

Abstract
Aims
Previously published pharmacokinetic (PK) models for sunitinib and its active metabolite SU12662 were based on a limited dataset or lacked important elements such as correlations between sunitinib and its metabolite. The current study aimed to develop an improved PK model that circumvented these limitations and to prove the utility of the PK model in treatment optimization in clinical practice.
Methods
One thousand two hundred and five plasma samples from 70 cancer patients were collected from three PK studies with sunitinib and SU12662. A semi-physiological PK model for sunitinib and SU12662 was developed incorporating pre-systemic metabolism using non-linear mixed effects modelling (nonmem). Allometric scaling based on body weight was applied. The final model was used for simulation of the PK of different treatment regimens.
Results
Sunitinib and SU12662 PK were best described by a one and two compartment model, respectively. Introduction of pre-systemic formation of SU12662 strongly improved model fit, compared with solely systemic metabolism. The clearance of sunitinib and SU12662 was estimated at 35.7 (relative standard error (RSE) 5.7%) l h−1 and 17.1 (RSE 7.4%) l h−1, respectively for 70 kg patients. Correlation coefficients were estimated between inter-individual variability of both clearances, both volumes of distribution and between clearance and volume of distribution of SU12662 as 0.53, 0.48 and 0.45, respectively. Simulation of the PK model predicted correctly the ratio of patients who did not reach proposed PK targets for efficacy.
Conclusions
A semi-physiological PK model for sunitinib and SU12662 in cancer patients was presented including pre-systemic metabolism. The model was superior to previous PK models in many aspects.
Keywords: modelling, pharmacokinetics, semi-physiological model, SU12662, sunitinib, therapeutic drug monitoring
What is Already Known about this Subject
The necessity and evidence of therapeutic drug monitoring has been reported for sunitinib and its active metabolite SU12662.
Pharmacokinetic modelling has proved its utility in therapeutic drug monitoring.
Limitations existed for a previous published pharmacokinetic model for sunitinib and SU12662 in the application of therapeutic drug monitoring.
What this Study Adds
We presented a superior pharmacokinetic model for sunitinib and SU12662 including pre-systemic metabolism.
The simulation of the current model obtained the correct concentration range for sunitinib and SU12662.
The model predicted correctly the ratio of patients who did not reach proposed PK targets for efficacy in clinic observations.
Introduction
Sunitinib (SU11248) is a tyrosine kinase inhibitor that has proven efficacy in the treatment of several tumours. It is currently approved for the treatment of metastatic renal cell carcinoma (mRCC), gastro-intestinal stromal tumours (GIST) and pancreatic neuroendocrine tumours (pNET) 1. Patients receive either an intermittent dosing schedule with 4 week dosing followed by 2 week rest (4/2 treatment, 50 mg once daily) or a continuous dosing regimen (37.5 mg once daily) based on the indication. N-desethyl sunitinib (SU12662), the active metabolite of sunitinib, shows similar activity to sunitinib. Both parent and metabolite compounds reach peak concentrations at 6–12 h after intake 1–6. Sunitinib is mainly metabolized by cytochrome P450 3A4 (CYP3A4) to SU12662, which is then further metabolized by CYP3A4 to inactive metabolites (Figure 1) 6. Sunitinib is primarily eliminated via the faeces (61%) with 16% of the dose excreted unchanged renally 6.
Figure 1.
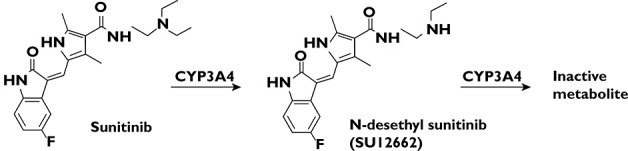
Structure of sunitinib and N-desethyl sunitinib (SU12662). Sunitinib is mainly metabolized by cytochrome P450 3A4 (CYP3A4) to SU12662 by N-de-ethlation. A further N-de-ethylation step mediated by CYP3A4 forms the inactive metabolite 6
In clinical practice, the necessity for dose individualization of sunitinib, for instance by using therapeutic drug monitoring (TDM), has gained increasing attention 7. Based on multiple environmental factors (like comedication) and (pharmaco-)genetic factors, plasma concentrations of sunitinib may vary widely between individual patients 8,9. The proposed pharmacokinetic (PK) target (total plasma trough concentration (Cmin) of sunitinib and SU12662) to obtain sufficient efficacy is 50 ng ml−1 for the 4/2 treatment regimen and 37.5 ng ml−1 for continuous dosing 7,10. TDM sampling of Cmin concentrations after once daily dosing is very impractical for clinical practice. However, by using appropriate extrapolation from a well evaluated population PK model, TDM can be applied using more or less randomly collected samples 11,12. In addition, such a PK model may help to improve quantitative understanding of the complex PK of sunitinib and SU12662 and their determinants 13–15.
Three PK models for sunitinib and SU12662 have been described thus far 16–18. Houk's model, albeit based on a rich dataset (590 subjects), disregarded irrationally the correlation between the parent and metabolite compounds which share similar PK pathways 16. Another PK model was developed based on PK data from 12 healthy volunteers 17. This limited and potentially non-representative population led to potentially biased parameter estimates. Besides, no covariates were considered. Previously, our group has described an empirical PK model for sunitinib and SU12662 which served sufficiently to find putative phenotypic and genotypic determinants of sunitinib PK in two clinical studies 18,19. However, this model may miss the information in pre-systemic formation of the metabolite which could lead to explaining the PK of sunitinib and SU12662 in a mechanistic way. Therefore, the objective of current study was to develop an improved semi-physiological PK model for sunitinib and SU12662 to overcome the shortcomings of published PK models.
Methods
Clinical studies
A total 1205 plasma samples (602 sunitinib samples and 603 SU12662 samples) from 70 cancer patients with complete PK data of sunitinib and SU12662 were obtained from three previously conducted clinical studies in multi-medical centres (Table 1) 18,20,21. All subjects received daily doses of sunitinib ranging from 25–50 mg orally. However, the complete dosing history was not clear for each patient. Samples of study 1 were collected on 2 days, 1 day during the period before steady-state and 1 after, at 0, 4, 8 and 24 h after dosing. Subjects from study 2 were sampled only at steady-state at 0, 1, 2, 4, 6, 8, 12 and 24 h post-dose. In study 3, plasma samples were also collected at steady-state at time points 0, 10, 20, 40 min and 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 24 h after sunitinib dosing.
Table 1.
Characteristics of patients and studies
| Number of patients | Number of samples | Dose (mg) | Weight (kg) median (range) | Sampling time points post-dose (h) | |
|---|---|---|---|---|---|
| All | 70 | 1205 | 25, 37.5, 50 | 82 (39–157) | |
| Study 1 [18] | 50 | 703 | 25, 37.5, 50 | 85 (39–157) | 0, 4, 8, 24 |
| Study 2 20 | 7 | 112 | 25, 37.5, 50 | 96 (53–105) | 0, 1, 2, 4, 6, 8, 12, 24 |
| Study 3 21 | 13 | 390 | 37.5, 50 | 70 (57–98) | 0, 0.17, 0.33, 0.67, 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 24 |
Software
Model estimation was performed using nonmem (version 7.3.0) 22 together with a gfortran compiler, using first order conditional estimation with interaction as estimation method. Piraña was used as modelling environment 23, and R (version 3.0.0) was used for processing of the data 24. Furthermore, the nonmem toolkit psn 25 and the R-package Xpose 26 and deSolve 27 were used.
Model development
For description of the PK of both sunitinib and SU12662 one and two compartment models were evaluated. Previously, significant pre-systemic metabolism of sunitinib has been suggested 1–6. A semi-physiological model to account for this phenomenon has been proposed for other compounds 15. In brief, a hypothetical enzyme compartment as part of the central compartment 15 is introduced to account for conversion of sunitinib to SU12662 both pre-systemically and systemically. The liver blood flow was assumed to be 80 l h−1 28. To evaluate the impact of this assumption on final parameter estimates, a sensitivity analysis was conducted in which a 25% lower or higher liver blood flow was assumed.
Bioavailability of sunitinib (F) is unknown. Therefore, all parameters of sunitinib were estimated relative to F. The fraction of sunitinib converted to SU12662 (fm) was assumed to be 21% 16. Due to the lack of full dose histories, pre-dose concentrations were handled using the ‘missing dose method’ as proposed by Soy et al. 29. This was performed by assuming and estimating the amount of drug in the central compartment for sunitinib and SU12662.
Inter-individual variability (IIV) was modelled using an exponential distribution according to (equation 1).
| 1 |
where Pi represents the individual parameter estimate for individual i, P represents the typical population parameter estimate and ηi the IIV effect distributed following N (0, ω2). Correlation between IIVs was estimated using the variance-covariance matrix. Starting with a full omega matrix including IIV on clearance and central volume distribution of both parent and metabolite compounds, correlations were eliminated sequentially (cut-off P = 0.01).
The residual errors were described by a proportional error model (equation 2).
| 2 |
where Cobs,ij or Cpred,ij represents, for the ith subject and the jth measurement, the observed concentration or predicted concentration. Proportional error εp,ij was assumed distributed following N (0, σ2). Different values for σ2 were estimated for the different studies to account for potential differences in residual variability between studies.
Body weight (WT) was included in the model a priori using allometric scaling for clearance (including Qh and Qi) and volume of distribution for both sunitinib and SU12662 (equations 3 and 4) 30.
| 3 |
| 4 |
where P represents WT-adjusted population parameter, and P0 represents baseline population parameter estimated for parameter P. For less than 6% of patients, WT was not available. These were imputed by taking the population mean weight of 70 kg.
Model selection and evaluation
Discrimination between models was guided by plausibility and precision of parameter estimates, correlation matrices (<0.85), shrinkage of ω and σ (<25%), goodness-of-fit (GOF) plots and drop of objective function value (OFV) with significance level of P < 0.01 (dOFV >6.63) for hierarchical models.
Graphical model assessment of the final model was conducted by evaluation of GOF plots 31 and prediction-corrected visual predictive check (pcVPC) 32. PcVPC was suggested to a be good diagnostic tool when there is large variability in data 32. Due to the involvement of data before and after steady-state which had considerable variability in our study, the pcVPC was used in model evaluation (1000 simulations).
Simulations of dosing regimens
Stochastic simulations with the final model were performed to demonstrate its utility. WT was simulated from a truncated log-normal distribution, with a mean of 82.3 kg, and a standard deviation of 19.4 kg, truncated between 39–157 kg. An intermittent dosing regimen (4/2 treatment, 50 mg once daily) and a continuous dosing regimen (37.5 mg once daily) were simulated separately for 1000 patients. Steady-state Cmin concentrations were recorded. The fraction of patients reaching the reported thresholds for Cmin was calculated. In addition, the time to reach >90% of the theoretical steady-state concentration was evaluated as this is important for clinical practice since reported target concentrations are related to steady-state concentrations. Therefore, the time to reach steady-state is crucial for implementing TDM of sunitinib. The theoretical steady-state concentration was calculated by equation 5.
| 5 |
where D represents the dose, CL represents clearance and Vc represents the central volume of distribution.
Results
The semi-physiological PK model including pre-systemic metabolism and correlation between parameters of sunitinib and SU12662 was successfully developed. Introduction of the pre-systemic metabolism in the model induced a strong improvement in model fit (dOFV >400) compared with a model lacking this formation. The final model consisted of a single compartment for sunitinib and two compartments for SU12662. Introduction to a peripheral compartment for sunitinib resulted in over-parameterization. A schematic representation of the model is depicted in Figure 2 (control stream in Appendix). It was assumed that the sunitinib concentration in the hypothetical enzyme site (Ch) was in equilibrium with the plasma concentration in the sunitinib central compartment as parameterized in equation 6. Sunitinib is firstly absorbed (equation 7) to the hypothetical enzyme compartment, where sunitinib remains either unchanged or is metabolized. Subsequently, unchanged sunitinib and metabolite distribute to the central compartment (equations 8 and 9). Sunitinib is further metabolized from the systemic circulation by the same enzyme. SU12662 distributes to its peripheral compartment and can also be metabolized from the systemic circulation (equations 9,10).
| 6 |
| 7 |
| 8 |
 |
9 |
 |
10 |
where A represents the amount of drug in a certain compartment, CL represents clearance, Ka represents the first order absorption rate constant, Vc and Vp represent the central and peripheral volumes of distribution, fm represents the fraction of sunitinib converted to SU12662, which was fixed to 0.21, Qh represents hepatic blood flow which was fixed at a value of 80 l h−1 as a population estimate for 70 kg individuals 28 and Qi represents inter-compartmental clearance between the central and peripheral compartment.
Figure 2.
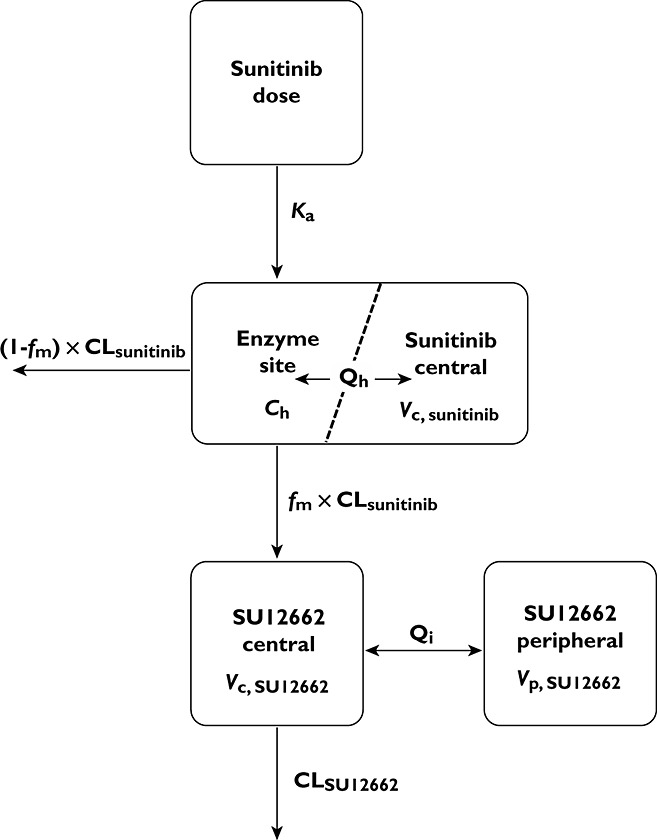
Schematic representation of semi-physiological pharmacokinetic (PK) model for sunitinib and N-desethyl sunitinib (SU12662). Ch, concentration at enzyme site; CL, clearance; fm, the fraction of sunitinib converted to SU12662; Ka, absorption rate constant; Qh, hepatic blood flow; Qi, inter-compartmental clearance; Vc, volume of distribution of central compartment; Vp, volume of distribution of peripheral compartment
The parameter estimates are summarized in Table 2. The CL of parent and metabolite drug was estimated at 35.7 (relative standard error (RSE) 5.7%) l h−1 and 17.1 (RSE 7.4%) l h−1 for 70 kg patients. The IIV of CL was found to be around 34% and 42%, respectively, for parent and metabolite drug. Correlation coefficients were estimated between inter-individual variability of both CL, both Vc, and between CL and Vc of SU12662 as 0.53, 0.48 and 0.45, respectively.
Table 2.
Parameter estimates for pharmacokinetic models for sunitinib and its active metabolite SU12662
| Parameter | Integrated models | Separate models | ||||
|---|---|---|---|---|---|---|
| Current model (n = 70) | Lindauer et al. 17 (n = 12) | Houk et al. 16 (n = 590) | ||||
| Estimate (RSE,%) | IIV, %CV (RSE,%) | Estimate (RSE,%)* | IIV, %CV (RSE,%)* | Estimate (RSE,%) | IIV, %CV (RSE,%) | |
| Sunitinib | ||||||
| RPS | – | – | 1.91(46.8) | – | – | – |
| MTT (h) | – | – | 1.48 (16.6) | – | – | – |
| N | – | – | 1.46 (20.6) | 61 (27) | – | – |
| Ka (h−1) | 0.34 (10.8) | – | 0.54 (10.1) | 26 (44) | 0.195 (6.8) | 81.2 (13) |
| CL (l h−1) | 35.7 (5.7) | 33.9 (12.0) | 32.4 (8.4) | 28 (17) | 37.7 (1.8) | 37.9 (8.8) |
| Vc (l) | 1360 (6.0) | 32.4 (10.6) | 1720 (10.7) | 33 (18) | 1940 (3.8) | 44.7 (13) |
| Qi (l h−1) | – | – | 3.3 (118) | – | 6.37 (18) | – |
| Qh (l h−1) | 80 fix | – | – | – | – | – |
| Vp (l) | – | – | 221 (27.0) | – | 588 (8.7) | – |
| σprop | 0.06 (13.5)† | – | 0.11 (9) | – | 0.147 (9.6) | – |
| 0.01 (10.5)‡ | – | |||||
| σadd (ng ml−1) | 0.07 (65.1) | – | – | – | ||
| SU12662 | ||||||
| Ka (h−1) | – | – | – | – | 0.287 (6.6) | 89.1 (13) |
| CL (l h−1) | 17.1 (7.4) | 42.1 (7.0) | 14.6 (12.5) | 43 (22) | 20.2 (2.3) | 52.2 (8.5) |
| Vc (l) | 635 (13.1) | 57.9 (8.8) | 1410 (15.4) | 47 (18) | 2710 (3.9) | 65.1 (8.7) |
| Qi (l h−1) | 20.1 (32.6) | – | – | – | 27.7 (26) | – |
| Vp (l) | 388 (14.9) | – | – | – | 345 (22) | – |
| σprop | 0.03 (14.1)† | – | 0.11 (10) | – | 0.0913 (7.3) | – |
| 0.01 (12.1)‡ | – | |||||
| σadd (ng ml−1) | 0.36 (22.0) | – | – | – | ||
| Correlations | ||||||
| ρ(CLsunitinib, Vc,sunitinib) | – | – | 0.88 (5.5) | – | – | – |
| ρ(CLsunitinib, CLSU12662) | 0.53 | – | 0.70 (18.7) | – | – | – |
| ρ(CLsunitinib, Vc,SU12662) | – | – | 0.67 (23.6) | – | – | – |
| ρ(Vc,sunitinib, CLSU12662) | – | – | 0.90 (5.1) | – | – | – |
| ρ(Vc,sunitinib, Vc,SU12662) | 0.48 | – | 0.90 (2.0) | – | – | – |
| ρ(CLSU12662, Vc,SU12662) | 0.45 | – | 0.99 (0.3) | – | – | – |
90% confidence intervals (CIs) were originally reported, RSEs were calculated according to 90%CIs.
σprop for study 1.
σprop for study 2 and study 3. ρ, correlation coefficient; σadd, additive residual error; σprop, proportional residual error; CL, clearance; CV, coefficient of variation; IIV, inter-individual variability; Ka, absorption rate constant; MTT, mean transit time; N, number of transit compartments; Qh, hepatic blood flow; Qi, intercompartment clearance; RPS, ratio of pre-systemic to systemic metabolite formation; RSE, relative standard error; Vc, volume of distribution of central compartment; Vp, volume of distribution of peripheral compartment.
In the sensitivity analysis for the liver blood flow (Qh), there was no change in estimates of both CL, less than 10% change in parent Vc, less than 20% change in metabolite Vc, less than 15% for metabolite Qi, and less than 5% for metabolite Vp, when Qh changed ± 25% from 80 l h−1. There was almost no change in estimate of IIV and residual errors.
The GOF plots showed that the developed final model adequately described the data (Figure 3). The pcVPC indicated our model appropriately captured the PK and variability of observed plasma concentrations for both sunitinib and SU12662 (Figure 4).
Figure 3.
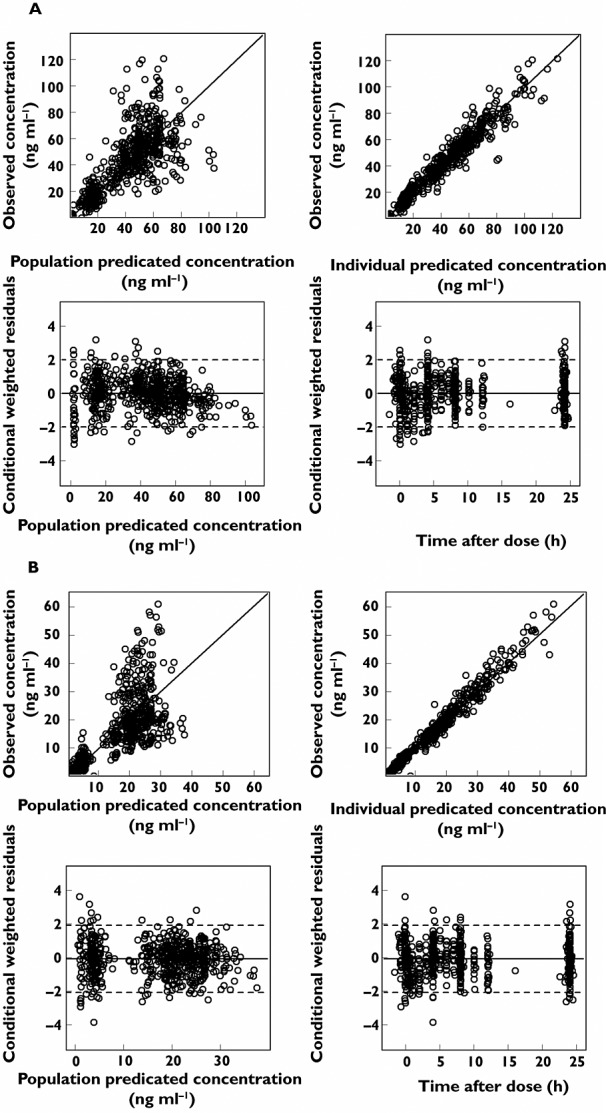
Goodness-of-fit plots for model predicted sunitinib (A) and SU12662 (B) concentrations. The plots include observed vs. population predicted concentration, observed vs. individual model predicted concentration, conditional weighted residuals (CWRES) vs. population predicted concentration and CWRES vs. time after dose
Figure 4.
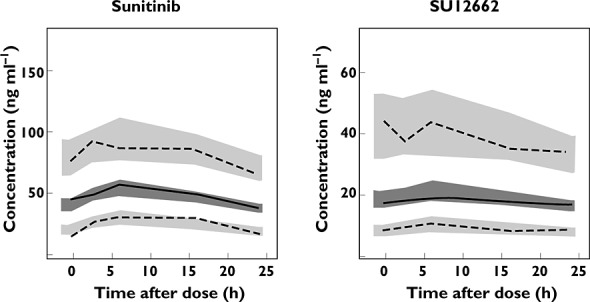
Prediction-corrected visual predictive check of sunitinib and SU12662 concentration. Solid lines and dark grey areas represent the median observed values and simulated 95% confidence intervals (CIs). Dashed lines and light grey areas represent the 5% and 95% percentiles of the observed values and 95% CIs of the simulated percentiles
Simulation of concentration−time profiles for 6 weeks with both intermittent and continuous dosing regimens using the final PK model are shown in Figure 5. At steady-state, for the intermittent dosing regimen, the median (25th−75th percentile) of combined Cmin concentration for sunitinib and SU12662 was at 64 (48–82) ng ml−1. 27% of simulated patients failed to reach the PK target of 50 ng ml−1 for combined Cmin concentration of sunitinib and SU12662. For continuous dosing, the median (25th−75th percentile) of combined Cmin concentration was 48 (36–61) ng ml−1. Also, 27% of simulated patients failed to reach the PK target of 37.5 ng ml−1 for combined Cmin concentration, while 53% of the patients did not reach the threshold of 50 ng ml−1 for combined Cmin concentration. The time to reach >90% of the theoretical steady-state concentration was approximately 6 days for sunitinib and 8 days for SU12662.
Figure 5.
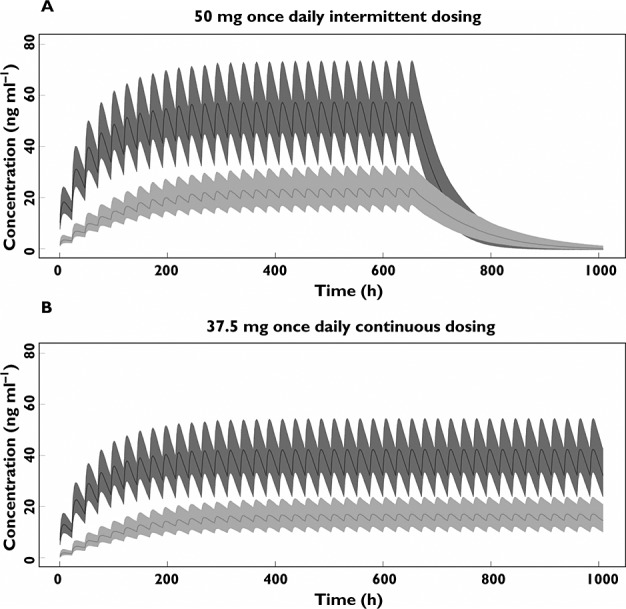
Simulation of pharmacokinetic model for repeated intermittent dosing regimen (50 mg once daily) (A) and continuous dosing regimen (37.5 mg once daily) (B) (n = 1000). The lines represent the median concentration; the areas represent 25th-75th percentile intervals. The dark grey shows concentration-time profile of sunitinib; the light grey shows SU12662.  , median sunitinib;
, median sunitinib;  , median SU12662;
, median SU12662;  , 25th-75th percentile sunitinib;
, 25th-75th percentile sunitinib;  25th-75th percentile SU12662
25th-75th percentile SU12662
Discussion
In this study, an integrated semi-physiological PK model for sunitinib and its active metabolite in cancer patients was successfully developed and evaluated. Table 2 shows a comparison between the parameter estimates of the developed model and two different previously developed models 16,17.
In several PK studies of sunitinib it has been observed that the majority and considerable amount of SU12662 is formed in the absorption phase of sunitinib 2–6. In the empirical model presented by Houk et al., a separate absorption rate of SU12662 was estimated which was found to be larger than the parent drug, suggesting a possible existence of a first pass effect of sunitinib 16, which was also pointed out by Lindauer et al. 17.
Considerable correlation between inter-individual variability of both CL, both Vc and between CL and Vc of SU12662 was observed which has also been suggested by Lindauer et al. 17. This could be attributed to the fact that all these parameter estimates were relatively scaled by the bioavailability. Besides, it is reasonable to consider the correlation of parameters, such as CL and Vc, between parent and metabolite compounds, due to the similarity in metabolism between the two compounds 6. This is also a very important observation for subsequent simulation studies, as neglecting correlation may result in the simulation of unrealistic PK data.
In the model presented by Houk et al., sunitinib and SU12662 were not considered simultaneously 16. As we discussed above, our model is superior in combining the parent and metabolite compounds in PK model with considering the correlation between the important parameters of both compounds. In the model developed by Lindauer et al. 17, the data was short in sample size (n = 12), inclusion of healthy volunteers only who are potentially non-representative for cancer patients and only data before steady-state. Our model was based on a much larger population of real cancer patients from multi-medical centres with both plasma concentrations determined before and after steady-state. These benefits resulted in a better reflection of clinical circumstances.
We applied our model in simulation of approved dosing regimens from which it was proved that the simulation of current PK model was consistent with the observation in published clinical studies for sunitinib and SU12662. Recently, we performed a study which evaluated the feasibility of TDM for sunitinib (n = 29) 10. At day 15 ± 1 day with continuous dosing of sunitinib 37.5 mg once daily, the median (25th−75th percentile) of the combined Cmin concentration of sunitinib and SU12662 was 49.5 (41.8–64.0) ng ml−1, which was in the similar range from the simulation study of the current model (48 (36–61) ng ml−1). The same study found that at day 15 ± 1 day, 52% of patients failed to reach the threshold of 50 ng ml−1 10, which is in agreement with the findings of the current model simulation (53% of the patients not reaching this target). Furthermore, the combined median Cmin concentrations after day 14 (64 ng ml−1 for intermittent dosing and 48 ng ml−1 for continuous dosing) are in the same range as the combined mean/median Cmin concentrations reported at steady-state by previous clinical studies from different groups 1,5,33–38. These findings suggested that the simulation of the current model reflected closely the clinical observation. We previously reported that based on the information collected from clinical practice, around half of the patients (n = 31), who received 25 mg (n = 2), 37.5 mg (n = 12), 50 mg (n = 15) or 62.5 mg (n = 2) of sunitinib daily, failed to reach the proposed PK target of 50 ng ml−1 39. This is remarkably higher than the fraction of 27% reported in our simulation study. This might be explained by several reasons. In the simulation study dose reductions and interruptions due to toxicity were not considered, while in clinical practice this frequently occurs. In addition, the drug plasma concentrations can also be influenced by different situations, e.g. comedication, food, smoking, etc. 8,9, in clinical practice, which were not considered in the simulation study.
Based on the parameter estimates from the current model, the theoretical Css,min concentrations were calculated. We have found that both sunitinib and SU12662 reached >90% of steady-state within 6–8 days. However, a period between 10–14 days for sunitinib and SU12662 before reaching steady-state has been reported 1.
The current PK model can be further used to study, for example, the influence of dose reduction and extrapolation of current plasma concentrations to future plasma concentrations, to predict the sufficiency of plasma concentrations in patients at the early stage of treatment. In addition, the PK model can be used in PK−pharmacodynamic (PD) modelling to understand further the toxicity-related/treatment-related exposure−response relationship in sunitinib treatment 40,41.
We presented here a semi-physiological PK model for sunitinib and its active metabolite SU12662 in cancer patients including pre-systemic metabolism. The model was superior to previous PK models of sunitinib and SU12662 in multiple aspects. It also adequately captured the PK of sunitinib and its metabolite as demonstrated by a simulation study.
Conflict of Interest
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work. Dr Nielka van Erp reports grants from Novartis, grants from GSK, grants from Janssen-Cilag in the previous 3 years. There are no other relationships or activities that could appear to have influenced the submitted work.
No sources of funding were used to assist in the preparation of this study.
Appendix
$PROBLEM sunitinib PK model
$INPUT ID TIME CMT DV AMT TLDOS ADDL II STUDY SS MDV PDOS DOSE SST STATE FLAG WT
$DATA PKsun.csv IGNORE = @
$SUBROUTINES ADVAN6 TRANS1 TOL = 4
$MODEL
NCOMP = 4
COMP = (DEPOT, DEFDOSE) ; Dosing compartment
COMP = (OBSLIV) ; Sunitinib central compartment
COMP = (CENTRALM) ; SU12662 central compartment
COMP = (PERIM) ; SU12662 peripheral compartment
$PK
ASCL = (WT/70)**0.75
ASV = WT/70
K12 = THETA(1)
V2 = THETA(2)*ASV * EXP(ETA(1))
QH = THETA(3)*ASCL
CLP = THETA(4)*ASCL* EXP(ETA(4))
CLM = THETA(5)*ASCL* EXP(ETA(3))
V3 = THETA(6)*ASV * EXP(ETA(2))
Q34 = THETA(7)*ASCL
V4 = THETA(8)*ASV
FM = THETA(9)
F2 = 0
IF(FLAG.EQ.0) F2 = THETA(10)
F3 = 0
IF(FLAG.EQ.0) F3 = THETA(11)
K34 = Q34/V3
K43 = Q34/V4
S2 = V2
S3 = V3
S4 = V4
$DES
Cliv = (K12*A(1) + QH/V2*A(2))/(QH+CLP)
DADT(1) = -K12*A(1)
DADT(2) = QH*Cliv-QH/V2*A(2)
DADT(3) = FM*CLP*Cliv-CLM/V3*A(3)-K34*A(3) + K43*A(4)
DADT(4) = K34*A(3)-K43*A(4)
$ERROR
TY = F
IF(F.LT.0.001) TY = 0.001
IPRED = TY
IF (CMT.EQ.2) Y = IPRED * (1 + EPS(1))
IF (CMT.EQ.3) Y = IPRED * (1 + EPS(2))
IF (STUDY.EQ.1.AND.CMT.EQ.2) Y = IPRED * (1 + EPS(3))
IF (STUDY.EQ.1.AND.CMT.EQ.3) Y = IPRED * (1 + EPS(4))
IRES = DV-IPRED
IWRES = IRES/IPRED
$THETA
(0, 0.6) ; 1 = K12 – Absorption rate constant
(0, 2000) ; 2 = V2 – Central volume of distribution of sunitinib
(80 FIX) ; 3 = QH – Hepatic blood flow
(0, 38) ; 4 = CLP – Clearance of sunitinib
(0, 20) ; 5 = CLM – Clearance of SU12662
(0, 1000) ; 6 = V3 – Central volume of distribution of SU12662
(0,30) ; 7 = Q34 – Inter-compartmental clearance of SU12662
(0, 800) ; 8 = V4 – Peripheral volumes of distribution of SU12662
(0.21 FIX) ; 9 = FM – Fraction of sunitinib converted to SU12662
(0, 20) ; 10 = F2 – Dealing with missing dose history for sunitinib
(0, 100) ; 11 = F3 – Dealing with missing dose history for SU12662
$OMEGA BLOCK (4)
0.09; IIV V2
0.05 0.09; IIV V3
0 0.05 0.09; IIV CLM
0 0 0.05 0.08; IIV CLP
$SIGMA
0.0188; Proportional error sunitinib for study 2&3
0.0102; Proportional error SU12662 for study 2&3
0.02 ; Proportional error sunitinib for study 1
0.02 ; Proportional error SU12662 for study 1
$EST METHOD = 1 INTER MAXEVAL = 9999 NOABORT SIG = 3 PRINT = 1 POSTHOC
$COV PRINT = E
References
- SUTENT 12.5 mg hard capsules. Summary of product characteristics. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000687/WC500057737.pdf (last accessed 23 December 2013)
- Bello C, Sherman L, Zhou J. Effect of food on the pharmacokinetics of sunitinib malate (SU11248), a multi-targeted receptor tyrosine kinase inhibitor: results from a phase I study in healthy subjects. Anticancer Drugs. 2006;17:353–358. doi: 10.1097/00001813-200603000-00015. [DOI] [PubMed] [Google Scholar]
- Sweeney CJ, Chiorean EG, Verschraegen CF, Lee FC, Jones S, Royce M, Tye L, Liau KF, Bello A, Chao R, Burris HA. A phase I study of sunitinib plus capecitabine in patients with advanced solid tumors. J Clin Oncol. 2010;28:4513–4520. doi: 10.1200/JCO.2009.26.9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starling N, Vázquez-Mazón F, Cunningham D, Chau I, Tabernero J, Ramos FJ, Iveson TJ, Saunders MP, Aranda E, Countouriotis AM, Ruiz-Garcia A, Wei G, Tursi JM, Guillen-Ponce C, Carrato A. A phase I study of sunitinib in combination with FOLFIRI in patients with untreated metastatic colorectal cancer. Ann Oncol. 2012;23:119–127. doi: 10.1093/annonc/mdr046. [DOI] [PubMed] [Google Scholar]
- Britten C, Kabbinavar F, Hecht J. A phase I and pharmacokinetic study of sunitinib administered daily for 2 weeks, followed by a 1-week off period. Cancer Chemother Pharmacol. 2008;61:515–524. doi: 10.1007/s00280-007-0498-4. [DOI] [PubMed] [Google Scholar]
- Speed B, Bu H, Pool WF, Peng GW, Wu EY, Patyna S, Bello C, Kang P. Pharmacokinetics, distribution, and metabolism of sunitinib in rats, monkeys, and humans. Drug Metab Dispos. 2012;40:539–555. doi: 10.1124/dmd.111.042853. [DOI] [PubMed] [Google Scholar]
- Yu H, Steeghs N, Nijenhuis CM, Schellens JHM, Beijnen JH, Huitema ADR. Practical guidelines for therapeutic drug monitoring of anticancer tyrosine kinase inhibitors: focus on the pharmacokinetic targets. Clin Pharmacokinet. 2014;53:305–325. doi: 10.1007/s40262-014-0137-2. [DOI] [PubMed] [Google Scholar]
- Mathijssen RHJ, Sparreboom A, Verweij J. Determining the optimal dose in the development of anticancer agents. Nat Rev Clin Oncol. 2014;11:272–281. doi: 10.1038/nrclinonc.2014.40. [DOI] [PubMed] [Google Scholar]
- Van Leeuwen RWF, van Gelder T, Mathijssen RHJ, Jansman FGA. Drug–drug interactions with tyrosine-kinase inhibitors: a clinical perspective. Lancet Oncol. 2014;15:e315–326. doi: 10.1016/S1470-2045(13)70579-5. [DOI] [PubMed] [Google Scholar]
- Lankheet NAG, Kloth JSL, Gadellaa-van Hooijdonk CGM, Cirkel GA, Mathijssen RHJ, Lolkema MPJK, Schellens JHM, Voest EE, Sleijfer S, de Jonge MJA, Haanen JBAG, Beijnen JH, Huitema ADR, Steeghs N. Pharmacokinetically guided sunitinib dosing: a feasibility study in patients with advanced solid tumours. Br J Cancer. 2014;110:2441–2449. doi: 10.1038/bjc.2014.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotta V, Widmer N, Montemurro M, Leyvraz S, Haouala A, Decosterd LA, Csajka C, Buclin T. Therapeutic drug monitoring of imatinib: Bayesian and alternative methods to predict trough levels. Clin Pharmacokinet. 2012;51:187–201. doi: 10.2165/11596990-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Zhao W, Cella M, Della Pasqua O, Burger D, Jacqz-Aigrain E. Population pharmacokinetics and maximum a posteriori probability Bayesian estimator of abacavir: application of individualized therapy in HIV-infected infants and toddlers. Br J Clin Pharmacol. 2012;73:641–650. doi: 10.1111/j.1365-2125.2011.04121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordi T, Xie R, Huong NV, Huong DX, Karlsson MO, Ashton M. A semiphysiological pharmacokinetic model for artemisinin in healthy subjects incorporating autoinduction of metabolism and saturable first-pass hepatic extraction. Br J Clin Pharmacol. 2005;59:189–198. doi: 10.1111/j.1365-2125.2004.02321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frechen S, Junge L, Saari TI, Suleiman AA, Rokitta D, Neuvonen PJ, Olkkola KT, Fuhr U. A semiphysiological population pharmacokinetic model for dynamic inhibition of liver and gut wall cytochrome P450 3A by voriconazole. Clin Pharmacokinet. 2013;52:763–781. doi: 10.1007/s40262-013-0070-9. [DOI] [PubMed] [Google Scholar]
- Abduljalil K, Frank D, Gaedigk A, Klaassen T, Tomalik-Scharte D, Jetter A, Jaehde U, Kirchheiner J, Fuhr U. Assessment of activity levels for CYP2D6*1, CYP2D6*2, and CYP2D6*41 genes by population pharmacokinetics of dextromethorphan. Clin Pharmacol Ther. 2010;88:643–651. doi: 10.1038/clpt.2010.137. [DOI] [PubMed] [Google Scholar]
- Houk BE, Bello CL, Kang D, Amantea M. A population pharmacokinetic meta-analysis of sunitinib malate (SU11248) and its primary metabolite (SU12662) in healthy volunteers and oncology patients. Clin cancer Res. 2009;15:2497–2506. doi: 10.1158/1078-0432.CCR-08-1893. [DOI] [PubMed] [Google Scholar]
- Lindauer A, Di Gion P, Kanefendt F, Tomalik-Scharte D, Kinzig M, Rodamer M, Dodos F, Sörgel F, Fuhr U, Jaehde U. Pharmacokinetic/pharmacodynamic modeling of biomarker response to sunitinib in healthy volunteers. Clin Pharmacol Ther. 2010;87:601–608. doi: 10.1038/clpt.2010.20. [DOI] [PubMed] [Google Scholar]
- Kloth JSL, Klümpen H-J, Yu H, Eechoute K, Samer CF, Kam BLR, Huitema ADR, Daali Y, Zwinderman AH, Balakrishnar B, Bennink RJ, Wong M, Schellens JHM, Mathijssen RHJ, Gurney H. Predictive value of CYP3A and ABCB1 phenotyping probes for the pharmacokinetics of sunitinib: the ClearSun study. Clin Pharmacokinet. 2014;53:261–269. doi: 10.1007/s40262-013-0111-4. [DOI] [PubMed] [Google Scholar]
- Diekstra MHM, Klümpen HJ, Lolkema MPJK, Yu H, Kloth JSL, Gelderblom H, van Schaik RHN, Gurney H, Swen JJ, Huitema ADR, Steeghs N, Mathijssen RHJ. Association analysis of genetic polymorphisms in genes related to sunitinib pharmacokinetics, specifically clearance of sunitinib and SU12662. Clin Pharmacol Ther. 2014;96:81–89. doi: 10.1038/clpt.2014.47. [DOI] [PubMed] [Google Scholar]
- Kloth JS, Binkhorst L, de Bruijn P, Burger H, Hamberg P, Wiemer E. Effect of dosing time on sunitinib pharmacokinetics. Eur J Cancer. 2013;49 : 692. [Google Scholar]
- De Wit D, Gelderblom H, Sparreboom A, den Hartigh J, den Hollander M, König-Quartel JMC, Hessing T, Guchelaar HJ, van Erp NP. Midazolam as a phenotyping probe to predict sunitinib exposure in patients with cancer. Cancer Chemother Pharmacol. 2014;73:87–96. doi: 10.1007/s00280-013-2322-7. [DOI] [PubMed] [Google Scholar]
- Beal SL, Sheiner LB. NONMEM User Guides. Ellicott City, MD, USA: Icon Development Solutions; 1989. [Google Scholar]
- Keizer RJ, van Benten M, Beijnen JH, Schellens JHM, Huitema ADR. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101:72–79. doi: 10.1016/j.cmpb.2010.04.018. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. [Google Scholar]
- Lindbom L, Pihlgren P, Jonsson EN, Jonsson N. PsN-Toolkit-a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79:241–257. doi: 10.1016/j.cmpb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Jonsson EN, Karlsson MO. Xpose-an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- Soetaert K, Petzoldt T, Setzer R. Solving Differential Equations in R: package deSolve. J Stat Softw. 2010;33:1–25. [Google Scholar]
- Carlisle KM, Halliwell M, Read AE, Wells PN. Estimation of total hepatic blood flow by duplex ultrasound. Gut. 1992;33:92–97. doi: 10.1136/gut.33.1.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soy D, Beal SL, Sheiner LB. Population one-compartment pharmacokinetic analysis with missing dosage data. Clin Pharmacol Ther. 2004;76:441–451. doi: 10.1016/j.clpt.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Holford NH. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30:329–332. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24:2187–2197. doi: 10.1007/s11095-007-9361-x. [DOI] [PubMed] [Google Scholar]
- Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13:143–151. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S, Blay JY, Casali PG, Le Cesne A, Stephenson P, Deprimo SE, Harmon CS, Law CNJ, Morgan JA, Ray-Coquard I, Tassell V, Cohen DP, Demetri GD. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumours after imatinib failure. Eur J Cancer. 2009;45:1959–1968. doi: 10.1016/j.ejca.2009.02.011. [DOI] [PubMed] [Google Scholar]
- Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol. 2010;66:357–371. doi: 10.1007/s00280-009-1170-y. [DOI] [PubMed] [Google Scholar]
- Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, Bello C, Deprimo S, Brega N, Massimini G, Armand J-P, Scigalla P, Raymond E. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol. 2006;24:25–35. doi: 10.1200/JCO.2005.02.2194. [DOI] [PubMed] [Google Scholar]
- Shirao K, Nishida T, Doi T, Komatsu Y, Muro K, Li Y, Ueda E, Ohtsu A. Phase I/II study of sunitinib malate in Japanese patients with gastrointestinal stromal tumor after failure of prior treatment with imatinib mesylate. Invest New Drugs. 2009;28:866–875. doi: 10.1007/s10637-009-9306-9. [DOI] [PubMed] [Google Scholar]
- Novello S, Scagliotti GV, Rosell R, Socinski MA, Brahmer J, Atkins J, Pallares C, Burgess R, Tye L, Selaru P, Wang E, Chao R, Govindan R. Phase II study of continuous daily sunitinib dosing in patients with previously treated advanced non-small cell lung cancer. Br J Cancer. 2009;101:1543–1548. doi: 10.1038/sj.bjc.6605346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- Lankheet NAG, Knapen LM, Schellens JHM, Beijnen JH, Steeghs N, Huitema ADR. Plasma concentrations of tyrosine kinase inhibitors imatinib, erlotinib, and sunitinib in routine clinical outpatient cancer care. Ther Drug Monit. 2014;36:326–334. doi: 10.1097/FTD.0000000000000004. [DOI] [PubMed] [Google Scholar]
- Van Hasselt JGC, Boekhout AH, Beijnen JH, Schellens JHM, Huitema ADR. Population pharmacokinetic-pharmacodynamic analysis of trastuzumab-associated cardiotoxicity. Clin Pharmacol Ther. 2011;90:126–132. doi: 10.1038/clpt.2011.74. [DOI] [PubMed] [Google Scholar]
- Van Hasselt JGC, Gupta A, Hussein Z, Beijnen JH, Schellens JHM, Huitema ADR. Population pharmacokinetic−pharmacodynamic analysis for eribulin mesilate-associated neutropenia. Br J Clin Pharmacol. 2013;76:412–424. doi: 10.1111/bcp.12143. [DOI] [PMC free article] [PubMed] [Google Scholar]