mit.edu/nd/ | 42 | |
| Global silencing of indirect correlations | Silencing | 40 | ||
| Network deconvolution | Network deconvolution | 41 | ||
| Weighted correlation network | Pearson correlation | WGCNA | http://labs.genetics.ucla.edu/horvath/Coexpression Network/Rpackages/WGCNA/ | 117 |
| Differential | Pearson correlation | CoXpress | http://coxpress.sourceforge.net/code.R | 56 |
| co-expression | Pearson correlation | Dapfinder | http://exon.niaid.nih.gov/dapfinder/ | 26 |
| Data integration | Bicluster | cMonkey | http://bonneaulab.bio.nyu.edu/software.html#cmonkey | 118 |
| Itemset mining | DISTILLER | Request from authors | 113 | |
| Meta-analysis | Fisher’s combined probability test | metap’ in software ‘stata’ | http://www.stata.com/support/faqs/statistics/ meta-analysis/ | |
| OpenMeta | http://www.cebm.brown.edu/open_meta | |||
| Visualization | Cytoscape | http://www.cytoscape.org/ | 119 | |
| Gephi | http://gephi.github.io/ | 119 | ||
| Circos | http://circos.ca/ | 121 | ||
| Network interrogation | ||||
| Module finding | Vertex weighting by local neighborhood density | MCODE | http://baderlab.org/Software/MCODE | 64 |
| Union of k-cliques | cfinder | http://www.cfinder.org/ | 65 | |
| Markov Cluster Algorithm | mcl | http://micans.org/mcl/ | 66 | |
| Function analysis/gene set enrichment | Fisher’s Exact | DAVID | http://david.abcc.ncifcrf.gov/summary.jsp | 69 |
| Kolmogorov-Smirnov statistic modification | GSEA | http://www.broadinstitute.org/gsea/index.jsp | 122 | |
| Fisher’s Exact | GoMiner | http://discover.nci.nih.gov/gominer/index.jsp | 123 | |
| Hypergeometric | GeneMerge | http://www.oeb.harvard.edu/faculty/hartl/old_site/lab/publications/GeneMerge.html | 124 | |
| Fisher’s Exact | FuncAssociate | http://llama.mshri.on.ca/funcassociate/ | 125 | |
| Dimension reduction (independent component analysis or fixed effect meta-estimate) followed by weighted pearson correlation | ProfileChaser | http://profilechaser.stanford.edu/ | 126 | |
| Hypergeometric test | Bingo | http://apps.cytoscape.org/apps/bingo | 70 | |
| Jaccard coefficient | EnrichmentMap | http://baderlab.org/Software/EnrichmentMap/ | 70 | |
| Hypergeometric distribution | SubpathwayMiner | http://www.inside-r.org/packages/cran/SubpathwayMiner | 68 | |
| Identify Key regulators | Network topology properties | Cytoscape | toolsⵆnetworkAnalyzerⵆAnalyze network | 119 |
| Intramodular connectivity, causality testing | WGCNA | http://labs.genetics.ucla.edu/horvath/Coexpression Network/Rpackages/WGCNA/ | 117 | |
| Pathway crosstalk | Crosstalk enrichment Eigen vector | CrossTalkZ Eigengene | http://sonnhammer.sbc.su.se/download/software/CrossTalkZ/ | 84 |
| http://labs.genetics.ucla.edu/horvath/htdocs/CoexpressionNetwork/EigengeneNetwork/ | 127 | |||
| Gene function prediction | Bayesian network | MEFIT | http://mefit.joydownload.com/ | 128 |
| Fast heuristic algorithm from ridge regression | GeneMANIA | http://www.genemania.org/ | 129 | |
| New gene ontology | Hierarchical clustering | NeXo | http://www.nexontology.org/ | 130 |
Discovery of differentially expressed genes (selecting nodes)
A crucial step in network reconstruction is the identification of the relevant subset of variables/genes that will constitute the nodes in the network; for a transcriptome profiling study, these would be genes for which there is significant differential expression between the sample groups. A variety of statistical tests are commonly used for the identification of differentially expressed genes (DEGs), including Welch’s t-test, moderated t-test, and permutation tests. For parametric tests, accurate estimation of intra-sample-group variance is a critical issue; two improved variance estimation techniques are the locally pooled error27 and empirical Bayes methods.28 To find DEGs, we usually use the t-test with the ordered set of P-values converted to cumulative false discovery rate (FDR) estimates, for which a typical cutoff would be 10%. Both statistical functions are implemented in BRB ArrayTools.29 During the last two decades, multiple statistical approaches have been proposed for differential expression testing.30 Overall, they provide similar results with small differences.30 Thus, careful study design (rather than trash in, trash out) and the use of meta-analysis techniques to integrate multiple datasets are likely to be more important for reliable DEG discovery than a choice of one or another statistical test. Because omics data analysis typically involves tens of thousands of statistical tests, the correction for multiple hypotheses is essential.31
Correlation analysis for network reconstruction (finding links between nodes)
The central biological principles underlying correlation network analysis are 1) that DEGs reflect functional changes, and 2) that DEGs do not work individually but interact (eg, at the protein or pathway level) to functionally alter the biological system. In gene expression networks, nodes represent genes and edges represent significant pairwise associations between gene expression profiles. The central mathematical/statistical principle that allows us to use correlation networks for analysis of biological systems is that the correlation between two variables, if statistically significant, is always a result of causation. Specifically, correlation results from regulatory relations between the two variables, or from a common causal regulator to the two variables, or both, as in the case of a feed-forward loop.32 To reconstruct the network, the Pearson or Spearman correlation coefficient can be used to obtain an association (similarity) measure for each possible pair of DEGs, with a cutoff for statistical significance (an FDR cutoff of 10% for the possible pairwise associations tested) and for a minimum correlation level. Together with the nodes, the edges whose similarity measures exceed this cutoff constitute a network. In practice, normalized expression data for DEGs are retrieved and pairwise correlations are calculated for each class (biological state) separately using the R statistical analysis software, with the function cor.test; FDR is calculated using the function p.adjust. Several other software programs that can be used for calculating gene–gene associations (correlations, mutual information and others) are listed in Table 1. Note that correlations should be calculated within a group of samples that belong to one class/biological state (pooling samples from different states/classes to compute the correlation coefficient leads to significant bias).
Discriminating between direct and indirect links
Covariation gene networks in general consist of connections that result from a combination of direct and indirect effects between genes. For example, if a gene Y strongly depends on gene X and gene Z also depends on X, it is likely that a high association (eg, correlation) will exist between Y and Z even if there is no direct dependence between them (Fig. 2). Moreover, even if a true dependence exists between a pair of genes/nodes, its strength estimation can be biased by additional indirect relationships.33 For this reason, correlation networks in general have many edges that reflect indirect relationships between pairs of genes, where no direct relationship exists. Finding direct relationships between genes is important when one attempts to identify causal gene regulators of a given biological process.
Figure 2.
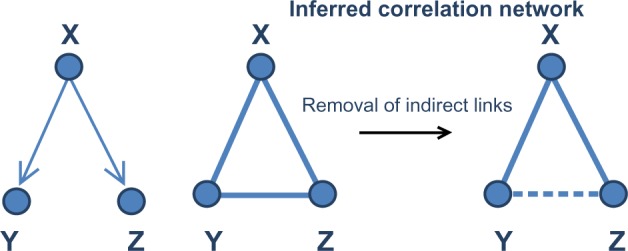
Removal of indirect links. As a demonstration, gene X can regulate the expression of both gene Y and Z. But there is no direct regulatory relationship between gene Y and Z. From the calculation of correlation of expression levels of three genes, correlations between gene X and Y, Z are observed as expected. However, genes Y and Z are also significantly correlated since they are both directly regulated by gene X. This correlation from common cause is called indirect link and can be removed by techniques, such as partial correlation, generating a network reflecting regulatory relationships.
Mathematically, direct effects can be defined as the association between two genes, holding the remaining genes constant.34 An effect that is not direct is called an indirect effect. The identification of direct links is an important goal of network reverse engineering.
To infer direct links between DEGs, we have been using the partial correlation coefficient.35,36 To calculate partial correlations, we use a method called the inverse method.37 Its implementation is straightforward in R using the function cor2pcor from the package “corpcor”. The detailed algorithm is described in Supplementary File. After calculation of partial correlation, the network can be built using links with absolute value of the partial correlation larger than a user-defined threshold.
Several other methods have been proposed to discriminate between direct and indirect links in covariation networks.38–41 For example, a variant of the partial correlation, which we call the local partial correlation, can be used in order to overcome the limitations of other methods.42
Proportion of unexpected correlations (improvement of reconstruction and error evaluation)
A fundamental problem of the standard correlation network approach is that practical limitations in the numbers of sample measurements can lead to an unacceptably high error rate. Recently, our group has proposed a method called proportion of unexpected correlations (PUC), which allows identifying and removing approximately half of false positive edges from a covariation network with no reduction in statistical power.43 The method takes into account a relation between the direction of regulation of two DEGs and the sign of correlation between the two genes. Thus, two up-and two downregulated genes must correlate positively; and a pair of oppositely regulated genes (one up-regulated and one down-regulated) should have negative correlation. Any deviation from this rule represents unexpected/erroneous edges and is removed from the network (Fig. 3). The proportion of these unexpected edges provides an error estimate for the whole network. For network reconstruction, each edge in a network can be evaluated and removed if it is unexpected.
Figure 3.

Illustration of expected and unexpected correlations. (A) When expression of two genes (gene x and gene y) are regulated toward the same direction when comparing two states, eg, both upregulated in disease (upper two panels), we should expect their expression levels to be positively correlated within each state if there exists regulatory relationship between gene x and gene y. When two genes are oppositely regulated when transiting from normal to disease (in the lower two panels, gene x is upregulated while gene z is down regulated), we should expect negative correlation between those two genes in each state. (B) Different combinations of between states and sign of correlations used to define expected or unexpected correlation.
Meta-analysis (improvement of reconstruction and error evaluation)
In omics-based network reconstruction, because of the large number of genes or variables measured (up to tens of thousands) and the limited number of samples (typically tens or hundreds), it is critical to assess the reproducibility of results. Although widely used methods (eg, FDR44) enable accounting for multiple hypothesis tests, the discrepancy between the number of samples and variables inherent to omics datasets limits the sensitivity and specificity for detecting edges through network reconstruction.
In order to overcome this problem and to augment the statistical significance for the nodes and links in a network, meta-analysis can be employed. This statistical approach combines results from different studies in order to achieve reproducibility.
The studies can be obtained from standardized omics data repositories. Good examples of such repositories are the Gene Expression Omnibus (GEO)45 and Array Express46 (for transcriptomics and epigenomics datasets); PRIDE47 (for proteomics datasets), the Human Metabolome Database48 (for metabolomics datasets), and lipid MAPS49 (for lipidomics datasets). Additionally, molecular interaction data from the BioGRID50 or BioCyc databases51 can be used as a prior for edge reconstruction.
In meta-analysis of multiple datasets – whether from publicly available datasets or experiments produced in the same lab – the strategy is usually the same. The datasets to be co-analyzed in a meta-analysis should be selected on the basis of their congruence with the central biological question of interest, and they should pass some predefined sample size and quality requirements (eg, number of measured/detected genes). After choosing the datasets, as a first step for meta-analysis we apply two filters: 1) the same sign of statistic (mean, covariance, or correlation) throughout all datasets (ie, if gene A is upregulated in case over control in data-set 1, it should have the same direction of regulation in all other datasets to pass the filter); 2) P-value thresholds across all datasets. These filters provide consistency and control for heterogeneity across datasets for a given gene (or gene pair in case of correlation). The next step is an actual statistical evaluation. In this step, meta-analysis combines common statistical measures, such as P-values, and calculate a weighted average for such measures. As a weighted average, we frequently use the Fisher’s P-value calculation. Let p1,…,pk be the P-values of one measure into k datasets (studies). For example, pi can be the t-Student test p value for gene A to be differentially expressed in study i. Then the Fisher’s P-value pFisher summarizes all these P-values p1,…, pk into one average P-value by the formula
where is a random variable with chi-square distribution with 2k degrees of freedom. After calculating Fisher’s P-values for all genes, the standard FDR procedure can be used to adjust for multiple hypothesis testing. Several other approaches have been proposed for meta-analysis of gene expression data (Table 1).52,53 In Supplementary File we describe in more detail the algorithm that we have employed for integrating differential expression, correlations, and differential associations/correlations.4
Differentially coexpressed gene pairs (evaluating network changes)
The networks discussed above model static correlations between genes that change their expression when the biological system transits from one state to another. However, the sets of edges within a gene covariation network can themselves vary from state to state, for example, when two genes are highly correlated in a subset of conditions but not across all conditions.54 Such a gene pair is called a differentially coexpressed gene pair (Fig. 4). It has been shown that differentially coexpressed gene pairs frequently play critical roles in pathogenesis. Several studies have explored gene coexpression changes in cancer, revealing known cancer genes that were top-ranked among coexpression changes but not necessary (separately) among differentially expressed genes.26,55
Figure 4.

(A) Gene 2 and gene 7 correlate with each other in both normal and disease conditions, but the signs of the correlation coefficient are opposite. (B) In normal condition, there is no correlation between gene 4 and gene 5, but they gain positive correlation when the biological system transitioned to disease. (C) Example of visualization of a network transitioning between normal and disease conditions. Red lines represent positive correlation, blue line represent negative correlation, and dotted gray lines represent nonexisting correlations in one condition that strongly appear in the other condition (on this case, becomes positively correlated).
In order to search for differentially coexpressed gene pairs, our group adapted a simple approach called differentially associated pairs (DAPs).26 The DAPs algorithm is described in Supplementary File. In addition to DAPs, multiple methods/software have been developed to find the changing edges in gene expression networks (Table 1).26,56
Integrating heterogeneous omics data types: inter-omics networks
The integration of different omics data types holds great promise for enabling more robust network reconstruction and detection of causal interactions in a particular biological context. For example, genome-wide measurements of epigenetic marks and transcriptome data can be combined to elucidate mechanisms of gene regulation.57–59 In cancer bioinformatics, integration of gene copy number data (chromosomal aberrations) and gene expression measurements has enabled the discovery of key drivers.4,60 And integration of metagenomics data from gut microbiota with intestinal gene expression can reveal new mechanisms of crosstalk between microbes and their hosts.6
Approaches for omics data integration generally fall into one of two modalities: first (and most prevalent) is integrating different types of data generated for a given gene/gene product.61 In other words, a given node pertains to more than one network (eg, measurements of the copy numbers of gene A and transcript levels of gene A pertain to genomic and transcriptomic networks, respectively) (Fig. 5A).
Figure 5.
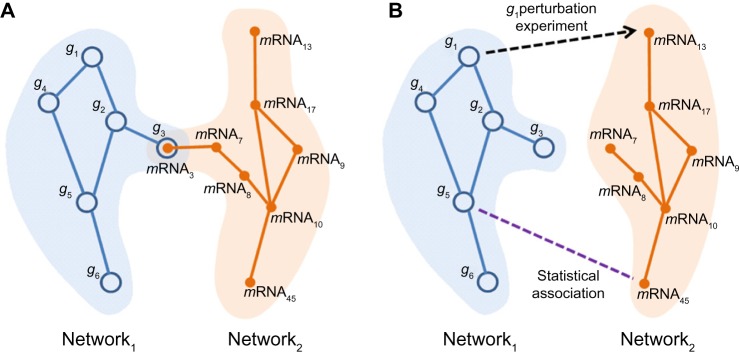
Data integration for inter-omics network. (A) Networks are constructed from different data types (eg, network 1 for gene genetic interaction network and network 2 for mRNA coexpression network). These two networks then can be integrated into one network by overlapping the nodes that are correspondent between two networks (eg, gene 3 and its transcript mRNA 3 are merged into one node). (B) In another type of integration, links are created between nodes by different evidence of interaction, either experimentally proved relationship (eg, knockout of gene 1 altered the expression level of mRNA13) or statistical association between features of two nodes (eg, gene 5 and mRNA45).
The other type of integration makes an edge/link between two nodes from different omics networks. We call the result of such integration an inter-omics network. An inter-omics network is a bipartite network in which each edge connects two nodes of different omics types (Fig. 5B). There are two different approaches to infer such inter-omics links/edges. The first one is based on bringing into reconstruction an experimental result supporting a link between nodes of different omics. For example, nodes from proteomics and metabolomics networks can be connected on the basis of the experiment showing that a specific protein is an enzyme necessary for the production of a given metabolite. The second approach, which infers edges between different omics, establishes connection between two different (knowledge-wise unrelated) quantitative variables based on their statistical association (eg, correlation between gene expression and abundance of metabolites measured in the same samples). Thus, the entire reconstruction procedure consists of inference on networks of each omics type separately, and then integration of these two networks into the inter-omics network. This is a straightforward and easily implementable algorithm. Furthermore, there is a popular tool, integr-Omics, that is used for heterogeneous data integration using partial least squares regression.62
Network interrogation
To gain maximal insights from a biological network that has been reconstructed as described above, systematic analysis of the network (network interrogation) is essential. In this section we describe several network interrogation techniques for investigating specific types of biological questions.
Revealing potential mechanisms of a biological process or disease
This goal is achieved by identification of pathways involved in the process, key regulatory nodes of those pathways, and interactions between identified pathways (including identification of nodes in network responsible for the interaction).
Which functional pathways are involved?
Finding dense subnetworks (ie, modules or clusters) Figure 6A. From a functional standpoint, subsets of genes that are highly interconnected in the correlation network (modules63) are often involved in similar biological processes. Tools for identification of modules include MCODE,64 cfinder,65 and graph clustering (MCL).66 A key advantage of network module analysis (vs direct clustering of genes from the data) is that, while modules would include genes up- and downregulated that correspond to potential stimulatory and inhibitory relations within a given functional pathway, traditional clustering approaches would group genes with similar behavior, thus separating up- and downregulated genes from the same pathway into different clusters. In addition, network reconstruction has an advantage over traditional gene-level clustering analysis in that the network provides insight into which subnetworks interact with each other and which nodes/genes might mediate such interactions.67
Figure 6.

Network interrogation. (A) Densely connected subnetworks (modules) are detected, and enriched functions of those modules are detected. (B) Genes with unknown function (gray) can be annotated based on the function of its neighbors in the network or the functions of the genes in the same module. (C) New gene ontologies can be generated by analyzing the hierarchical organization of gene clusters. (D) Multiple data types can be integrated to help infer the direction of regulation and identify key regulators based on their network topological features. (E) Crosstalks between pathways can be studied by extracting eigengenes or analyzing enriched interactions between networks. Key regulators for pathway crosstalk can also be identified based on their between-module topology properties.
Enrichment analysis with external data. (eg, Gene Ontology) Figure 6A
Once genes that work together (modules) are identified, the next step is to infer their biological functions. This is usually performed by using literature-curated, gene-centric biological knowledge bases that connect genes to functional categories (terms) such as the functional terms in the Gene Ontology. If a module is enriched for genes that are associated with a particular biochemical pathway, a location in a genome, or a location in cellular compartment, that finding can provide a basis for a hypothesis about the function of the module. A plethora of tools are available for gene functional enrichment analysis (Table 1). For example, gene sets can be annotated by pathways using tools like SubpathwayMiner68 or by gene ontology terms using tools such as DAVID.69 Other tools such as Bingo70 and EnrichmentMap71 can further construct a functional network, ie, a network in which nodes are genes and an edge between two genes is present if those genes share functional annotations.
Key regulators of pathways/modules
Identifying the key molecular regulators of the biological response or system under study is often a primary goal in omics studies, especially those with a tractable cellular model where molecular or genetic perturbations can be introduced. There are two major complementary strategies for finding key regulators in covariation networks: 1) using network topological properties, and 2) incorporating additional data into networks that provides information about causes of regulation for some nodes in a network Figure 6D.
Topological properties that have been described to date as pointing to key regulators mostly define different measures of the connectivity of a node. Those properties are the degree and centrality measures, such as betweenness centrality, closeness centrality, and eigenvector centrality. Nodes with high betweenness centrality (the so-called bottlenecks) have been shown to be predictive of gene essentiality.72 For example, such topological characteristics have been found to be associated with genes that are critical for pathogen virulence73 and with genes that are targets for hepatitis C virus.74 The estimation of these parameters is a straightforward and be easily accomplished using the Cytoscape plug-in called NetworkAnalyzer.75 Importantly, these properties need not be analyzed in isolation but can complement another approach we discuss below.2
Integrating additional information in order to find causes of regulation
It is axiomatic that a gene–gene network that has been reconstructed based on correlation analysis does not discriminate between direct regulation and common cause.32 Therefore, it is common to incorporate into a covariation network several types of complementary biological data that can directly or indirectly indicate that one gene regulates another.5,76 By overlaying such information on a coexpression network, one can establish the directionality of some edges, which improves the precision of identification of key regulators. The types of biological information include genetic variants (aberrations, mutations, gene polymorphisms, etc), epigenetic modifications, transcription factors, and other types of gene expression regulation such as microRNA (miRNA). For example, integrating genomic aberrations with global gene expression led to the discovery of key drivers of melanoma60 and breast7 and cervical cancers.4 Similarly, eQTLs (expression quantitative trait loci) were integrated with networks associated with diabetes and obesity, revealing causal genes of specific molecular pathways operating in these diseases.8
Integration of information about binding sites (or computationally predicted binding sites) of transcription factors into covariation networks is a particularly powerful approach,77 because the direction of causality for a connection between a transcription factor and a target gene is presumed to be known. While computational analysis of transcription factor binding site (TFBS) databases (such as TRANSFAC) can suggest the possibility of regulation by a given transcription factor, omics approaches for identification transcription factor binding sites such as ChIP-Seq provide more definitive genome-wide location information for the transcription factor in an investigated sample. The directionality information provided by those methods can be incorporated into network interrogation to generate more accurate prediction of key regulators.78 miRNAs are another important class of gene expression regulators that modulate (primarily downregulate) expression of target genes either by inhibiting translation or promoting mRNA degradation. In the past few years, ~1,881 miRNA genes have been identified in humans (according to miRBase, http://www.mirbase.org/cgi-bin/mirna_summary.pl?org=hsa), and knowledge of miRNA–target interactions is accumulating both by experimental validation and computational prediction.79,80 More accurate genome-wide miRNA target sequence location information allows the possibility of generating an miRNA–mRNA regulatory network, which could provide a more complete view of regulatory relationship in biological process. In a recent work, Sumazin et al integrated gene and miRNA expression data from sample-matched datasets and constructed a comprehensive miRNA–gene interaction network, inferring that phosphatase and tensin homolog (PTEN) is a key regulator of gliomagenesis.2
Integrating multiple types of data simultaneously can increase the precision of computational predictions. For example, one of us has reported that “using motif scanning and Histone acetylation local minima, improves the sensitivity for TF binding site prediction by approximately 50% over a model based on motif scanning alone”.57 In another work, Yang et al integrated gene expression with gene copy number alternation, DNA methylation, associated miRNA expression, and miRNA target prediction to identify key regulatory miRNA genes that regulate ovarian cancer development and then experimentally validated the function of one predicted miRNA gene.3 In practice, multiple tools have been developed for the integration of different resources of information to infer network and/or identification of key regulators (Table 1).
How the pathways interact
As networks represent models of global changes in biological system, they usually contain several groups of genes exerting specific biological functions. Cooperation of these functions/pathways plays an important role in regulating biological processes. Thus, a transcriptional network can be viewed as a group of interacting pathways/modules (meta-modules) rather than interacting individual genes.81 Studying the interaction between modules, thus, will provide us with a higher order view of biological system (see forest, not just trees) and understanding of causal relationship between functions.
In order to investigate the behavior of the pathways, a dimension reduction procedure can be used that transforms expression values of all genes in a given module into one representative value for each sample. One such procedure is to reduce the expression profiles of all of the genes within a module into a single eigengene profile that summarizes dominant mode of covariation of the genes in the module.82 Evaluation of statistical association between eigengenes tests a hypothesis of interaction between two pathways represented by corresponding eigengenes Figure 6E.
As an alternative to the eigengene approach, multiple methods have been proposed to calculate enrichment of links between members of separate pathways to identify cross-talking pathways based on diverse types of interactions such as protein interactions, coexpression, etc.83–85 Once a relationship between modules has been established, the next question is which nodes or genes are responsible for the interaction. Although multiple genes could act as mediators of interaction between two pathways, their relative importance can be different. Few approaches have been developed to find which nodes are critical for crosstalk between different modules in a network. Multiple sources of data are integrated to identify interactions between cancer-related pathways, and key regulators are identified (genes that are significantly altered for at least one molecular level) mediating those interactions. We have developed an approach that identifies nodes in a network responsible for interactions between modules that potentially correspond to genes regulating crosstalk between pathways represented by these modules. The approach is based on the idea that the genes that are in the shortest paths between modules should be more important in controlling perturbation from one pathway to another, mediating inter-module signaling or regulation. Several centrality measures have been proposed to evaluate the importance of nodes in a network.86 Among those, betweenness centrality measures the importance of a node in acting as a bridge between any nodes within a network.74 We modified standard betweenness centrality87 to adapt to the case of interaction between two defined subnetworks and to specifically address the question of which nodes belonging to subnetwork 1 have a higher probability to be bottlenecks in the transfer of signal to the nodes in subnetwork 2, and vice versa. For this metric, the shortest paths are calculated only between nodes of two subnetworks and not between any nodes within a network. This bipartite betweenness centrality can be calculated as follows:
where s belongs to subnetwork 1 and t belongs to subnetwork 2, σst is the total number of shortest paths from node s to node t, and σst(v) is the number of those paths that pass through vertex v (node for which the metric is calculated). Thus, this measurement represents the importance of a node in mediating information flow between two connected modules in a network. In our recent work, we found that this approach allows finding not only bottlenecks of interaction between different pathways within the same organism but even microbial genes critical for mediating interaction between gut microbiota and their host.6
Revealing function of individual node in the network
While most of our knowledge about gene functions is based on detailed and thorough gene-centered laboratory research, there are still genes whose functions have been less studied; network biology offers a novel way to infer functions for such genes. It uses an idea that genes that are located closely in a network may share a function. This principle is frequently called guilt by association Figure 6B.88 There are two major approaches that implement guilt by association for prediction of node function. The first is the so-called direct approach. Although there are a few slightly different methods using this approach (neighbor counting, graphic algorithm, probabilistic methods), they all assign a function to a node based on the functions of its direct neighbors.89–91 The second approach, the modular approach, is to guide the assignment of a function to a gene by the collective function of other genes that belong to a given module in which the investigated gene is located.61,92
Besides identifying functions of individual nodes, generations of new ontology systems based on networks or pairwise similarities were proposed Figure 6C.93 Interestingly, besides demonstrating a high level of consistency with existing ontologies, they provide solutions for situations when standard approaches (ie, knowledge-based approaches) fail to reflect comprehensive biology.94 Indeed, some terms/categories that were missing in the standard GO and inferred by a network approach were submitted to the GO Consortium and incorporated into the ontology.93,95
Network cross-species conservation
An important facet of network interrogation is the assessment of evidence for network function. Just as cross-species comparison is a core strategy for elucidating novel protein function (eg, BLAST), cross-species comparison of network structure can reveal functions for network subgraphs that might not have been evident from sequence-level conservation of individual network components. In practice, subgraphs of the novel network (and in some approaches, constituent protein sequences) are used as keys to search for structural and component-sequence similarity to subgraphs in another species by searching for parsimonious subgraph-to-subgraph mappings (called a local network alignment). Alternatively, gene coexpression networks from two species can be compared in their entirety, to obtain a global alignment. A successful alignment enables all available functional annotations in the orthologous subgraph to bring to bear on the functional interpretation of the novel network’s subgraph. Various local and global network alignment algorithms have been proposed, including NetworkBLAST,96 PINALOG,97 IsoRankN,98 and the Narayanan–Karp99 and Hodgkinson–Karp100 algorithms.
Different biological problems and some perspectives
Some biological questions that can be addressed within the framework of network analysis remained beyond of the scope of this review. For example, one can try to evaluate the number of nodes needed to be perturbed in order to achieve a transition from one state of biological system to another. This measure of network controllability101 (number of needed nodes), although seemingly theoretical, can have very practical implications. On one hand, if a few nodes can govern a regulatory network modeling a disease, a gene perturbation/gene silencing approach can be a good strategy for treatment. On the other hand, if a large proportion of nodes in a network have to be modified in order to achieve recovery, then a different pharmaceutical strategy using compounds that can simultaneously affect multiple molecular targets should be followed.
Furthermore, some mathematical properties observed in biological networks such as small world, scale-free, assortative mixing,102 and several others103 warrant further investigation to comprehend what types of environmental pressures led to selection of these properties during evolution and how they contribute to fitness and resilience of biological systems.
Biological example: transkingdom network for interrogation of host–microbe interactions
In our recent work,6 by applying network analysis we studied the effects of antibiotics on the gut microbial community (microbiota) and on the host (mouse). The major outcome of this study – which was based on network analysis – was the identification of specific mammalian processes that are affected by antibiotics (ABx) and the identification of the microbes (including some microbial genes) that contributed to these effects. Importantly, a big part of the critical findings of mechanisms of effects of ABx was revealed using network analysis and could not be predicted based on existent knowledge in the field.
Below we have outlined step by step the analysis employed in this study, which consisted in the reconstruction of mammalian transcriptomic and microbial genomics networks, integration of these two networks into one transkingdom network, and its interrogation that led to biological insights that have been validated experimentally.
-
Finding differentially expressed mammalian genes
The gene expression raw data were normalized using BRB Array Tools using the LOWESS smother. Next we compared gene expression between control and ABx-treated mice on two genetic backgrounds and found 1,583 differentially expressed genes with an FDR cutoff of 10% (see section Discovery of differentially expressed genes).
-
Reconstruction of gene expression network
To reconstruct the transcriptomic network, we calculated correlations in four groups of control mice. We performed meta-analysis (see section Meta-analysis) of gene–gene correlations and removed unexpected correlations (see section Proportion of unexpected correlations) and obtained a network of 1,275 nodes and 13,714 links with an FDR cutoff of 5%.
-
Identification of subnetworks
MCODE network clustering identified two major subnetworks: one (631 genes) that was dependent on ABx-resistant microbes, and the other (77 genes) dependent on microbiota.6
-
Data mining of gene expression subnetworks
Functional annotation enrichment analysis using the web tool DAVID revealed that the first subnetwork was enriched for mitochondrial functions including genes coding for electron transport chain, oxidation–reduction, ATP biosynthesis, and cellular and mitochondrial ribosomes, while second one was enriched for annotations related to immune function.
-
Finding microbial genes enriched by antibiotics
We have compared copy numbers of microbial genes (annotated in SEED104) between control ABx-treated mice and found 4,523 bacterial genes with differential abundance between ABx and controls.
-
Reconstruction of microbial gene network
In order to identify the ABx-resistant microbes or microbial genes that influence the host, a covariance network was constructed for the 1,689 microbial genes that were enriched by antibiotics in two mouse strains (Swiss Webster, C57BL6/J). This analysis resulted in a network with 1,143 nodes connected by 23,429 edges (combined FDR <0.0001).
-
Reconstruction of transkingdom network
In order to reveal ABx-resistant microbes and their genes that affect the host, we reconstructed transkingdom network. For this, we calculated the correlations between microbial genes that were part of the microbial network with mouse gene expression from the second subnetwork (steps 3, 4). The correlation was calculated using measurements in the two ABx-treated groups of mice separately (C57BL6/J and Swiss Webster) and the resulting P-values were combined as described above (see section Meta-analysis). The resulting transkingdom network consisted of 513 microbial and 334 mouse genes linked by 708 edges (FDR 0.01, Fig. 7).
-
Finding microbial subnetworks
Using MCODE, we found two major microbial sub-networks (101 and 60 nodes) linked to the host part of the transkingdom network. The genomes enrichment analysis (see Materials and Methods of REF for details)6 indicated that two microbes (Pseudomonas aeruginosa and Escherichia coli) as potential sources of the genes of these subnetworks.
-
Finding bottleneck microbial genes (betweenness centrality)
By applying bipartite betweenness centrality analysis,105 we have revealed five top microbial genes as potentially critical for ability of microbes to affect the host.
Figure 7.
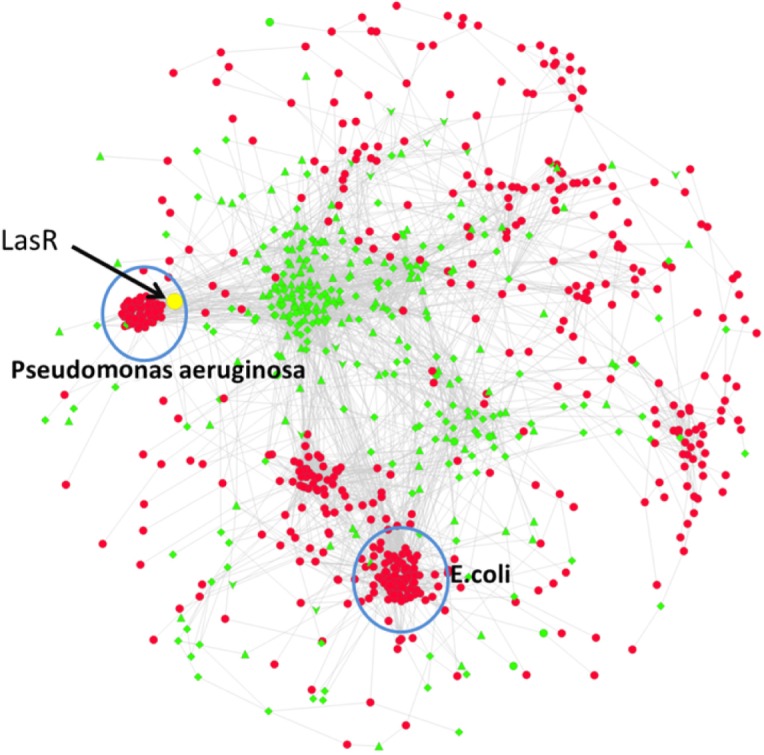
Transkingdom network resulting from network analysis. Transkingdom network includes microbial genes (red) and host (mouse) genes (green). A key regulator is identified as a gene within top 1% of bipartite betweenness centrality is LasR (yellow). Two microbial gene subnetworks, indicated by blue circles, are enriched with genes from Pseudomonas aeruginosa and Escherichia coli.
Note: One of the microbes (P. aeruginosa) and one gene (LasR) have been experimentally tested, confirming the predicted effect on mammalian cells and validating the efficiency of transkingdom network analysis.
Conclusion
In this review, we have described how network analysis can help us to answer different questions commonly asked in biological research. We have also provided a detailed algorithm for this analysis, including approaches employed by our group as well as frequently used by the network-biology community (Table 1).
Supplementary Material
Algorithm for the Calculation Partial Correlations.
Algorithm for Meta-Analysis Scheme.
Algorithm for Calculation P-values for DAPs.
Acknowledgments
We thank Khiem Lam for editing this paper.
Footnotes
ACADEMIC EDITOR: Thomas Dandekar, Associate Editor
FUNDING: AM was funded from NIH (AI109695; AI107485); AY was supported by FAPESP (grant 2013/24516), LT was supported by FAPESP (grants 2013/14722-8 and 2013/06223-1). SAR was funded by NIH HL098807. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE). Provenance: the authors were invited to submit this paper.
Author Contributions
NS and AM contributed to the conception of the work. XD AY, SR, LT, AM contributed to the design of the work. XD, AY, SR, LT and AM contributed to the acquisition, analysis, and interpretation of data for the work. XD, AY, SR, LT and AM drafted the work. XD, AY, SR, LT, NS and AM revised the paper. XD, AY, SR, LT, NS and AM approved the final version to be published.
REFERENCES
- 1.Amit I, Garber M, Chevrier N, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326(5950):257–63. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sumazin P, Yang X, Chiu HS, et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011;147(2):370–81. doi: 10.1016/j.cell.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang D, Sun Y, Hu L, et al. Integrated analyses identify a master microRNA regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer Cell. 2013;23(2):186–99. doi: 10.1016/j.ccr.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mine KL, Shulzhenko N, Yambartsev A, et al. Gene network reconstruction reveals cell cycle and antiviral genes as major drivers of cervical cancer. Nat Commun. 2013;4:1806. doi: 10.1038/ncomms2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen JC, Alvarez MJ, Talos F, et al. Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell. 2014;159(2):402–14. doi: 10.1016/j.cell.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgun A, Dzutsev A, Dong X, et al. Uncovering effects of antibiotics on the host and microbiota using transkingdom gene networks. Gut gutjnl-2014-308820. 2015 doi: 10.1136/gutjnl-2014-308820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran LM, Zhang B, Zhang Z, et al. Inferring causal genomic alterations in breast cancer using gene expression data. BMC Syst Biol. 2011;5:121. doi: 10.1186/1752-0509-5-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Zhu J, Lum PY, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452(7186):429–35. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olson NE. The microarray data analysis process: from raw data to biological significance. NeuroRx. 2006;3(3):373–83. doi: 10.1016/j.nurx.2006.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anders S, McCarthy DJ, Chen Y, et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc. 2013;8(9):1765–86. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 11.Hamady M, Knight R. Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res. 2009;19(7):1141–52. doi: 10.1101/gr.085464.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernandez P, Müller M, Appel RD. Automated protein identification by tandem mass spectrometry: issues and strategies. Mass Spectrom Rev. 2006;25(2):235–54. doi: 10.1002/mas.20068. [DOI] [PubMed] [Google Scholar]
- 13.Berger JA, Hautaniemi S, Jarvinen AK, Edgren H, Mitra SK, Astola J. Optimized LOWESS normalization parameter selection for DNA microarray data. BMC Bioinformatics. 2004;5:194. doi: 10.1186/1471-2105-5-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 15.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon R, Lam A, Li M-C, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-array tools. Cancer Inform. 2007;3:11. [PMC free article] [PubMed] [Google Scholar]
- 17.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim WK, Wang K, Lefebvre C, Califano A. Comparative analysis of microarray normalization procedures: effects on reverse engineering gene networks. Bioinformatics. 2007;23(13):i282–8. doi: 10.1093/bioinformatics/btm201. [DOI] [PubMed] [Google Scholar]
- 19.Dillies MA, Rau A, Aubert J, et al. French StatOmique Consortium. A comprehensive evaluation of normalization methods for illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 2013;14(6):671–83. doi: 10.1093/bib/bbs046. [DOI] [PubMed] [Google Scholar]
- 20.Jauhiainen A, Madhu B, Narita M, Narita M, Griffiths J, Tavare S. Normalization of metabolomics data with applications to correlation maps. Bioinformatics. 2014;30(15):2155–61. doi: 10.1093/bioinformatics/btu175. [DOI] [PubMed] [Google Scholar]
- 21.McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10(4) doi: 10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borra RC, Andrade PM, Silva ID, et al. The Th1/Th2 immune-type response of the recurrent aphthous ulceration analyzed by cDNA microarray. J Oral Pathol Med. 2004;33(3):140–6. doi: 10.1111/j.0904-2512.2004.00089.x. [DOI] [PubMed] [Google Scholar]
- 23.Perez-Diez A, Morgun A, Shulzhenko N. Microarrays for cancer diagnosis and classification. Adv Exp Med Biol. 2007;593:74–85. doi: 10.1007/978-0-387-39978-2_8. [DOI] [PubMed] [Google Scholar]
- 24.Shulzhenko N, Morgun A, Hsiao W, et al. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med. 2011;17(12):1585–93. doi: 10.1038/nm.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shulzhenko N, Yambartsev A, Goncalves-Primo A, Gerbase-DeLima M, Morgun A. Selection of control genes for quantitative RT-PCR based on microarray data. Biochem Biophys Res Commun. 2005;337(1):306–12. doi: 10.1016/j.bbrc.2005.09.048. [DOI] [PubMed] [Google Scholar]
- 26.Skinner J, Kotliarov Y, Varma S, et al. Construct and compare gene coexpression networks with DAPfinder and DAPview. BMC Bioinformatics. 2011;12:286. doi: 10.1186/1471-2105-12-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jain N, Thatte J, Braciale T, Ley K, O’Connell M, Lee JK. Local-pooled-error test for identifying differentially expressed genes with a small number of replicated microarrays. Bioinformatics. 2003;19(15):1945–51. doi: 10.1093/bioinformatics/btg264. [DOI] [PubMed] [Google Scholar]
- 28.Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21(9):2067–75. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y, Simon R. BRB-ArrayTools Data Archive for human cancer gene expression: a unique and efficient data sharing resource. Cancer Inform. 2008;6:9–15. doi: 10.4137/cin.s448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan W. A comparative review of statistical methods for discovering differentially expressed genes in replicated microarray experiments. Bioinformatics. 2002;18(4):546–54. doi: 10.1093/bioinformatics/18.4.546. [DOI] [PubMed] [Google Scholar]
- 31.Dudoit S, Shaffer JP, Boldrick JC. Multiple hypothesis testing in microarray experiments. Stat Sci. 2003;18(1):71–103. [Google Scholar]
- 32.Pearl J. Causality: Models, Reasoning and Inference. Vol. 29. Cambridge University Press; Cambridge, England: 2000. [Google Scholar]
- 33.Pearl J. Direct and indirect effects; Paper presented at: Proceedings of the Seventeenth Conference on Uncertainty in Artificial Intelligence; 2001; Seattle, WA. August 2–5 2001. [Google Scholar]
- 34.Pearl J. An introduction to causal inference. Int J Biostat. 2010;6(2):7. doi: 10.2202/1557-4679.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De La Fuente A, Bing N, Hoeschele I, Mendes P. Discovery of meaningful associations in genomic data using partial correlation coefficients. Bioinformatics. 2004;20(18):3565–74. doi: 10.1093/bioinformatics/bth445. [DOI] [PubMed] [Google Scholar]
- 36.Marbach D, Costello JC, Küffner R, et al. DREAM5 Consortium. Wisdom of crowds for robust gene network inference. Nat Methods. 2012;9(8):796–804. doi: 10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whittaker J. Graphical Models in Applied Multivariate Statistics. New York: Wiley; 1990. [Google Scholar]
- 38.Margolin AA, Nemenman I, Basso K, et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006;7(suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jang IS, Margolin A, Califano A. hARACNe: improving the accuracy of regulatory model reverse engineering via higher-order data processing inequality tests. Interface focus. 2013;3(4):20130011. doi: 10.1098/rsfs.2013.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barzel B, Barabási AL. Network link prediction by global silencing of indirect correlations. Nat Biotechnol. 2013;31(8):720–5. doi: 10.1038/nbt.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feizi S, Marbach D, Médard M, Kellis M. Network deconvolution as a general method to distinguish direct dependencies in networks. Nat Biotechnol. 2013;31(8):726–33. doi: 10.1038/nbt.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomas LD, Fossaluza V, Yambartsev A. Building complex networks through classical and Bayesian statistics-A comparison Paper presented at: XI Brazilian Meeting on Bayesian Statistics. Amparo–SP–Brazil: EBEB; 2012. [Google Scholar]
- 43.Yambartsev A, Perlin M, Kovchegov Y, Shulzhenko N, Mine KL, Morgun A. Unexpected links reflect the noise in networks. arXiv. 2013;1310:8341. doi: 10.1186/s13062-016-0155-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19(3):368–75. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- 45.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brazma A, Parkinson H, Sarkans U, et al. ArrayExpress – a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003;31(1):68–71. doi: 10.1093/nar/gkg091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martens L, Hermjakob H, Jones P, et al. PRIDE: the proteomics identifications database. Proteomics. 2005;5(13):3537–45. doi: 10.1002/pmic.200401303. [DOI] [PubMed] [Google Scholar]
- 48.Wishart DS, Tzur D, Knox C, et al. HMDB: the human metabolome database. Nucleic Acids Res. 2007;35(suppl 1):D521–6. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fahy E, Subramaniam S, Murphy RC, et al. Update of the LIPID MAPS comprehensive classification system for lipids. J Lipid Res. 2009;50(suppl):S9–14. doi: 10.1194/jlr.R800095-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34(suppl 1):D535–9. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caspi R, Altman T, Dreher K, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012;40(D1):D742–53. doi: 10.1093/nar/gkr1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rhodes DR, Yu J, Shanker K, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A. 2004;101(25):9309–14. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hwang D, Rust AG, Ramsey S, et al. A data integration methodology for systems biology. Proc Natl Acad Sci U S A. 2005;102(48):17296–301. doi: 10.1073/pnas.0508647102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schafer J, Strimmer K. A shrinkage approach to large-scale covariance matrix estimation and implications for functional genomics. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1175. Article32. [DOI] [PubMed] [Google Scholar]
- 55.Kostka D, Spang R. Finding disease specific alterations in the co-expression of genes. Bioinformatics. 2004;20(suppl 1):i194–9. doi: 10.1093/bioinformatics/bth909. [DOI] [PubMed] [Google Scholar]
- 56.Watson M. CoXpress: differential co-expression in gene expression data. BMC Bioinformatics. 2006;7:509. doi: 10.1186/1471-2105-7-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramsey SA, Knijnenburg TA, Kennedy KA, et al. Genome-wide histone acetylation data improve prediction of mammalian transcription factor binding sites. Bioinformatics. 2010;26(17):2071–5. doi: 10.1093/bioinformatics/btq405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–69. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 59.Ramsey, Stephen A, et al. Epigenome-guided analysis of the transcriptome of plaque macrophages during atherosclerosis regression reveals activation of the Wnt signaling pathway. PLoS genetics. 2014;10(2):e1004828. doi: 10.1371/journal.pgen.1004828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akavia UD, Litvin O, Kim J, et al. An integrated approach to uncover drivers of cancer. Cell. 2010;143(6):1005–7. doi: 10.1016/j.cell.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peña-Castillo L, Tasan M, Myers CL, et al. A critical assessment of Mus musculus gene function prediction using integrated genomic evidence. Genome Biol. 2008;9(suppl 1):S2. doi: 10.1186/gb-2008-9-s1-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Le Cao KA, Gonzalez I, Dejean S. integrOmics: an R package to unravel relationships between two omics datasets. Bioinformatics. 2009;25(21):2855–6. doi: 10.1093/bioinformatics/btp515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402(6761 suppl):C47–52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 64.Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4(1):2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palla G, Derényi I, Farkas I, Vicsek T. Uncovering the overlapping community structure of complex networks in nature and society. Nature. 2005;435(7043):814–18. doi: 10.1038/nature03607. [DOI] [PubMed] [Google Scholar]
- 66.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30(7):1575–84. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Butte AJ, Kohane IS. Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. Honolulu, Hawaii: 2000. (Paper presented at: Pac Symp Biocomput). [DOI] [PubMed] [Google Scholar]
- 68.Li C, Li X, Miao Y, et al. SubpathwayMiner: a software package for flexible identification of pathways. Nucleic Acids Res. 2009;37(19):e131–e131. doi: 10.1093/nar/gkp667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Da Wei Huang BTS, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 70.Maere S, Heymans K, Kuiper M. BiNGO: a cytoscape plugin to assess over-representation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–9. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 71.Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5(11):e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu H, Kim PM, Sprecher E, Trifonov V, Gerstein M. The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput Biol. 2007;3(4):e59. doi: 10.1371/journal.pcbi.0030059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McDermott JE, Taylor RC, Yoon H, Heffron F. Bottlenecks and hubs in inferred networks are important for virulence in Salmonella typhimurium. J Comput Biol. 2009;16(2):169–80. doi: 10.1089/cmb.2008.04TT. [DOI] [PubMed] [Google Scholar]
- 74.Diamond DL, Syder AJ, Jacobs JM, et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010;6(1) doi: 10.1371/journal.ppat.1000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Assenov Y, Ramirez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24(2):282–4. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- 76.Zhu J, Zhang B, Smith EN, et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat Genet. 2008;40(7):854–61. doi: 10.1038/ng.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Geertz M, Maerkl SJ. Experimental strategies for studying transcription factor-DNA binding specificities. Brief Funct Genomics. 2010;9:5–6. 362–73. doi: 10.1093/bfgp/elq023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463(7279):318–25. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lewis BP, Shih I-H, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 80.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human microRNA targets. PLoS Biol. 2004;2(11):e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oldham MC, Konopka G, Iwamoto K, et al. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11(11):1271–82. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li Y, Agarwal P, Rajagopalan D. A global pathway crosstalk network. Bioinformatics. 2008;24(12):1442–7. doi: 10.1093/bioinformatics/btn200. [DOI] [PubMed] [Google Scholar]
- 84.McCormack T, Frings O, Alexeyenko A, Sonnhammer EL. Statistical assessment of crosstalk enrichment between gene groups in biological networks. PLoS One. 2013;8(1):e54945. doi: 10.1371/journal.pone.0054945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang T, Gu J, Yuan J, Tao R, Li Y, Li S. Inferring pathway crosstalk networks using gene set co-expression signatures. Mol Biosyst. 2013;9(7):1822–8. doi: 10.1039/c3mb25506a. [DOI] [PubMed] [Google Scholar]
- 86.Freeman LC. Centrality in social networks conceptual clarification. Soc Networks. 1979;1(3):215–39. [Google Scholar]
- 87.Newman ME. Fast algorithm for detecting community structure in networks. Phys Rev E. 2004;69(6):066133. doi: 10.1103/PhysRevE.69.066133. [DOI] [PubMed] [Google Scholar]
- 88.Oliver S. Guilt-by-association goes global. Nature. 2000;403(6770):601–3. doi: 10.1038/35001165. [DOI] [PubMed] [Google Scholar]
- 89.Chua HN, Sung WK, Wong L. Exploiting indirect neighbours and topological weight to predict protein function from protein-protein interactions. Bioinformatics. 2006;22(13):1623–30. doi: 10.1093/bioinformatics/btl145. [DOI] [PubMed] [Google Scholar]
- 90.Karaoz U, Murali TM, Letovsky S, et al. Whole-genome annotation by using evidence integration in functional-linkage networks. Proc Natl Acad Sci U S A. 2004;101(9):2888–93. doi: 10.1073/pnas.0307326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee H, Tu Z, Deng M, Sun F, Chen T. Diffusion kernel-based logistic regression models for protein function prediction. OMICS. 2006;10(1):40–55. doi: 10.1089/omi.2006.10.40. [DOI] [PubMed] [Google Scholar]
- 92.King AD, Przulj N, Jurisica I. Protein complex prediction via cost-based clustering. Bioinformatics. 2004;20(17):3013–20. doi: 10.1093/bioinformatics/bth351. [DOI] [PubMed] [Google Scholar]
- 93.Dutkowski J, Kramer M, Surma MA, et al. A gene ontology inferred from molecular networks. Nat Biotechnol. 2013;31(1):38–45. doi: 10.1038/nbt.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dutkowski J, Ono K, Kramer M, et al. NeXO Web: the NeXO ontology database and visualization platform. Nucleic Acids Res. 2014;42(Database issue):D1269–74. doi: 10.1093/nar/gkt1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kalaev M, Smoot M, Ideker T, Sharan R. NetworkBLAST: comparative analysis of protein networks. Bioinformatics. 2008;24(4):594–6. doi: 10.1093/bioinformatics/btm630. [DOI] [PubMed] [Google Scholar]
- 97.Phan HT, Sternberg MJ. PINALOG: a novel approach to align protein interaction networks--implications for complex detection and function prediction. Bioinformatics. 2012;28(9):1239–45. doi: 10.1093/bioinformatics/bts119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liao CS, Lu K, Baym M, Singh R, Berger B. IsoRankN: spectral methods for global alignment of multiple protein networks. Bioinformatics. 2009;25(12):i253–8. doi: 10.1093/bioinformatics/btp203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Narayanan M, Karp RM. Comparing protein interaction networks via a graph match-and-split algorithm. J Comput Biol. 2007;14(7):892–907. doi: 10.1089/cmb.2007.0025. [DOI] [PubMed] [Google Scholar]
- 100.Hodgkinson L, Karp RM. Algorithms to detect multiprotein modularity conserved during evolution. IEEE/ACM Trans Comput Biol Bioinform. 2012;9(4):1046–58. doi: 10.1109/TCBB.2011.125. [DOI] [PubMed] [Google Scholar]
- 101.Liu YY, Slotine JJ, Barabasi AL. Controllability of complex networks. Nature. 2011;473(7346):167–73. doi: 10.1038/nature10011. [DOI] [PubMed] [Google Scholar]
- 102.Piraveenan M, Prokopenko M, Zomaya A. Assortative mixing in directed biological networks. IEEE/ACM Trans Comput Biol Bioinform. 2012;9(1):66–78. doi: 10.1109/TCBB.2010.80. [DOI] [PubMed] [Google Scholar]
- 103.Newman ME. The structure and function of complex networks. SIAM Rev. 2003;45(2):167–256. [Google Scholar]
- 104.Overbeek R, Begley T, Butler RM, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33(17):5691–702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Borgatti SP, Everett MG. Network analysis of 2-mode data. Soc Networks. 1997;19(3):243–69. [Google Scholar]
- 106.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy – analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–15. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 107.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Romualdi C, Bortoluzzi S, d’Alessi F, Danieli GA. IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiol Genomics. 2003;12(2):159–62. doi: 10.1152/physiolgenomics.00096.2002. [DOI] [PubMed] [Google Scholar]
- 109.Mordelet F, Vert JP. SIRENE: supervised inference of regulatory networks. Bioinformatics. 2008;24(16):i76–82. doi: 10.1093/bioinformatics/btn273. [DOI] [PubMed] [Google Scholar]
- 110.Ernst J, Beg QK, Kay KA, Balázsi G, Oltvai ZN, Bar-Joseph Z. A semi-supervised method for predicting transcription factor-gene interactions in Escherichia coli. PLoS Comput Biol. 2008;4(3):e1000044. doi: 10.1371/journal.pcbi.1000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Faith JJ, Hayete B, Thaden JT, et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5(1):e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Villaverde AF, Ross J, Morán F, Banga JR. Mider: network inference with mutual information distance and entropy reduction. PLoS One. 2014;9(5):e96732. doi: 10.1371/journal.pone.0096732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lemmens K, De Bie T, Dhollander T, et al. DISTILLER: a data integration framework to reveal condition dependency of complex regulons in Escherichia coli. Genome Biol. 2009;10(3):R27. doi: 10.1186/gb-2009-10-3-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Michoel T, De Smet R, Joshi A, Van de Peer Y, Marchal K. Comparative analysis of module-based versus direct methods for reverse-engineering transcriptional regulatory networks. BMC Syst Biol. 2009;3(1):49. doi: 10.1186/1752-0509-3-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ciofani M, Madar A, Galan C, et al. A validated regulatory network for Th17 cell specification. Cell. 2012;151(2):289–303. doi: 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schaefer J, Opgen-Rhein R, Strimmer K. Corpcor: Efficient Estimation of Covariance and (Partial) Correlation. 2007. (R Package Version 1(4)). Available at: http://www.strimmerlab.org/software/corpcor/
- 117.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9(1):559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Reiss DJ, Baliga NS, Bonneau R. Integrated biclustering of heterogeneous genome-wide datasets for the inference of global regulatory networks. BMC Bioinformatics. 2006;7(1):280. doi: 10.1186/1471-2105-7-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. ICWSM. 2009;8:361–2. [Google Scholar]
- 121.Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zeeberg BR, Feng W, Wang G, et al. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4(4):R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Castillo-Davis CI, Hartl DL. GeneMerge – post-genomic analysis, data mining, and hypothesis testing. Bioinformatics. 2003;19(7):891–2. doi: 10.1093/bioinformatics/btg114. [DOI] [PubMed] [Google Scholar]
- 125.Berriz GF, King OD, Bryant B, Sander C, Roth FP. Characterizing gene sets with FuncAssociate. Bioinformatics. 2003;19(18):2502–4. doi: 10.1093/bioinformatics/btg363. [DOI] [PubMed] [Google Scholar]
- 126.Engreitz JM, Chen R, Morgan AA, Dudley JT, Mallelwar R, Butte AJ. ProfileChaser: searching microarray repositories based on genome-wide patterns of differential expression. Bioinformatics. 2011;27(23):3317–8. doi: 10.1093/bioinformatics/btr548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Huttenhower C, Hibbs M, Myers C, Troyanskaya OG. A scalable method for integration and functional analysis of multiple microarray datasets. Bioinformatics. 2006;22(23):2890–7. doi: 10.1093/bioinformatics/btl492. [DOI] [PubMed] [Google Scholar]
- 128.Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(suppl 1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Algorithm for the Calculation Partial Correlations.
Algorithm for Meta-Analysis Scheme.
Algorithm for Calculation P-values for DAPs.