


Abstract
Statistical and clustering analyses of gene expression results from high-density microarray experiments produce lists of hundreds of genes regulated differentially, or with particular expression profiles, in the conditions under study. Independent of the microarray platforms and analysis methods used, these lists must be biologically interpreted to gain a better knowledge of the patho-physiological phenomena involved. To this end, numerous biological annotations are available within heterogeneous and widely distributed databases. Although several tools have been developed for annotating lists of genes, most of them do not give methods for evaluating the relevance of the annotations provided, or for estimating the functional bias introduced by the gene set on the array used to identify the gene list considered. We developed Genome Functional INtegrated Discoverer (GFINDer), a web server able to automatically provide large-scale lists of user-classified genes with functional profiles biologically characterizing the different gene classes in the list. GFINDer automatically retrieves annotations of several functional categories from different sources, identifies the categories enriched in each class of a user-classified gene list and calculates statistical significance values for each category. Moreover, GFINDer enables the functional classification of genes according to mined functional categories and the statistical analysis is of the classifications obtained, aiding better interpretation of microarray experiment results. GFINDer is available online at http://www.medinfopoli.polimi.it/GFINDer/.
INTRODUCTION
The post-genomic era has led to high-throughput methodologies that generate a massive amount of experimental data at an exponential rate. While in the past biologists studied single genes at a time, nowadays both the genomic sequences of many organisms (e.g. human, mouse, rat and many other animals and plants) and the high-throughput technologies that allow the investigation of gene expressions and mutations on a whole genomic scale are available. Among the latter, the most promising is microarray technology, which enables the analysis of tens of thousands of genes simultaneously generating a great amount of data.
Many efforts are being made to develop statistical analysis and clustering methods to analyse microarray experiment results and to identify groups of genes with similar expression patterns. However, independent of the microarray platform and data processing method used to identify differentially expressed genes, the common task any researcher faces is to translate the identified lists of genes into a better understanding of the biological phenomena involved. This, which was initially done via tedious searches through the literature and a number of public databases, prompted the development of automatic methods that could help in biologically interpreting microarray experiment results.
Several tools have been developed for annotating lists of genes identified in microarray experiments with biological information increasingly available from heterogeneous and widely distributed public databases [e.g. Unigene (1), LocusLink (2), Swiss-Prot (3), PFAM (4), KEGG (5), OMIM (6)]. However, most of these do not provide any means to evaluate the relevance of the annotations retrieved for the list of genes considered. Recently, a few tools have been proposed that use gene annotations provided through the Gene Ontology (GO) controlled vocabularies (7) to enrich lists of genes with biological information. Some of these [e.g. DAVID (8), Affymetrix (9), FatiGO (10), GoMiner (11), MAPPFinder (12) and GOTM (13)] also present the GO categories more relevant for a given set of genes according to the number of genes of the considered set belonging to a given category.
To enable the performance of statistical functional evaluations of user-classified sequence data, we developed the GFINDer web server. It allows the annotation of large numbers of user-classified sequence identifiers with the information present in different databases, functionally classifying them according to several functional categories (i.e. biological processes, molecular functions, cellular components, biochemical pathways, protein domains and genetic diseases) and statistically analysing the classifications obtained. The statistical analyses provided enable the evaluation of the functional bias of lists of candidate-regulated genes identified through microarray experiments and the highlighting of significant biological characteristics of the analysed gene sets. Moreover, they allow the detection of patterns of differential expression in classes of genes with specific functional characteristics, hence facilitating a genomic approach to the understanding of the fundamental biological processes and complex cellular patho-physiological mechanisms.
MATERIALS AND METHODS
Using information technologies, which allow the management and analysis of a vast quantity of biological data with a simple user interface, we developed GFINDer, a web server that enables the performance of statistical functional evaluations of user-classified sequence data.
System architecture
The GFINDer web server system is implemented in a three-layer architecture based on a multi-database structure (Figure 1). In the first layer, the data layer, a MySQL DBMS server manages all different types of annotations and data results provided. The core system engine is based on a relational database, Master DB, that maintains information about the web server users and their uploaded lists of classified sequence data. Another relational database keeps information about the GO structure (i.e. terms and relationships between them), and a third relational database stores many different gene annotations, including associations between genes and GO categories. These last two databases are kept updated by automatic procedures, implemented in the Java programming language, that automatically retrieve gene annotations and GO information from several online databanks as soon as new releases become available.
Figure 1.

The three-layer GFINDer system architecture.
In the second layer, the processing layer, a web server manages the requests coming from client computers and runs all system processing and analyses. This is the main layer of the GFINDer system. It conststs of Active Server Page scripts and uses Microsoft ActiveX Data Object technology and Standard Query Language to communicate with the DBMS server on the data layer, which is connected through a fast Local Area Network (LAN).
The third layer, the user layer, is composed of any client computer connected to the web server on the processing layer through an Internet/intranet communication network and that loads in its web browser the GFINDer graphic user interface, which is implemented as web pages using Hyper Text Markup Language.
This choice of a three-layer architecture maximizes the GFINDer system performance because it enables the subdivision of the required computational power between the two web and DBMS servers. Moreover, any user can easily utilize our system through a friendly web interface from any where an Internet connection is available.
Statistical analysis
To investigate and better interpret the relevance of biological annotations of a group of genes, statistical descriptions and analyses of the annotations should be used. When the considered genes are selected from a predefined set or subdivided into classes, to evaluate the statistical significance of specific annotation categories provided through controlled vocabularies, for each considered gene class the number of genes and their frequency, distribution and probability of occurrence in each category can be considered. Several different statistical approaches can be used to calculate a probability value for a given annotation category.
If we consider a group of genes, for instance the N genes on a microarray, any of these genes either belongs to a given category or not, e.g. M of the N genes are in category A and N − M are not. Moreover, independent of the statistical analysis or clustering method applied to the results of an experiment using that microarray, at least a subset K of the N genes on the microarray is selected and assigned to a given class (e.g. class 1, regulated genes). Of these K genes, x will be in category A and it is important to find out what the probability is of this happening by chance. This probability is appropriately modelled by a hypergeometric distribution with parameters (N, M, K) (14,15). Based on this, the P-value of having x genes or fewer of category A can be calculated by summing the probabilities of a random list of K genes having 1, 2,…, x genes of category A (14,15):
![]()
This corresponds to a one-sided test in which small P-values relate to under-represented categories. A one-sided test for over-represented categories can also be performed. In this case, the P-value for over-represented categories can be calculated as 1 − P.
Nevertheless, the hypergeometric distribution is rather difficult and time consuming to calculate when the total number N of considered genes is high. Currently, this occurs in many microarrays that include tens of thousands of genes. For example, the HG-U133 (A + B) set from Affymetrix Inc. contains 44 759 unique probes, which represent 42 731 unique sequences from the GenBank database corresponding to 25 516 unique UniGene clusters and 17 820 individual genes. However, it is well known that the hypergeometric distribution tends to the binomial distribution when N is large. If a binomial distribution is used, the probability of having x genes of category A in a set of K randomly picked genes is given by the formula of the binomial probability in which the probability of extracting a gene from category A is estimated by the ratio M/N of the category A genes present on the microarray, and the P-value for over-represented categories can be calculated as
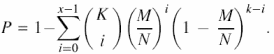
Alternative approaches to easily calculate the probability of having x genes of category A if we pick randomly K of the N genes include the Chi-square test (χ2) or test for equality of two proportions, and the Fisher's exact test (16). Both these tests are based on data arranged in a 2 × 2 table for a particular gene category and class of interest (e.g. category A, class 1). Thus, according to the above example, this 2 × 2 table must have marginal row and column totals N1., N2., N.1, and N.2 representing the total number of genes in the considered category A, in all the other categories, in the considered class 1 and in all the other classes, respectively (i.e. N1. = K, N2. = N − K, N.1 = M, N.2 = N − M).
Unfortunately, the χ2 test for equality of proportions cannot distinguish between under- and over-represented gene categories and cannot be used for small samples. All expected frequencies Eij = Ni. · N.j/N should be ≥5 for the test to provide valid conclusions. When this is not the case, the Fisher's exact test can be used (16,17). In Fisher's exact test the marginal totals N1., N2., N.1, N.2 of the 2 × 2 table rows and columns are considered to be fixed and the hypergeometric distribution is used to calculate the probability of observing an individual table combination. The P-value of a particular occurrence is the sum of all probabilities lower than or equal to the probability corresponding to the observed combination (16). If this P-value is <0.05, the null hypothesis of equal proportions can be rejected and the observed combination can be affirmed to be different from that expected by chance alone. However, as no-parametric tests, the χ2 and Fisher's exact tests have less power than the hypergeometric and binomial distribution tests.
In GFINDer we implemented the hypergeometric and binomial distribution tests and the Fisher's exact test to assess the statistical significance of the biological annotations over-represented in a group of genes. Because its characteristics are not completely appropriate to our application, we did not implement the χ2 test for equality of proportions. Therefore, using GFINDer the user can select any of the three implemented tests. Nevertheless, differences in the resulting P-values using the three statistics are observable for small values of the number N of considered genes. In fact, only when N is large, the binomial distribution does approximate the hypergeometric well. However, because the hypergeometric distribution test requires a greater number of combinatorial operations than the binomial test, it appears more appropriate especially for samples that require a reasonable computational time, i.e. with a total number N of genes that is not high.
Web interface
The GFINDer user interface is meant to increase, at maximum system usage, ease and friendliness, allowing the evaluation of the functional significance of microarray experiment results through graphical views and statistical indexes in a web browser environment accessible from anywhere an Internet connection is available. Our implemented web user interface is organized in modules allowing users to study the distribution of different classes of genes among GO categories, KEGG biochemical pathways, PFAM protein domains or OMIM diseases. Each module provides a specific task, as follows.
Uploading and Annotation modules
Through the Uploading module the user can input a list of genes (e.g. selected by means of microarray experiments and specified by either GenBank accession numbers, RefSeq IDs, Affymetrix probe IDs, UniGene cluster IDs or LocusLink IDs) to the GFINDer web server. In the list, each gene can appear classified within predefined classes identified by any symbol (e.g. 1, −1, 0). For example, these classes can represent gene expression regulations obtained from microarray experiments, or user classifications that resulted from any clustering method, or different experimental biological conditions.
The Annotation module enables production of a tabular output of the uploaded gene list enriched with several annotations, including gene names and symbols, LocusLink identifiers, protein product identifiers (from the NCBI LocusLink database) and GO categories with their evidence. Clicking on an annotated gene name opens a new window and displays more useful annotations about that gene. These include Unigene Cluster ID, Affymetrix ID, UniSTS ID and Swiss-Prot ID; the organism the gene belongs to; its cytogenetic localization, EC Number, biochemical pathways (from the KEGG database), protein product domains (from the PFAM database), genetic diseases (from the OMIM database), citations in scientific literature (i.e. PubMed links) and links to other databases such as the GDB Human Genome Data Base (18), Mouse Genome Informatics (MGI) Database (19) and Rat Genome Database (RGD) (20). Each of these annotations is linked to the corresponding original resource to display more information about that gene.
Exploration modules
The Gene Ontology module exploits the GO semantic network to perform analyses on the GO categories to which the genes in the loaded list belong. By choosing the level of ontology tree coverage (low levels provide high coverage but low term specificity; high levels lead to low coverage but to high term specificity), the module shows the GO categories represented by the input gene list from the ontology root down to the specified level of the ontology tree. For each GO category, the category name and the specific ontology to which it belongs (i.e. biological process, molecular function or cellular component), the absolute and percentage number of genes in the input list that belong to the category and the list of these genes (Figure 2), and links to external viewers of the ontology structure from the category up to the ontology root are provided. A histogram graphical representation of the distribution of the genes in the loaded list among the represented GO categories is also given. Therefore, this module enables the user to easily and graphically understand either how many and which GO categories are related to the considered genes, or how many genes refer to each GO category, also providing many useful annotations on each gene and the tools to graphically understand the semantic relations among the represented categories.
Figure 2.

The gene list window.
The Pathway module performs a functional taxonomy of the genes in the input list on the basis of the KEGG biochemical pathways the genes are involved in. The result shows the gene distribution among biochemical pathways and provides for each gene a link to additional annotations available through the KEGG's DBGET system. The user can also get the list of the considered genes that belong to each pathway.
The Protein Domain module produces a functional classification of the input genes according to the protein domains present in the gene protein products, as given by the PFAM databank (4). The result illustrates the distribution of input genes among protein domains and provides for each gene a link to additional annotations available through the PFAM website. As in the Pathway Exploration module, in addition the user can get the list of the considered genes that belong to each protein domain.
The Disease module shows the distribution of input genes among the genetic diseases and disorders they are involved in, as given by the OMIM databank, and provides for each gene a link to additional annotations available through the OMIM website. Similarly to the other exploration modules, the user can get the list of the considered genes that are related to each disease.
Categorization module
This module enables the definition of groups of input genes according to their membership of specific annotation categories and in relation to user-selected terms. User-defined keywords can be input and searched within the controlled vocabularies of selected annotations (i.e. GO biological processes, cellular components, molecular functions; KEGG biochemical pathways; PFAM protein domains; and OMIM diseases). The annotations related to the user keywords are shown and the input genes with these annotations are grouped in a category represented by those keywords. Then, the defined categorizations can be statistically analysed.
Statistical modules
If in the loaded input list genes are grouped in classes or a reference gene list is also loaded (e.g. the list of all the genes in the microarray used to produce the loaded list of genes to analyse), GFINDer allows statistical analyses to be performed on the GO, KEGG, PFAM and OMIM categorizations of the input genes. This enables the highlighting of which biological processes, molecular functions, cellular components, biochemical pathways, protein domains and genetic diseases the genes in the whole input list, or in each class contained, are related to, and with what probability. Thus, a plain list of gene identifiers is enriched with biological meaning and statistical significances.
In the GFINDer web interface, specific modules are available to statistically estimate the relevance of the GO, KEGG, PFAM and OMIM annotations provided for the input gene list. To this end, the annotated genes are grouped accordingly to their annotation categories, and their distribution among the categories considered is statistically evaluated as previously illustrated in the Statistical analysis section.
The Gene Ontology module (Figure 3) allows statistical analyses of the GO categories represented in the input gene list, defining the level of specificity and coverage of the GO hierarchy to be considered. After selecting a specific gene class, the module automatically and recursively considers each GO category represented in that class and provides a result table containing the observed number of input genes, their expected number and the significance P-value of each GO category in the selected class.
Figure 3.

Statistical analysis of the GO biological process categories represented in the RA-repressed genes considered. Red and blue P-values, and corresponding vertical lines on histogram bars, indicate the 1% and 5% significance levels, respectively.
Similarly, the Pathway, Protein Domain and Disease modules provide statistical analyses of the biochemical pathways, protein domains and genetic diseases, respectively, of a user-selected gene class in the input gene list. They show a result table containing the observed number of input genes, their expected number and the significance P-value of each biochemical pathway, protein domain and genetic disease of the selected gene class.
RESULTS
Using the GFINDer web server, the typical analysis steps that can be performed are as follows:
input classified sequence IDs (e.g. probes of genes identified as regulated in a microarray experiment);
determination of the individual genes represented in the considered probe set (i.e. present on the used microarray) and in the user-identified classes to analyse (e.g. up- and down-regulated genes);
- dynamic mining of available annotations from different online databases;
- functional categorization of the identified genes according to the retrieved annotations (e.g. biological processes, cellular components, molecular functions, biological pathways, protein domains and diseases);
- determination of gene functional categorizations according to user-selected terms within the controlled vocabularies of the retrieved annotations;
evaluation of statistically significant categories for each user gene class in relation to the experimental functional bias induced by the genes included in the considered reference gene set (e.g. all the genes in the microarray used);
output tabular and graphical visualization of resulting significant gene functional categories within the user gene classes.
To demonstrate GFINDer's potentialities, we used it to functionally analyse the results of a microarray experiment aimed at identifying genes that are differentially expressed in U937 cells after 4 h of treatment with 10−6 M retinoic acid (RA). In this experiment, two copies of the Affymetrix HG-U133 chip set (HG-U133A and HG-U133B) containing 44 759 unique probes were used. Absolute and comparative evaluations of the microarray gene expression results were performed through classical replica analyses and statistical methods, and only those genes that were differentially expressed in both RA treated samples compared to both controls were considered significantly regulated. This led to the identification of 805 unique probes, which were classified into 386 RA-induced and 419 RA-repressed genes. These probes were submitted to the GFINDer web server together with the initial pool of 44 759 probes as a reference set.
GFINDer determined that the 805 submitted probes represented 718 unique UniGene clusters and 594 individual genes, while the reference probe set included 17 820 genes, which were automatically annotated. We discovered that 179 genes out of the 594 were related to biological process annotations (86 genes highly expressed in the RA-induced class versus 93 highly expressed in the RA-repressed class), 162 to cellular component annotations (81 RA-induced and 81 RA-repressed), 202 to molecular function annotations (98 induced and 104 repressed by RA treatment), 79 to biochemical pathways (44 induced and 35 repressed by RA treatment), 302 to protein domains (165 RA-induced and 137 RA-repressed) and 346 to genetic diseases (169 induced and 177 repressed by RA treatment).
Following, we used GFINDer to statistically evaluate the relevance of the biological process GO categories within the identified RA-induced and RA-repressed genes. We concentrated on those functional categories significant at 5% (P < 0.05) and represented by at least two genes. The highlighted categories (Figures 3 and 4) agree with the functions that can be presumably induced or repressed in the experimental condition considered. In fact, the RA treatment of U937 cells results in partial differentiation along the myelomonocytic lineage, and the analysis of differential gene expression at an early time point (4 h) of the RA treatment aims at identifying genes that are involved in the early phases of the differentiation process. These are genes with functions related to the early phases of the RA response, such as control of cell differentiation, development and proliferation processes. Such findings validate the approach implemented and made available through the GFINDer web server.
Figure 4.

Hierarchical Gene Ontology tree of the most statistically significant biological process category (i.e. cell cycle checkpoint) identified for the RA-repressed genes considered. As clearly appears, this is a child and a more specific category of the cell proliferation category.
DISCUSSION
The development of high-throughput technologies has generated the need for bioinformatics approaches that can help in biologically interpreting microarray experiment results. Although several tools have been proposed for annotating lists of genes identified in microarray experiments, most of them give no methods for evaluating the significance of the annotations provided for a considered gene list. Such evaluation is related to the analysis of the functional bias introduced by the set of genes present on the array used to identify the specific list of genes, and it is particularly useful for the interpretation of functional annotations, which can lead to a better understanding of the biological phenomena involved in a specific experimental condition.
The GFINDer web server we developed includes an annotation module as well as a number of data mining and analysis modules, which enable the most relevant functional annotations within user-defined classes of genes to be highlighted, independently of the methods used to define them. GFINDer automatically translates lists of differentially regulated genes into functional profiles of the following categories: biological processes, cellular components, molecular functions, biochemical pathways, protein domains and genetic diseases, providing statistical significance values for each category. The controlled vocabularies representing these categories enable functional annotations of a given set of genes on a genomic scale and across different species. Moreover, the GO categories, through their hierarchical tree structure, allow the description of a very wide range of biological specificity, from very general to very precise concepts, using the exact correspondent terms. For this reason, GO terms are often used to give semantic biological classifications of genes. However, GO classifications can be usefully complemented with the biochemical pathways, protein domains and genetic diseases a gene is known to be involved in, which are provided by our web server.
Allowing the user to upload gene lists with predefined classifications (e.g. groups of genes obtained by applying clustering algorithms on gene expression values), GFINDer also enables the performance of functional statistical analyses of these classifications according to the membership of each gene in a class to specific functional categories. To our knowledge, this important feature is not available in other similar tools.
The Exploration and Statistical modules implemented in GFINDer allow the user to easily and rapidly observe the difference in the distribution of functional categories among different sets of genes (e.g. the different gene sets that resulted to be regulated in different microarray experiments, or belonging to distinct gene classes identified through expression profile clustering). Moreover, the statistical significance of the distribution of a gene set among different functional categories enables the immediately identification of the most relevant biological categories for that set of genes. This helps in better interpreting microarray experiment results and in highlighting new biological knowledge about the genes considered.
Finally, it is important to note that the annotations and analyses provided by GFINDer can only be as accurate as the underlining online databases from which the annotations are retrieved. The GFINDer web server is freely available online for academic and non-profit use at http://www.medinfopoli.polimi.it/GFINDer/.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Myriam Alkalay and Natalia Meani for providing the experimental data used to validate GFINDer.
REFERENCES
- 1.Schuler G.D. (1997) Pieces of the puzzle: expressed sequence tags and the catalog of human genes. J. Mol. Med., 75, 694–698. [DOI] [PubMed] [Google Scholar]
- 2.Pruitt K., Tatusov,T. and Maglott,D. (2001) RefSeq and LocusLink: NCBI gene-cantered resources. Nucleic Acids Res., 29, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boeckmann B., Bairoch,A., Apweiler,R., Blatter,M.C., Estreicher,A. and Gasteiger,E. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res., 31, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sonnhammer E.L.L., Eddy,S.R. and Durbin,R. (1997) PFAM: a comprehensive database of protein domain families based on seed alignments. Proteins, 28, 405–420. [DOI] [PubMed] [Google Scholar]
- 5.Kanehisa M. and Goto,S. (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acid Res., 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKusick V.A. (1998) Mendelian Inheritance in Man. A Catalog of Human Genes and Genetic Disorders, 12th edn. Johns Hopkins University Press, Baltimore, MD. [Google Scholar]
- 7.The Gene Ontology Consortium (2000) Gene Ontology: tool for the unification of biology. Nature Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glynn D. Jr, Sherman,B.T., Hosack,D.A., Yang,J., Gao,W. and Lane,H.C. (2003) DAVID: Database for Annotation, visualization, and Integrated Discovery. Genome Biol., 4, R60. [PubMed] [Google Scholar]
- 9.Liu G., Loraine,A.E., Shigeta,R., Cline,M., Cheng,J., Valmeekam,V., Sun,S., Kulp,D. and Siani-Rose,M.A. (2003) NetAffx: Affymetrix probesets and annotations. Nucleic Acids Res., 31, 82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al-Shahrour A.F., Díaz-Uriarte,R. and Dopazo,J. (2004) FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics, 20, 578–580. [DOI] [PubMed] [Google Scholar]
- 11.Zeeberg B.R., Feng,W., Wang,G., Wang,M.D., Fojo,A.T. and Sunshine,M. (2003) GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol., 4, R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doniger S.W., Salomonis,N., Dahlquist,K.D., Vranizan,K., Lawlor,S.C. and Conklin,B.R. (2003) MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol., 4, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang B., Schmoyer,D., Kirov,S. and Snoddy,J. (2004) GOTree Machine (GOTM): a web-based platform for interpreting sets of interesting genes using Gene Ontology hierarchies. BMC Bioinformatics, 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tavazoie S., Hughes,J.D., Campbell,M.J., Cho,R.J. and Church,G.M. (1999) Systematic determination of genetic network architecture. Nature Genet., 22, 281–285. [DOI] [PubMed] [Google Scholar]
- 15.Casella G. and Berger,R.L. (2002) Statistical Inference. 2nd edn. Duxbury Press, Belmont, CA. [Google Scholar]
- 16.Fisher L.D. and van Belle,G. (1993) Biostatistics: A Methodology for the Health Sciences. John Wiley & Sons, New York. [Google Scholar]
- 17.Stokes M.E., Davis,C.S. and Koch,G.G. (2001) Categorical Data Analysis Using the SAS System. 2nd edn. John Wiley & Sons, New York. [Google Scholar]
- 18.Letovsky S.I., Cottingham,R.W., Porter,C.J. and Li,P.W. (1998) GDB: the Human Genome Database. Nucleic Acids Res., 26, 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blake J.A., Richardson,J.E., Davisson,M.T. and Eppig,J.T. (1997) The Mouse Genome Database (MGD). A comprehensive public resource of genetic, phenotypic and genomic data. The Mouse Genome Informatics Group. Nucleic Acids Res., 25, 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Twigger S., Lu,J., Shimoyama,M., Chen,D., Pasko,D., Long,H., Ginster,J., Chen,C.F., Nigam,R., Kwitek,A. et al. (2002) Rat Genome Database (RGD): mapping disease onto the genome. Nucleic Acids Res., 30, 125–128. [DOI] [PMC free article] [PubMed] [Google Scholar]