

Abstract
Ornithine aminotransferase (OAT) and γ-aminobutyric acid aminotransferase (GABA-AT) are classified under the same evolutionary subgroup and share a large portion of structural, functional, and mechanistic features. Therefore, it is not surprising that many molecules that bind to GABA-AT also bind well to OAT. Unlike GABA-AT, OAT had not been viewed as a potential therapeutic target until recently; consequently, the number of therapeutically viable molecules that target OAT is very limited. In this review the two enzymes are compared with respect to their active site structures, catalytic and inactivation mechanisms, and selective inhibitors. Insight is offered that could aid in the design and development of new selective inhibitors of OAT for the treatment of cancer.
Keywords: Ornithine aminotransferase (OAT), Gamma-aminobutyric acid aminotransferase (GABA-AT), Hepatocellular carcinoma (HCC), Irreversible enzyme inhibition
Introduction
Aminotransferases are pyridoxal 5′-phosphate (PLP)-dependent enzymes that catalyze the reversible transfer of an amino group from a substrate to an acceptor α-keto acid1,2.All aminotransferases share a similar catalytic mechanism, in which two coupled half-reactions are required to complete one transamination cycle. Both reactions involve the tautomerization of an aldimine formed between the substrate and PLP, catalyzed by a basic lysine residue. In the first half-reaction, the amine remains with the cofactor after hydrolysis, leaving it in the pyridoxamine 5′-phosphate (PMP) form, and the substrate becomes the corresponding carbonyl compound. The second half-reaction involves the reactivation of the enzyme in a similar manner, by transferring the amino group from PMP to a suitable carbonyl-containing acceptor compound, such as α-ketoglutarate or pyruvate.3, 4
γ-Aminobutyric acid aminotransferase (GABA-AT; EC 2.6.1.19) and ornithine δ-aminotransferase (OAT; EC 2.6.1.13) are similar PLP-dependent aminotransferases that belong to the same evolutionary subgroup.5 GABA-AT is found in many organs, including brain, liver, kidney, and pancreas; however, it is present at higher specific activity in glial cells and presynaptic neurons.6 It catalyzes the transfer of the amino group of γ-aminobutyric acid (GABA) to α-ketoglutarate, producing succinic semialdehyde and L-glutamate (Scheme 1).7 OAT, on the other hand, is found in the mitochondrial matrix of most human and animal tissues;8 it catalyzes the transfer of the δ-amino group of L-ornithine to α-ketoglutarate, forming glutamate γ-semialdehyde and L-glutamate (Scheme 1).9
Scheme 1.
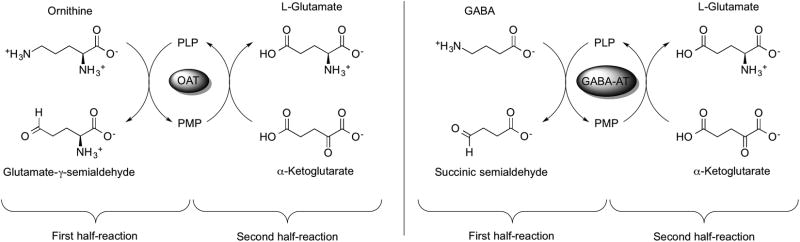
Catalytic mechanisms of OAT (left) and GABA-AT (right), including both half-reactions.
Being closely related, these two enzymes share not only high secondary structural homology, but also possess very similar catalytic mechanisms.10,11 Unexpectedly, OAT and GABA-AT have only 17% overall sequence identity,12 but the residues at the active sites of the two enzymes are strikingly similar.13 It is not surprising, then, that some potent inhibitors of GABA-AT also inhibit OAT.11 Thus, the development of selective inhibitors requires exploration of the activity at both enzymes and fine-tuning of the inhibitor's properties so that other PLP-dependent enzymes are not affected.
Inhibition of GABA-AT prevents the degradation of the inhibitory neurotransmitter GABA into inactive succinic semialdehyde, thus increasing GABA levels at the synapse. Because abnormally low levels of GABA in the brain are associated with serious neurological disorders such as epilepsy,14 Huntington's disease,15 Parkinson's disease,16 and Alzheimer's disease,17 inhibition of GABA-AT is considered a viable target for drugs aimed at treating these ailments. For example, potent inhibitors of GABA-AT have been developed to treat epilepsy18 and addictions.19 In addition to the beneficial effects on neurodegenerative diseases, inhibition of GABA-AT may be useful in immunomodulation20 and gastroesophageal reflux disease (GERD).21
Inhibition of OAT is proposed to be beneficial in treating hyperammonemias by enhancing the urea cycle to clear excess ammonia.22,23 Recently, human OAT was shown to play a role in regulating mitotic cell division in rapidly growing cells and became a potential target for the development of chemotherapeutic drugs.24 It was also found that OAT is over expressed in the cells of hepatocellular carcinoma (HCC),25 a proliferative disease, so its inhibition may be beneficial in treating HCC, in addition to hyperammonemias. The development of drugs that inhibit OAT has been hampered by the fact that the deficiencies of this enzyme in humans may result in gyrate atrophy (GA) of the choroid and retina, which may progressively lead to blindness.26,27,28 However, although inhibition of OAT may increase the plasma level of ornithine and cause hyperornithinemia (which can lead to gyrate atrophy), the exact mechanism of GA is still unknown.29 There are speculations that the excessive metabolites of ornithine released during inhibition of OAT by 5FMOrn, such as spermine, may be toxic to the retinal pigment epithelial (RPE) cells in GA.30 However, because GA is a slowly progressing disease, it may not be a problem in acute treatments of cancer patients.
In this review we will focus on targeting the selective inhibition of OAT by building on the knowledge acquired by studying GABA-AT. This study will be based on the structural differences and similarities of the enzymes (Figure 1),31 known inhibitors of the enzymes, and inhibitory mechanisms of these compounds. A structural homology of the two enzymes will be compared, and the substrate binding pockets will be analyzed to understand their specificity. Finally, their inhibitory mechanisms will be compared to aid in the design and development of new selective inhibitors of OAT. For more information about PLP-dependent enzymes and their role as therapeutic targets, there are excellent reviews on the subject by Eliot et al3 and Alessio et al.32
Figure 1.
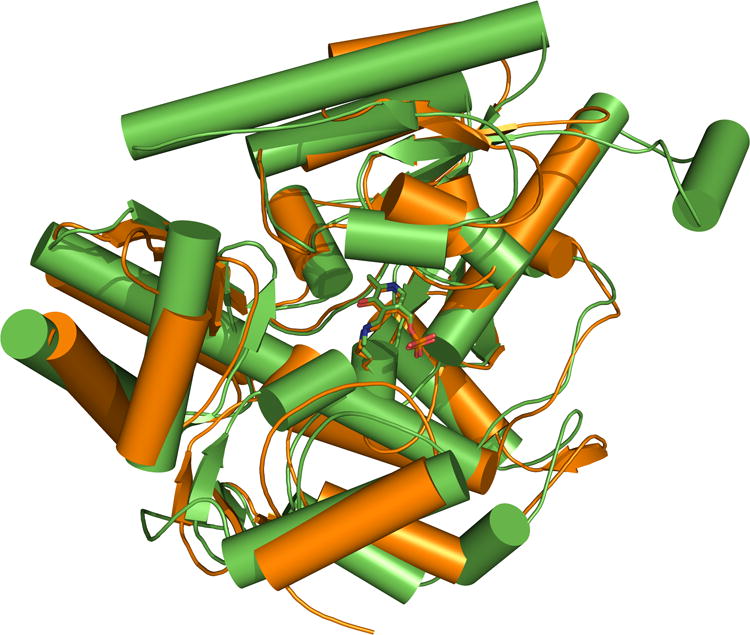
Superimposition of pig GABA-AT (green; PDB 1OHV) and human OAT (orange; PDB 1OAT). Active site similarity was found to be up to 57%, and the root mean square deviation (rmsd) value between the two internal (PLP-lysine) aldimines is 0.36 Å. Values were obtained from the Comparison of Protein Active Site Structures (CPASS) database.26
Sequence and Structurally Conserved Residues
Aligned sequences of aminotransferases revealed that there is only small sequence homology among them, even though many of them share very similar tertiary structures. Mehta et al.33 performed a sequence alignment of aminotransferases and found that there are only four residues known to be conserved among all of those investigated: a lysine, an aspartate, an arginine, and a glycine. The active site lysine acts as a covalent tether for the PLP cofactor via an aldimine linkage. The aspartate residue forms a hydrogen bond with the protonated nitrogen in the pyridine ring of the cofactor, acting as an anchoring site. The arginine residue is also conserved in the active site, where it anchors the carboxyl functional group on the substrate to facilitate catalytic turnover. The glycine residue is known to act as a roof for the substrate binding site but its function is relatively less well understood.10 However, it is known that the NH in the backbone of the glycine residue hydrogen bonds to the phosphate of the cofactor to assist in its stabilization. Shen et al.4 and Toney et al.34 performed a topographical alignment of active site residues of OAT against GABA-AT and Asp-AT (Table I).
Table I. Homologous residues in the active sites of OAT, GABA-AT and Asp-AT.
Structural similarities between OAT, GABA-AT, and Asp-AT
Because all of the aminotransferases share the same initial catalytic mechanism, it is understandable that they share a significant amount of structural homology. Visualization of the active site similarities among those aminotransferases has been aided by the publication of relevant crystal structures. Storici et al.35 first reported the crystal structure of GABA-AT from pig liver, which is known to be 96% identical to human brain GABA-AT.36 The crystal structure of human recombinant OAT was first reported by Shen et al.4 In its native form, the PLP cofactor is linked to a lysine residue in the active site, as discussed above. The crystal structures of the native enzymes show that the cofactor interacts similarly with the conserved active site residues between OAT (Figure 2), GABA-AT (Figure 3), and Asp-AT (Figure 4). Also, the phosphate group of the cofactor provides a firm anchor to the region in the active site known as the “phosphate-binding cup”.37 The crystal structures of ligand-bound GABA-AT38 and OAT12,39 were compared with their native unliganded forms, and no large-scale conformational changes were observed.40 it appears that these enzymes do not undergo domain closure when the ligand is bound, unlike aminotransferase subgroup I enzymes.41
Figure 2.

The active site of native OAT with internal PLP. Shown are the residues that interact noncovalently with the cofactor. Dashed lines indicate hydrogen bonds. Associated letters indicate the originating chain.
Figure 3.

The active site of native GABA-AT with internal PLP. Shown are the residues that interact noncovalently with the cofactor. Rounded lines indicate hydrophobic interactions. Dashed lines indicate hydrogen bonds.
Associated letters indicate the originating chain.
Figure 4.

The active site of native Asp-AT with internal PLP. Shown are the residues that interact noncovalently with the cofactor. Rounded lines indicate hydrophobic interactions. Dashed lines indicate hydrogen bonds.
Associated letters indicate the originating chain.
Substrate recognition
In the active sites, the natural substrates are aligned in such a way that their reacting amino groups are juxtaposed to C4′ of the cofactor, which is involved in the internal aldimine with the conserved lysine residue (K292 for OAT, K329 for GABA-AT, and K258 for Asp-AT). This orientation permits the external aldimine to be formed by exchanging the lysine for the substrate amine moiety. However, unlike Asp-AT and other aminotransferases, the active sites of GABA-AT and OAT stabilize their natural substrates, GABA and ornithine, respectively, in such a way that the terminal amine group is transiminated. In the case of OAT, the α-carboxylate of ornithine is recognized by R180 as R413 interacts with E235, correctly positioning the δ-amino group of ornithine for transimination.4 This is similar for GABA-AT, where E270 interacts with R445 via a salt bridge; the α-carboxylate of GABA is held electrostatically by R192, which correctly positions the γ-amino group toward the cofactor for transimination.35 In Asp-AT, however, a glutamate residue is not present, and therefore the second arginine residue (R386) is free to bind to the α-carboxylate of the aspartate substrate, making the transamination of the α-amino group possible.42
To understand the role of the active site glutamate residue, site-directed mutagenesis was performed by Markova et al.13 and Liu et al.43 for human recombinant OAT and E. coli GABA-AT, respectively. It was concluded that the glutamate residue is important for neutralizing the positive charge of the secondary arginine residue through a salt bridge during the first half reaction,40 when there is an internal PLP-lysine aldimine as the substrate approaches. This closed system allows productive binding of ornithine to OAT and GABA to GABA-AT and restricts the binding of dicarboxylated amine-acceptor substrates like α-ketoglutarate and pyruvate that are required for the second half reaction. On the other hand, when there is an external PMP during the second half reaction, this hydrogen bonding network is weakened, and the salt bridge opens up to allow α-ketoglutarate to interact with the second arginine residue. It was hypothesized by Markova et al.13 that this glutamate residue acts as the switch between the first and second half reactions of both enzymes. This is consistent with the known ping-pong mechanism for these enzymes34, 44 and rationalizes why it occurs.
Differences in the active sites of OAT and GABA-AT
Differences between the active site residues are listed in Table II. It is reasonable to assume that these differences can determine the substrate selectivity between the two enzymes, given their structural similarity. The major differences are Tyr55A and Tyr85A in OAT, which are replaced by Phe351B and Ile72A in GABA-AT. The phenylalanine and isoleucine residues in the active site of GABA-AT contribute to the arrowing of the active site and the increasing hydrophobicity by restricting the size of the substrate binding pocket.12,43 In OAT, however, the tyrosine residues serve as an anchor point for the charged amino group at the 2-position of the substrate,12 and through molecular modeling studies, we have found that Tyr85 possesses a significant degree of conformational flexibility that exposes an accessory binding pocket not present in other aminotransferases.
Table II. Differences between OAT and GABA-AT active site residues.
Irreversible Inhibitors (inactivators) of GABA-ATalso Inhibit OAT
Because of the structural similarities between GABA-AT and OAT, some inactivators of GABA-AT have also been found to inactivate OAT (Figure 6).11 Gabaculine and 4-amino-5-hexynoic acid, for example, were reported to inactivate GABA-AT45,46 while also inactivating OAT with equal potency, both in vitro and in vivo. Also, it was found that GABA is a competitive inhibitor of OAT with a Ki value of 3.4 mM.47 Vigabatrinon the other hand, is a selective inactivator of GABA-AT. It inactivates GABA-AT with a KI value of 0.85 mM, kinact value of 0.24 min-1, and kinact/KI of 0.28 min-1mM-1.48 For OAT, vigabatrin was found to be a very weak reversible inhibitor with a Ki value of 46 mM (unpublished data).
Figure 6.

Some known irreversible inhibitors of (a) OAT, (b) GABA-AT, and (c) both enzymes.
Irreversible Inhibitors of OAT
5-Fluoromethylornithine (5FMOrn) and L-canaline (Figure 6), analogs of the natural substrate ornithine, are irreversible inhibitors of OAT.49,50 Although L-canaline was shown to be a reversible competitive inhibitor of Asp-AT with about 1000 fold weaker affinity than for OAT, it has shown strong irreversible inhibition of OAT by forming a stable oxime with the PLP cofactor.51 On the other hand, 5FMOrn is a selective irreversible inhibitor of OAT. According to mechanistic studies done by Bolkenius et al.52, 5FMOrn forms an external aldimine with PLP, and subsequent enzyme-catalyzed reactions lead to a stable unsaturated ketone that includes a covalent link to the cofactor (see mechanism below).
Catalytic Mechanisms of GABA-AT and OAT
An understanding of the mechanistic differences between GABA-AT and OAT can help in the design of selective inhibitors of OAT over GABA-AT. The catalytic mechanisms for OAT and GABA-AT are shown in Schemes 2 and 3, respectively. Interestingly, the two mechanistic pathways are identical, except for the structure of the substrate that is recognized and the resulting product. Because of this similarity, further investigation of the inactivation mechanisms would be helpful to differentiate between the two enzymes with targeted inhibitors.
Scheme 2. Catalytic mechanism for OAT.

Scheme 3. Catalytic mechanism for GABA-AT.

Irreversible inhibitors or inactivators of GABA-AT are well studied and can be subdivided into four Classes on the basis of their inactivation mechanisms.53 Class I compounds inactivate GABA-AT through a Michael addition mechanism, leading to covalent modification of the active site residue. Class II inactivators disrupt GABA-AT via an enamine mechanism and give ternary adducts comprised of the inhibitor, the enzyme and the cofactor. Class III inactivators only modify PLP, which may involve enzyme-catalyzed aromatization of the inactivator. Class IV inactivators require the PMP form of the enzyme, and are not categorized based on their mechanism. Vigabatrin (Figure 6), for example, is both a category I and II inactivator, while gabaculine belongs to Class III. Unlike GABA-AT, the inactivation mechanisms of OAT have not been thoroughly studied, probably because of the small number of OAT inactivators. However, they are known to be similar to those of GABA-AT.11
Structural and mechanistic comparisons of irreversible inhibitors of GABA-AT and OAT
Structure- and mechanism-based selectivity can be better understood by studying the interactions of vigabatrin and 5FMOrn in the active site of GABA-AT and OAT, respectively. Storici et al. reported the crystal structure of OAT inactivated with 5FMOrn (PDB 2OAT)12 and suggested that the specificity of 5FMOrn and L-canaline towards OAT might be because of the α-amino group that they both bear. The interactions between the ligand and the active site residues of OAT and GABA-AT inactivated by 5FMOrn and vigabatrin are depicted in Figures 7 and 8, respectively. For 5FMOrn-inactivated OAT, it can be seen that the α-amino group of 5FMOrn interacts with Tyr55, and the α-carboxyl group is stabilized by Arg180. In GABA-AT, Phe321 is in place of Tyr55 at this position, as there is no α-amino group in the substrate that requires hydrogen bonding.
Figure 7.

Scheme of the active site of OAT inactivated by 5-fluoromethylornithine (PDB 2OAT). Shown are the residues that interact with the new adduct. Rounded lines indicate hydrophobic interactions. Dashed lines indicate hydrogen bonds or salt bridges. Associated letters indicate the originating subunit.
Figure 8.

Scheme of the active site of GABA-AT inactivated by vigabatrin (PDB 1OHW). Shown are the residues that interact with the new adduct. Rounded line indicates hydrophobic interaction. Dashed lines denote hydrogen bonds or salt bridges. Associated letters indicate the originating subunit.
Mechanism-based inactivators have a higher chance of being selective because of the distinct mechanistic pathway that is required for covalent modification of the enzyme.54 5FMOrn selectively inactivates OAT as shown in Scheme 4, by forming an enamine intermediate that forms a ternary adduct in a manner similar to Class II inactivators of GABA-AT.52 However, vigabatrin is categorized as both a Class I and Class II inactivator, because it displays both mechanisms.55 Vigabatrin can form either a Michael acceptor, or to a lesser extent, an enamine intermediate in the active site of GABA-AT; the former is attacked by the active site lysine residue for covalent modification (Scheme 5). The same type of intermediates may form with OAT, but they may not be in a proper position for the lysine residue of OAT to undergo covalent modification. On the other hand, when OAT catalyzes a reaction with 5FMOrn, it forms an enamine intermediate, which leads to a covalent adduct with the enzyme that slowly hydrolyzes to an unsaturated ketone, which is tightly bound to OAT. 5FMOrn does not bind to GABA-AT because the inhibitor is too large to fit in the active site, and/or it bears an α-amino group that does not interact well with the hydrophobic pocket of GABA-AT. Therefore, it is reasonable to suggest that the α-amino group on the inhibitor is important for OAT selectivity, and this residue difference could be utilized in the future in the design of inhibitors that are selective toward OAT over GABA-AT.
Scheme 4. Inactivation of OAT by 5FMOrn46.

Scheme 5. Inactivation of GABA-AT by vigabatrin49.

Gabaculine (Figure 6) has a very simple structure, with neither a longer (5-atom) main chain, nor an α-amino group. It is therefore not surprising that it is not selective for either GABA-AT, OAT, or other PLP-dependent enzymes. The mechanism of inactivation of PLP-dependent enzymes by gabaculine is shown in Scheme 6.
Scheme 6.

Inactivation of PLP-dependent enzymes by gabaculine.
4-Amino-5-hexynoate (gamma-ethynyl GABA or GEG), however, is an unusual case, because it has different mechanistic pathways for OAT and GABA-AT, even though it is known to inactivate both. Burke et al.56 reported the mechanism of inactivation of GABA-AT by GEG (Scheme 7), which was confirmed with X-ray crystallography by Storici et al.38 De Biase et al.57 reported the mechanism of inactivation of OAT by GEG (Scheme 8); there is no crystal structure available for the OAT-GEG inactivated complex. By comparing the mechanisms of inactivation of the two enzymes by GEG, differences in the properties of the two active sites can be identified that might account for the mechanistic differences. The first part of the mechanism is identical for both enzymes, where an external aldimine with PLP is formed and the γ-proton of the inhibitor is abstracted to give an alleneamine intermediate. In the case of GABA-AT, the amino group of a lysine residue undergoes a Michael addition to the penultimate carbon of the intermediate, which leads to three potential tautomers of covalently modified adducts (Figure 9). However for OAT an enamine mechanism ensues; the alleneamine intermediate undergoes transimination to restore the original internal aldimine with release of the alleneamine derivative of GEG. This species covalently modifies the enzyme by undergoing (all) eneamine alkylation to form a ternary complex with the lysine-bound cofactor. It is noteworthy that in GABA-AT, alleneamine alkylation does not occur, which may be the result of steric hindrance in the active site.56 It is thought that the active site of OAT may be more accommodating in terms of overall size (as discussed above), which may account for the exclusive enamine inactivation mechanism by both inactivators of OAT. Therefore, this mechanistic difference may be utilized in the design of a selective inhibitor of OAT. By comparing the crystal structures of OAT inactivated by 5FMOrn (pdb #2OAT) and that of GABA-AT inactivated by vigabatrin (1OHW) and GEG (1OHY), we found a critical residue difference that might explain the difference in inactivation mechanisms subsequent to γ-proton abstraction: Tyr85 (OAT) vs Ile71 (GABA-AT). Because of its noncyclic nature, Ile71 of GABA-AT can change conformation so the linear allene can adoptan orientation appropriate for Michael addition by Lys329, forming a covalent adduct with the penultimate carbon of the inactivator. In the case of OAT, the corresponding residue is Tyr85, which occupies more of the binding pocket space, both because of its larger footprint and because the backbone helix is displaced 1.7 Å toward the ligand. Furthermore, it appears that this residue might be conformationally locked in the presence of the ethynyl group. We believe the smaller binding space does not allow for the planar deprotonated intermediate to adopt a conformation where the penultimate carbon is close enough to the reactive lysine for Michael addition, thus preventing the covalent intermediate from forming directly; inactivation, then, is only achieved through the enamine pathway, where the linkage to Lys292 is made through C4 of the PLP cofactor, which would be more easily accessible.
Scheme 7.

Inactivation of GABA-AT by GEG.
Scheme 8.

Inactivation of OAT by GEG.
Figure 9.
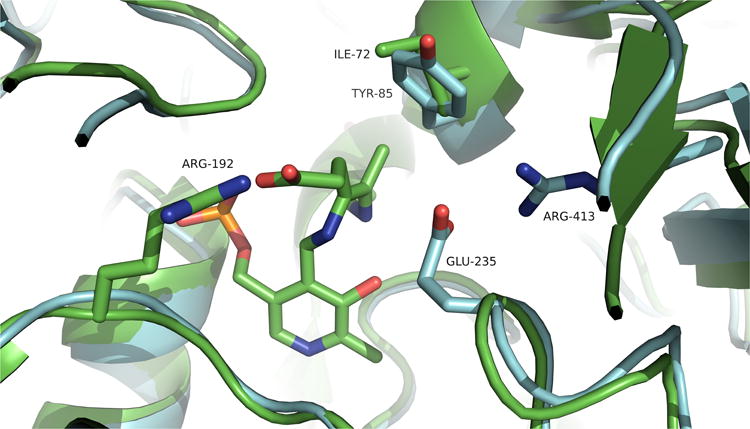
Binding site differences between GABA-AT inactivated with GEG (green) and native OAT (blue). The region near Ile72 has more sterically accessible volume than the corresponding area in OAT, occupied by Tyr85.
Insights regarding selectivity for OAT over GABA-AT
Because of their identical catalytic mechanisms, the same active moieties that exist in GABA-AT inactivators are applicable to OAT inactivators. For example, the δ-fluoromethyl group present in 5-fluoromethylornithine is also present in the GABA-AT inactivator S-4-amino-5-fluoropentanoic acid (AFPA, Figure 10).58 Similarly, the aminooxyl moiety found in L-canaline is also a part of the non-selective GABA-AT inactivator aminooxypropionic acid.59 These reactive groups, however, are not expected to have a great impact on enzyme selectivity. In fact, AFPA, L-canaline, and aminooxypropionic acid are fairly non-selective.49,60
Figure 10. Structural comparison between OAT inactivators (5-fluoromethylornithine and L-canaline) and GABA-AT inactivators (AFPA and aminooxypropionic acid).

The binding pockets of OAT and GABA-AT are similar as well, as noted earlier. One of the main reasons why many molecules can bind to both enzymes indiscriminately is the fact that the distance between the anchor points of the ligands, as measured between the PLP cofactor and the carboxylate-binding Arg residue (Arg180 in OAT and Arg192 in GABA-AT), is virtually identical. Bearing this in mind, however, there are some interesting differences, in addition to those mentioned above, that can help to explain substrate selectivity and provide avenues for selective ligand development.
First, it is obvious that ornithine is one carbon longer than GABA. The latter is fully extended in the catalytic site, as can be seen in the crystal structures of inactivated GABA-AT. To make space for the extra carbon on Orn, there are two major changes in OAT that provide the extra room required for the ligand to exist in a ‘compressed’ conformation (Figure 11). First, the backbone at Ser321 (Asn352 at GABA-AT), which corresponds roughly to C2 in ornithine, is about 1.3 Å farther away from the ligand, giving it some extra room. Most importantly, though, Phe351 in GABA-AT is a glycine residue (Gly320) in OAT, which frees up space for the carboxylate to extend into what would be sterically restricted space in GABA-AT, thereby allowing the ligand to form a slightly offset salt bridge with Arg180. In GABA-AT, Phe351 partially occupies a similar steric space as Tyr55 in OAT, but also intrudes into the binding pocket so that only the smaller GABA molecule will fit.
Figure 11.
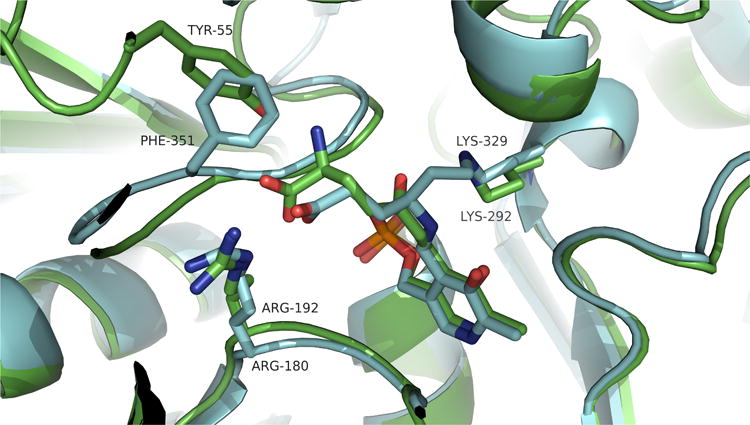
Comparison between the binding of the inactivated forms of vigabatrin at GABA-AT (blue) and 5-fluoromethylornithine at OAT (green). Vigabatrin displays a more ‘stretched’ conformation, whereas 5-FMOrn is more ‘compressed’, in order to fit into a similar space.
Finally, another aspect of ornithine that seems to be important for selectivity is the 2-amino group. As discussed earlier, in OAT, this moiety is stabilized by two tyrosine residues, Tyr55 and Tyr85; in GABA-AT, the corresponding residues are a phenylalanine and an isoleucine, which are more hydrophobic and less tolerant of charged groups. The inclusion of an amino group in this region has undoubtedly improved the selectivity of 5-fluoromethylornithine for OAT over GABA-AT, in addition to improving binding.
Conclusions
Although OAT and GABA-AT share only a small portion of their amino acid sequences, their overall structures and functions are surprisingly similar. Because of its increasing relevance as a therapeutic target, we have analyzed OAT on the basis of our knowledge of GABA-AT, with the ultimate goal of finding new avenues for selective inactivator design. This goal could be achieved by modifying existing GABA-AT inactivators, designing all new OAT inactivators, or improving the selectivity of current pan-inhibitors of PLP-dependent enzymes.
Because their catalytic mechanisms are essentially identical, the same mechanistically important groups that are contained in GABA-AT inactivators are applicable to new OAT-targeting molecules. In addition, because the substrate anchor points (the cofactor PLP and a conserved arginine residue) are approximately equidistant in both enzymes, the generally smaller GABA-AT inactivators can be used as templates for the design of selective inhibitors. However, there are certain differences in the active sites that can allow for some compounds to be selective for one enzyme over the other. We examined the particular case of gamma-ethynyl GABA (GEG), which inactivates both enzymes via different mechanisms; this is ostensibly the result of a more confined binding site for OAT in the region of the reactive alkyne. Finally, we identified aspects of substrates and drugs that seem to be important for selectivity for OAT over GABA-AT. These include the longer chain length of ornithine and its 2-amino group.
This short review should help in the search of a clinically viable inactivator of OAT that could be applicable to the treatment of proliferative disorders like hepatocellular carcinoma (HCC), an aggressive cancer in dire need of more effective treatments.
Figure 5.
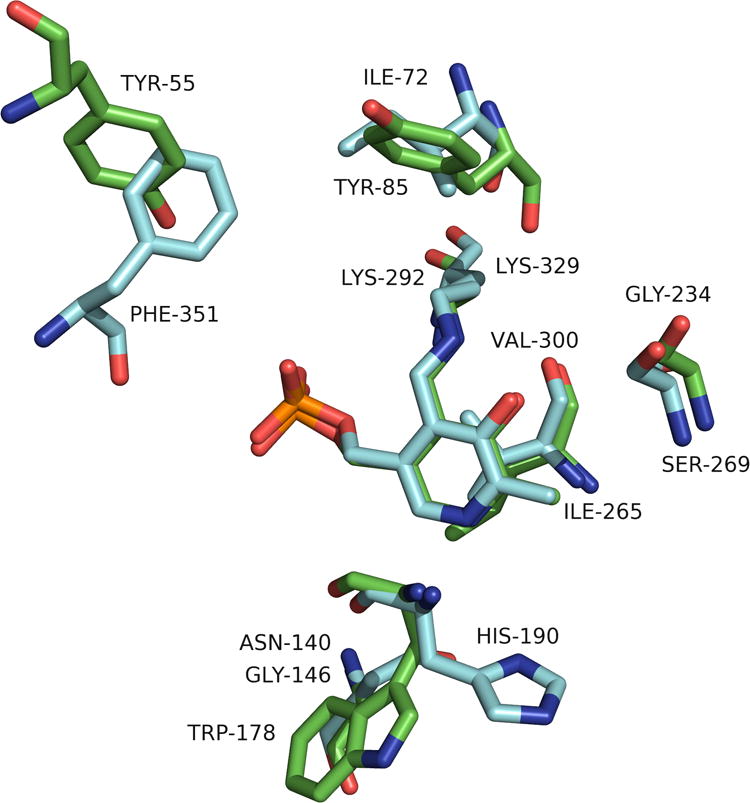
Superimposition of GABA-AT (cyan) and OAT (green) that highlights only the different residues in the active sites.
Biographies
Hyunbeom Lee earned his BS degree in chemistry in 2008 from Illinois Institute of Technology in Chicago, Illinois. He received his Ph.D. degree in organic chemistry in 2014 at Northwestern University, under the supervision of Professor Richard B. Silverman. Hyunbeom's research was mainly focused on the investigation and elucidation of the mechanism of inactivation of gamma-aminobutyric acid aminotransferase as a target for the treatment of epilepsy. He also investigated several other PLP-dependent enzymes, namely ornithine aminotransferase, glutamate decarboxylase, aspartate aminotransferase, and alanine aminotransferase. He will be pursuing his research endeavors at Korea Institute of Science and Technology in the field of molecular recognition research.
Jose I. Juncosa, Jr. obtained his License in Chemistry (summa cum laude) at Simon Bolivar University in Caracas, Venezuela, in 2005. That year, he also obtained his degree as a Performing Professor of Piano. He then obtained his Ph.D. degree in Medicinal Chemistry and Molecular Pharmacology at Purdue University, in West Lafayette, Indiana, under the supervision of Dr. David E. Nichols, partly supported by a Purdue Doctoral Fellowship. His graduate research involved the design and synthesis of novel dopamine D1 and serotonin 5-HT2A receptor agonists, and molecular modeling of dopamine and serotonin receptors. After obtaining his Ph.D. degree, he joined the group of Prof. Richard B. Silverman at Northwestern University as a postdoctoral fellow, where he currently designs, synthesizes, and evaluates irreversible inhibitors of GABA aminotransferase. In fall 2014, Dr. Juncosa will begin an appointment as Assistant Professor of Chemistry at Salisbury University in Salisbury, Maryland.
Richard B. Silverman received his B.S. degree in chemistry from The Pennsylvania State University in 1968 and his Ph.D. degree in organic chemistry from Harvard University in 1974 (with time off for a two-year military obligation from 1969-1971). After two years as a NIH postdoctoral fellow with the late Robert Abeles in enzymology at Brandeis University, he joined the chemistry faculty at Northwestern University. Since 2004 he has been the John Evans Professor of Chemistry. His recent awards include: Medicinal Chemistry Hall of Fame, American Chemical Society (2009), Perkin Medal (2009), Hall of Fame, Central High School of Philadelphia (2011), E.B. Hershberg Award for Important Discoveries in Medicinally Active Substances, American Chemical Society (2011), Fellow, American Chemical Society (2011), Sato Memorial International Award, Pharmaceutical Society of Japan (2012), BMS-Smissman Award, American Chemical Society (2013), Centenary Prize, Royal Society of Chemistry (2013), Excellence in Medicinal Chemistry Prize, Israel Chemical Society (2014), elected to the American Academy of Arts & Sciences (2014). Silverman is the inventor of Lyrica™, a drug marketed by Pfizer for epilepsy, neuropathic pain, and fibromyalgia and has completed Phase I clinical trials of another drug for infantile spasms. He has published over 325 research articles, holds 57 domestic and foreign patents, and has written five books; the 3rd edition of The Organic Chemistry of Drug Design and Drug Action was published in May 2014.
References
- 1.Alexander FW, Sandmeier E, Mehta PK, Christen P. Evolutionary relationships among pyridoxal-5′-phosphate-dependent enzymes. Regio-specific alpha, beta and gamma families. European journal of biochemistry / FEBS. 1994;219(3):953–960. doi: 10.1111/j.1432-1033.1994.tb18577.x. [DOI] [PubMed] [Google Scholar]
- 2.Jansonius JN. Structure, evolution and action of vitamin B6-dependent enzymes. Current opinion in structural biology. 1998;8(6):759–769. doi: 10.1016/s0959-440x(98)80096-1. [DOI] [PubMed] [Google Scholar]
- 3.Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 4.Shen BW, Hennig M, Hohenester E, Jansonius JN, Schirmer T. Crystal structure of human recombinant ornithine aminotransferase. J Mol Biol. 1998;277(1):81–102. doi: 10.1006/jmbi.1997.1583. [DOI] [PubMed] [Google Scholar]
- 5.Hwang BY, Cho BK, Yun H, Koteshwar K, Kim BG. Revisit of aminotransferase in the genomic era and its application to biocatalysis. J Mol Catal B: Enzym. 2005;37(1–6):47–55. [Google Scholar]
- 6.Jeremiah S, Povey S. The biochemical genetics of human gamma-aminobutyric acid transaminase. Ann Hum Genet. 1981;45(Pt 3):231–236. doi: 10.1111/j.1469-1809.1981.tb00334.x. [DOI] [PubMed] [Google Scholar]
- 7.Cooper AJ. Glutamate-gamma-aminobutyrate transaminase. Methods Enzymol. 1985;113:80–82. doi: 10.1016/s0076-6879(85)13019-3. [DOI] [PubMed] [Google Scholar]
- 8.Herzfeld A, Knox WE. The properties, developmental formation, and estrogen induction of ornithine aminotransferase in rat tissues. The Journal of biological chemistry. 1968;243(12):3327–3332. [PubMed] [Google Scholar]
- 9.Peraino C, Bunville LG, Tahmisian TN. Chemical, physical, and morphological properties of ornithine Aminotransferase from rat liver. The Journal of biological chemistry. 1969;244(9):2241–2249. [PubMed] [Google Scholar]
- 10.Mehta PK, Hale TI, Christen P. Evolutionary relationships among aminotransferases. Tyrosine aminotransferase, histidinol-phosphate aminotransferase, and aspartate aminotransferase are homologous proteins. European journal of biochemistry / FEBS. 1989;186(1-2):249–253. doi: 10.1111/j.1432-1033.1989.tb15202.x. [DOI] [PubMed] [Google Scholar]
- 11.Jung MJ, Seiler N. Enzyme-activated irreversible inhibitors of L-ornithine:2-oxoacid aminotransferase. Demonstration of mechanistic features of the inhibition of ornithine aminotransferase by 4-aminohex-5-ynoic acid and gabaculine and correlation with in vivo activity. J Biol Chem. 1978;253(20):7431–7439. [PubMed] [Google Scholar]
- 12.Storici P, Capitani G, Muller R, Schirmer T, Jansonius JN. Crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-fluoromethylornithine. J Mol Biol. 1999;285(1):297–309. doi: 10.1006/jmbi.1998.2289. [DOI] [PubMed] [Google Scholar]
- 13.Markova M, Peneff C, Hewlins MJE, Schirmer T, John RA. Determinants of Substrate Specificity in ω-Aminotransferases. J Biol Chem. 2005;280(43):36409–36416. doi: 10.1074/jbc.M506977200. [DOI] [PubMed] [Google Scholar]
- 14.Gale K. GABA in epilepsy: the pharmacologic basis. Epilepsia. 1989;30(3):S1–11. doi: 10.1111/j.1528-1157.1989.tb05825.x. [DOI] [PubMed] [Google Scholar]
- 15.Wu JY, Bird ED, Chen MS, Huang WM. Abnormalities of neurotransmitter enzymes in Huntington's chorea. Neurochem Res. 1979;4(5):575–586. doi: 10.1007/BF00964435. [DOI] [PubMed] [Google Scholar]
- 16.Perry TL, Hansen S, Lesk D. Plasma amino acid levels in children of patients with Huntington's chorea. Neurology. 1972;22(1):68–70. doi: 10.1212/wnl.22.1.68. [DOI] [PubMed] [Google Scholar]
- 17.Sherif FM, Ahmed SS. Basic aspects of GABA-transaminase in neuropsychiatric disorders. Clin Biochem. 1995;28(2):145–154. doi: 10.1016/0009-9120(94)00074-6. [DOI] [PubMed] [Google Scholar]
- 18.Lippert B, Metcalf BW, Jung MJ, Casara P. 4-amino-hex-5-enoic acid, a selective catalytic inhibitor of 4-aminobutyric-acid aminotransferase in mammalian brain. European journal of biochemistry / FEBS. 1977;74(3):441–445. doi: 10.1111/j.1432-1033.1977.tb11410.x. [DOI] [PubMed] [Google Scholar]
- 19.Pan Y, Gerasimov MR, Kvist T, Wellendorph P, Madsen KK, Pera E, Lee H, Schousboe A, Chebib M, Bräuner-Osborne H, Craft CM, Brodie JD, Schiffer WK, Dewey SL, Miller SR, Silverman RB. (1S, 3S)-3-Amino-4-difluoromethylenyl-1-cyclopentanoic Acid (CPP-115), a Potent γ-Aminobutyric Acid Aminotransferase Inactivator for the Treatment of Cocaine Addiction. J Med Chem. 2011 doi: 10.1021/jm201231w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin Z, Mendu S, Birnir B. GABA is an effective immunomodulatory molecule. Amino acids. 2013;45(1):87–94. doi: 10.1007/s00726-011-1193-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jirholt J, Asling B, Hammond P, Davidson G, Knutsson M, Walentinsson A, Jensen JM, Lehmann A, Agreus L, Lagerstrom-Fermer M. 4-aminobutyrate aminotransferase (ABAT): genetic and pharmacological evidence for an involvement in gastro esophageal reflux disease. PloS one. 2011;6(4):e19095. doi: 10.1371/journal.pone.0019095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seiler N. Ornithine aminotransferase as a therapeutic target in hyperammonemias. Advances in experimental medicine and biology. 1997;420:113–142. doi: 10.1007/978-1-4615-5945-0_8. [DOI] [PubMed] [Google Scholar]
- 23.Seiler N. Ornithine aminotransferase, a potential target for the treatment of hyperammonemias. Current drug targets. 2000;1(2):119–153. doi: 10.2174/1389450003349254. [DOI] [PubMed] [Google Scholar]
- 24.Wang G, Shang L, Burgett AWG, Harran PG, Wang X. Diazonamide toxins reveal an unexpected function for ornithine δ-amino transferase in mitotic cell division. Proceedings of the National Academy of Sciences. 2007;104(7):2068–2073. doi: 10.1073/pnas.0610832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyasaka Y, Enomoto N, Nagayama K, Izumi N, Marumo F, Watanabe M, Sato C. Analysis of differentially expressed genes in human hepatocellular carcinoma using suppression subtractive hybridization. British journal of cancer. 2001;85(2):228–234. doi: 10.1054/bjoc.2001.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Donnell JJ, Sandman RP, Martin SR. Gyrate atrophy of the retina: inborn error of L-ornithin:2-oxoacid aminotransferase. Science. 1978;200(4338):200–201. doi: 10.1126/science.635581. [DOI] [PubMed] [Google Scholar]
- 27.Shih VE, Berson EL, Mandell R, Schmidt SY. Ornithine ketoacid transaminase deficiency in gyrate atrophy of the choroid and retina. American journal of human genetics. 1978;30(2):174–179. [PMC free article] [PubMed] [Google Scholar]
- 28.Ueda M, Masu Y, Ando A, Maeda H, Del Monte MA, Uyama M, Ito S. Prevention of ornithine cytotoxicity by proline in human retinal pigment epithelial cells. Investigative Ophthalmology & Visual Science. 1998;39(5):820–7. [PubMed] [Google Scholar]
- 29.Weleber RG, Kurz DE, Trzupek KM. Treatment of retinal and choroidal degenerations and dystrophies: current status and prospects for gene-based therapy. Ophthalmology clinics of North America. 2003;16(4):583–593. doi: 10.1016/s0896-1549(03)00072-5. [DOI] [PubMed] [Google Scholar]
- 30.Kaneko S, Ueda-Yamada M, Ando A, Matsumura S, Okuda-Ashitaka E, Matsumura M, Uyama M, Ito S. Cytotoxic Effect of Spermine on Retinal Pigment Epithelial Cells. Investigative Ophthalmology & Visual Science. 2007;48(1):455–463. doi: 10.1167/iovs.06-0379. [DOI] [PubMed] [Google Scholar]
- 31.Powers R, Copeland JC, Germer K, Mercier KA, Ramanathan V, Revesz P. Comparison of protein active site structures for functional annotation of proteins and drug design. Proteins. 2006;65(1):124–135. doi: 10.1002/prot.21092. [DOI] [PubMed] [Google Scholar]
- 32.Alessio A, Amadasi A. Pyridoxal 5′-phosphate enzymes as targets for therapeutic agents. Current medicinal chemistry. 2007;14(12):1291–1324. doi: 10.2174/092986707780597899. [DOI] [PubMed] [Google Scholar]
- 33.Mehta PK, Hale TI, Christen P. Aminotransferases: demonstration of homology and division into evolutionary subgroups. European journal of biochemistry / FEBS. 1993;214(2):549–561. doi: 10.1111/j.1432-1033.1993.tb17953.x. [DOI] [PubMed] [Google Scholar]
- 34.Toney MD, Pascarella S, De Biase D. Active site model for gamma-aminobutyrate aminotransferase explains substrate specificity and inhibitor reactivities. Protein science : a publication of the Protein Society. 1995;4(11):2366–2374. doi: 10.1002/pro.5560041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Storici P, Capitani G, De Biase D, Moser M, John RA, Jansonius JN, Schirmer T. Crystal Structure of GABA-Aminotransferase, a Target for Antiepileptic Drug Therapy. Biochemistry. 1999;38(27):8628–8634. doi: 10.1021/bi990478j. [DOI] [PubMed] [Google Scholar]
- 36.De Biase D, Barra D, Simmaco M, John RA, Bossa F. Primary structure and tissue distribution of human 4-aminobutyrate aminotransferase. European journal of biochemistry / FEBS. 1995;227(1-2):476–480. doi: 10.1111/j.1432-1033.1995.tb20412.x. [DOI] [PubMed] [Google Scholar]
- 37.Denesyuk AI, Denessiouk KA, Korpela T, Johnson MS. Functional attributes of the phosphate group binding cup of pyridoxal phosphate-dependent enzymes. J Mol Biol. 2002;316:155–172. doi: 10.1006/jmbi.2001.5310. [DOI] [PubMed] [Google Scholar]
- 38.Storici P, De Biase D, Bossa F, Bruno S, Mozzarelli A, Peneff C, Silverman RB, Schirmer T. Structures of γ-Aminobutyric Acid (GABA) Aminotransferase, a Pyridoxal 5′-Phosphate, and [2Fe-2S] Cluster-containing Enzyme, Complexed with γ-Ethynyl-GABA and with the Antiepilepsy Drug Vigabatrin. J Biol Chem. 2004;279(1):363–373. doi: 10.1074/jbc.M305884200. [DOI] [PubMed] [Google Scholar]
- 39.Shah SA, Shen BW, Brünger AT. Human ornithine aminotransferase complexed with L-canaline and gabaculine: structural basis for substrate recognition. Structure (London, England : 1993) 1997;5(8):1067–1075. doi: 10.1016/s0969-2126(97)00258-x. [DOI] [PubMed] [Google Scholar]
- 40.Liu W, Peterson PE, Carter RJ, Zhou X, Langston JA, Fisher AJ, Toney MD. Crystal Structures of Unbound and Aminooxyacetate-Bound Escherichia coli γ-Aminobutyrate Aminotransferase. Biochemistry. 2004;43(34):10896–10905. doi: 10.1021/bi049218e. [DOI] [PubMed] [Google Scholar]
- 41.John RA. Pyridoxal phosphate-dependent enzymes. Biochim Biophys Acta. 1995;1248(2):81–96. doi: 10.1016/0167-4838(95)00025-p. [DOI] [PubMed] [Google Scholar]
- 42.Malashkevich VN, Toney MD, Jansonius JN. Crystal structures of true enzymatic reaction intermediates: aspartate and glutamate ketimines in aspartate aminotransferase. Biochemistry. 1993;32(49):13451–13462. doi: 10.1021/bi00212a010. [DOI] [PubMed] [Google Scholar]
- 43.Liu W, Peterson PE, Langston JA, Jin X, Zhou X, Fisher AJ, Toney MD. Kinetic and Crystallographic Analysis of Active Site Mutants of Escherichia coli γ-Aminobutyrate Aminotransferase. Biochemistry. 2005;44(8):2982–2992. doi: 10.1021/bi048657a. [DOI] [PubMed] [Google Scholar]
- 44.Kirsch JF, Eichele G, Ford GC, Vincent MG, Jansonius JN, Gehring H, Christen P. Mechanism of action of aspartate aminotransferase proposed on the basis of its spatial structure. J Mol Biol. 1984;174(3):497–525. doi: 10.1016/0022-2836(84)90333-4. [DOI] [PubMed] [Google Scholar]
- 45.Rando RR. Mechanism of the irreversible inhibition of gamma-aminobutyric acid-alpha-ketoglutaric acid transaminase by the neutrotoxin gabaculine. Biochemistry. 1977;16(21):4604–4610. doi: 10.1021/bi00640a012. [DOI] [PubMed] [Google Scholar]
- 46.Jung MJ, Lippert B, Metcalf BW, Schechter PJ, Bohlen P, Sjoerdsma A. The effect of 4-amino hex-5-ynoic acid (gamma-acetylenic GABA, gammma-ethynyl GABA) a catalytic inhibitor of GABA transaminase, on brain GABA metabolism in vivo. J Neurochem. 1977;28(4):717–723. doi: 10.1111/j.1471-4159.1977.tb10618.x. [DOI] [PubMed] [Google Scholar]
- 47.Seiler N, Spraggs H, Daune G. Interrelationships between ornithine, glutamate and GABA-I. Feed-back inhibition of ornithine aminotransferase by elevated brain GABA levels. Neurochem Int. 1987;10(3):391–397. doi: 10.1016/0197-0186(87)90115-x. [DOI] [PubMed] [Google Scholar]
- 48.Pan Y, Qiu J, Silverman RB. Design, synthesis, and biological activity of a difluoro-substituted, conformationally rigid vigabatrin analogue as a potent gamma-aminobutyric acid aminotransferase inhibitor. J Med Chem. 2003;46(25):5292–5293. doi: 10.1021/jm034162s. [DOI] [PubMed] [Google Scholar]
- 49.Rahiala EL, Kekomaki M, Janne J, Raina A, Raiha NC. Inhibition of pyridoxal enzymes by L-canaline. Biochim Biophys Acta. 1971;227(2):337–343. doi: 10.1016/0005-2744(71)90065-9. [DOI] [PubMed] [Google Scholar]
- 50.Daune G, Gerhart F, Seiler N. 5-Fluoromethylornithine, an irreversible and specific inhibitor of L-ornithine:2-oxo-acid aminotransferase. The Biochemical journal. 1988;253(2):481–488. doi: 10.1042/bj2530481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kito K, Sanada Y, Katunuma N. Mode of inhibition of ornithine aminotransferase by L-canaline. J Biochem. 1978;83(1):201–206. doi: 10.1093/oxfordjournals.jbchem.a131892. [DOI] [PubMed] [Google Scholar]
- 52.Bolkenius FN, Knodgen B, Seiler N. DL-canaline and 5-fluoromethylornithine. Comparison of two inactivators of ornithine aminotransferase. The Biochemical journal. 1990;268(2):409–414. doi: 10.1042/bj2680409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nanavati SM, Silverman RB. Design of potential anticonvulsant agents: mechanistic classification of GABA aminotransferase inactivators. J Med Chem. 1989;32(11):2413–2421. doi: 10.1021/jm00131a001. [DOI] [PubMed] [Google Scholar]
- 54.Silverman RB. Mechanism-based enzyme inactivators. Methods Enzymol. 1995;249:240–283. doi: 10.1016/0076-6879(95)49038-8. [DOI] [PubMed] [Google Scholar]
- 55.Nanavati SM, Silverman RB. Mechanisms of inactivation of .gamma.-aminobutyric acid aminotransferase by the antiepilepsy drug. gamma-vinyl GABA (vigabatrin) J Am Chem Soc. 1991;113(24):9341–9349. [Google Scholar]
- 56.Burke JR, Silverman RB. Mechanism of inactivation of .gamma.-aminobutyric acid aminotransferase by 4-amino-5-hexynoic acid (.gamma-ethynyl GABA) J Am Chem Soc. 1991;113(24):9329–9340. [Google Scholar]
- 57.De Biase D, Simmaco M, Barra D, Bossa F, Hewlins M, John RA. Mechanism of inactivation and identification of sites of modification of ornithine aminotransferase by 4-aminohex-5-ynoate. Biochemistry. 1991;30(8):2239–2246. doi: 10.1021/bi00222a029. [DOI] [PubMed] [Google Scholar]
- 58.Silverman RB, Levy MA. Irreversible inactivation of pig brain gamma-aminobutyric acid-alpha-ketoglutarate transaminase by 4-amino-5-halopentanoic acids. Biochem Biophys Res Commun. 1980;95(1):250–255. doi: 10.1016/0006-291x(80)90731-7. [DOI] [PubMed] [Google Scholar]
- 59.Vasel'ev VY, Eremin VP, Severin ES, Sytinskii IA. Comparative characterization of γ-aminobutyrate glutamate transaminases from pig kidney and liver. Biokhimiya (Moscow) 1973;38(2):355–364. [PubMed] [Google Scholar]
- 60.Worthen DR, Ratliff DK, Rosenthal GA, Trifonov L, Crooks PA. Structure-activity studies of L-canaline-mediated inhibition of porcine alanine aminotransferase. Chemical research in toxicology. 1996;9(8):1293–1297. doi: 10.1021/tx9600199. [DOI] [PubMed] [Google Scholar]