

Abstract
Aim
Apixaban is an orally active inhibitor of coagulation factor Xa and is eliminated by multiple pathways, including renal and non-renal elimination. Non-renal elimination pathways consist of metabolism by cytochrome P450 (CYP) enzymes, primarily CYP3A4, as well as direct intestinal excretion. Two single sequence studies evaluated the effect of ketoconazole (a strong dual inhibitor of CYP3A4 and P-glycoprotein [P-gp]) and diltiazem (a moderate CYP3A4 inhibitor and a P-gp inhibitor) on apixaban pharmacokinetics in healthy subjects.
Method
In the ketoconazole study, 18 subjects received apixaban 10 mg on days 1 and 7, and ketoconazole 400 mg once daily on days 4–9. In the diltiazem study, 18 subjects received apixaban 10 mg on days 1 and 11 and diltiazem 360 mg once daily on days 4–13.
Results
Apixaban maximum plasma concentration and area under the plasma concentration–time curve extrapolated to infinity increased by 62% (90% confidence interval [CI], 47, 78%) and 99% (90% CI, 81, 118%), respectively, with co-administration of ketoconazole, and by 31% (90% CI, 16, 49%) and 40% (90% CI, 23, 59%), respectively, with diltiazem.
Conclusion
A 2-fold and 1.4-fold increase in apixaban exposure was observed with co-administration of ketoconazole and diltiazem, respectively.
Keywords: anticoagulants, apixaban, CYP3A4 inhibitors, drug–drug interactions, P-glycoprotein inhibitors
What is Already Known about this Subject
Modulation of cytochrome P450 (CYP) metabolism or P-glycoprotein (P-gp)-mediated drug transport by co-administered drugs, herbal remedies or certain foods can cause clinically significant pharmacokinetic and pharmacodynamic effects.
Apixaban biotransformation occurs predominantly through oxidative metabolism via CYP3A4, although apixaban itself is neither an inducer nor an inhibitor of CYP enzymes.
What this Study Adds
Co-administration of ketoconazole, a strong inhibitor of CYP3A4 and P-gp, or diltiazem, a mechanism-based inhibitor of CYP3A4 and an inhibitor of P-gp, resulted in a ≤ 2-fold increase in apixaban exposure compared with apixaban alone.
Introduction
The coagulation factor Xa plays a pivotal role in the clotting cascade by prompting the conversion of prothrombin to thrombin, making factor Xa an important potential drug target for anticoagulation 1. Apixaban is an orally active, selective inhibitor of factor Xa 2,3 that is approved in a number of countries to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation 4,5, for thromboprophylaxis in patients who have undergone elective hip or knee replacement surgery 6–8 and for the treatment of venous thromboembolism, including deep vein thrombosis and pulmonary embolism 9,10.
Anticoagulants, including vitamin K antagonists and low molecular weight heparins, are associated with a risk of bleeding and require careful monitoring for signs and symptoms of bleeding. For example, dosing of vitamin K antagonists, such as warfarin, requires ongoing monitoring of the international normalized ratio (INR) for dose adjustment 11,12. Warfarin is subject to clinically significant interactions with food and drugs and requires even more frequent INR monitoring when starting or stopping concomitant interacting drugs. While clinical trials have shown that apixaban can be safely and effectively administered as a fixed dose without routine pharmacologic monitoring 4–8,10, apixaban also carries a potential increased risk of bleeding with increased exposure. Therefore, it is important to identify potential factors that could increase the exposure of apixaban.
Peak apixaban plasma concentration is reached approximately 3 h after oral administration in healthy subjects, with an average elimination half-life (t1/2) of approximately 12 h 13,14. Apixaban has a bioavailability of approximately 50% 15,16. Based on in vitro findings using a bidirectional permeability assay of P-glycoprotein (P-gp)–mediated drug transport (MDR-1 transfected LLC-PK1 cell monolayers), apixaban is a substrate for P-gp 17. Therefore, P-gp may play a role in limiting the oral bioavailability of apixaban through intestinal efflux 18.
Apixaban is eliminated by multiple pathways, including renal and non-renal elimination. Renal clearance approximates the glomerular filtration rate and accounts for approximately 27% of apixaban total systemic clearance 15,16. Non-renal elimination pathways consist of metabolism by cytochrome P450 (CYP) enzymes, with subsequent sulfation by sulfotransferases 19,20, as well as biliary and possible direct intestinal excretion 21,22. Apixaban metabolites account for approximately 25% of recovered radioactivity 19,23 and have no detectable pharmacological activity 24. A series of experiments with human cDNA-expressed CYP enzymes and CYP chemical inhibitors demonstrated that the oxidative metabolism of apixaban is predominantly catalyzed by CYP3A4, with only minor contributions from other CYP enzymes including CYP1A2, 2C8, 2C9, 2C19 and 2J2 20. Sulfate conjugation is mediated primarily by SULT1A1 24. In vitro studies indicate that apixaban, at concentrations corresponding to those in human subjects, is neither an inducer nor an inhibitor of CYP3A4 or other P450 enzymes 24. Thus, apixaban is unlikely to affect the CYP-mediated metabolism of other drugs. Furthermore, the multiple dose pharmacokinetics (PK) in human subjects is time independent, demonstrating that exposure to apixaban does not result in induction or inhibition of its own metabolism 13. Thus, on the basis of these data, modulation of P-gp and/or CYP3A4 activity by other concomitant medication appears to present the greatest potential for drug interactions with apixaban.
CYP3A4 is the major CYP enzyme expressed in the human intestine 25 and liver 26,27. CYP3A4 is known to be involved in the metabolism of a wide variety of xenobiotic compounds, including many therapeutic drugs 28–31. Drugs that are activated or eliminated by CYP3A4 metabolism can potentially have drug–drug interactions (DDI) with other therapies that may induce or inhibit CYP3A4, and these interactions may result in clinically significant PK effects. CYP3A4 modulators consist of prescription and non-prescription drugs as well as other xenobiotics found in certain herbal remedies and food products 32–34 and can also result in clinically significant effects on some substrates 35–38. When evaluating the impact of CYP3A4 modulators, the functional association between the drug efflux transporter, P-gp, and CYP3A4 should also be considered, since some drugs modulate both systems as inhibitors or inducers 39, and the net effect of P-gp and CYP3A4 modulation could be greater than the impact of modulation of either pathway alone. For example, ketoconazole, widely used in DDI studies as a representative strong inhibitor of both CYP3A4 and P-gp, increases midazolam exposure by up to 16-fold 40,41. Diltiazem, a mechanism-based moderate inhibitor of CYP3A4 42 and a P-gp inhibitor 41,43,44, increases midazolam exposure less than 5-fold 40.
Although a role for CYP3A4-mediated metabolism and P-gp-mediated transport has been shown for apixaban in vitro, clinical PK studies are necessary to determine the potential clinical impact of concomitant administration of CYP3A4 and P-gp modulators. This report presents data on the effect of CYP3A4 and P-gp inhibitors on apixaban PK. The first of the two studies described in this report (ketoconazole DDI study) investigated the interaction between ketoconazole 400 mg once daily dosed to steady-state and a single 10 mg dose of apixaban. To define further the potential for DDI between moderate CYP3A4 inhibitors and apixaban, effects of co-administered diltiazem (360 mg once daily) dosed to steady-state on the PK of a single 10 mg dose of apixaban were examined in the second study (diltiazem DDI study).
Methods
Patients and study design
These studies were open label, non-randomized, single sequence, crossover studies conducted in healthy subjects at the Bristol-Myers Squibb Clinical Research Center (Hamilton, New Jersey, USA). Non-smoking men and women, 18–45 years of age, with a body mass index of 18–30 kg m−2 were eligible for the studies. The main exclusion criteria included history of bleeding disorder, history of gastrointestinal disease or surgery that would interfere with absorption of study drug, use of any hormonal contraceptive within 3 months, use of any prescription drug or over-the-counter acid controller within 4 weeks and use of any other drug or dietary supplement known to increase the potential for bleeding within 2 weeks before dosing. Exclusion criteria in the diltiazem DDI study also included patients with a history of significant arrhythmia, sinus bradycardia, low blood pressure or orthostatic hypotension. The studies were conducted in accordance with the principles stated in the Declaration of Helsinki and were consistent with International Conference on Harmonization Good Clinical Practice and applicable regulatory requirements. The protocol and patient consent form for both studies were approved by the New England Institutional Review Board (Wellesley, Massachusetts, USA) prior to study start. All subjects provided informed, written consent prior to the initiation of any study specific procedures.
Treatments
Subjects in both studies were admitted to the study centre on the day before the first dose and remained there until study completion. A summary of the study design and treatment schedule for each study is shown in Figure 1.
Figure 1.
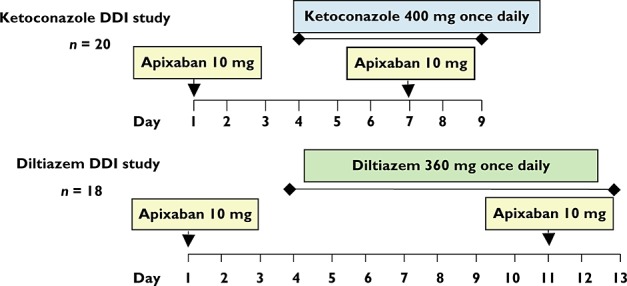
Study treatment schedules. Apixaban was administered as single doses on two occasions, ketoconazole and diltiazem as multiple daily doses for 6 and 10 days, respectively. DDI, drug–drug interaction
Subjects in the ketoconazole DDI study received a single 10 mg tablet of apixaban on days 1 and 7. In addition, from days 4–9, subjects received 400 mg once daily of ketoconazole (Nizoral®, Sanofi-aventis, Bridgewater, New Jersey, USA). Subjects in the diltiazem DDI study received a single 10 mg tablet of apixaban on days 1 and 11. Once daily doses of diltiazem 360 mg (Cardizem® LA, Abbott Laboratories, North Chicago, Illinois, USA) were given on days 4–13. The 10 mg single dose of apixaban was chosen for both studies because it represented the high end of the dose range tested in phase III trials. Subjects in both studies were required to fast (except for water) from 10 h before until 4 h after apixaban administration. In addition, subjects fasted from 1 h before until 1 h after diltiazem dosing in the diltiazem DDI study. Subjects were not permitted to consume alcohol-containing beverages, grapefruit, grapefruit-containing products, citrus juices or other citrus-containing products, or caffeine-containing food or beverages from 3 days prior to the first dose until study discharge. After dosing a mouth check was performed to ensure that the subject had swallowed each dose.
Sample collection
Blood samples for PK analysis of apixaban were collected immediately pre-dose and at 1, 2, 3, 4, 6, 8, 12, 24, 36, 48, 60 and 72 h after apixaban administration on days 1 and 7 in the ketoconazole DDI study, and on days 1 and 11 in the diltiazem DDI study. In the ketoconazole DDI study, blood samples for determining trough ketoconazole concentrations were collected immediately pre-dose on days 6–10, and in the diltiazem DDI study, pre-dose blood samples for determining trough diltiazem concentrations were collected on days 8–11. Blood and urine samples were collected over the course of both studies for routine clinical safety laboratory evaluations.
Sample analysis
Whole blood samples for apixaban PK analysis were collected into 4.5 ml sodium citrate tubes and centrifuged at 1000 g for 15 min to separate plasma. Samples of plasma were frozen at or below −20°C and shipped to the bioanalytical laboratory (Intertek Pharmaceutical Services [formerly known as Alta Analytical Laboratory], El Dorado Hills, California, USA). Apixaban concentrations were measured by a validated liquid chromatography atmospheric pressure ionization tandem mass spectrometry (LC-MS/MS) method using an acetonitrile protein precipitation extraction as the clean up step 45. The standard curves were well fitted by a 1/×2-weighted linear equation over the concentration range of 1 ng ml−1 to 1000 ng ml−1. Values for the between-run and within-run precision for the analytical quality control samples of apixaban were ≤6.64% coefficient of variation (CV) and ≤5.55% CV, respectively, for the ketoconazole DDI study and ≤6.15% CV and ≤12.9% CV, respectively, for the diltiazem DDI study. The deviations from the nominal concentration were ± 7.50%.
Blood samples (6 ml) for ketoconazole analysis were collected in tubes with ethylenediaminetetra-acetic acid as anticoagulant, and centrifuged at 1000 g for 15 min to separate plasma. Plasma samples were frozen at or below −20°C and shipped to the bioanalytical laboratory (Tandem Labs, Inc., Salt Lake City, Utah, USA) for analysis by a validated LC-MS/MS method using a methanol protein precipitation extraction as the clean up step. Samples for diltiazem analysis were collected in a similar manner but with heparin as anticoagulant, and sent for analysis by a validated LC-MS/MS method (Vimta Labs, Andhra Pradesh, India) using Oasis HLB solid phase extraction as the clean up step. Values for the within-run precision for the analytical quality control samples were ≤4.5% CV for ketoconazole and ≤12.6% CV for diltiazem. The between-run precision for ketoconazole was not calculated because sample analysis was completed in one run. The between-run precision for the analytical quality control samples for diltiazem was ≤9.10% CV. The deviations from the nominal concentration were ± 5.3% and ± 4.2% for ketoconazole and diltiazem, respectively. Ketoconazole and diltiazem at concentrations of 950 ng ml−1 and 1 μg ml−1, respectively, did not interfere with the analysis of apixaban.
Pharmacokinetic analysis
Single dose PK parameters were determined based on plasma concentrations over time. Maximum plasma concentration (Cmax) and time to maximum plasma concentration (tmax) were recorded from experimental observations. Individual subject PK parameters were derived by non-compartmental methods using Kinetica version 4.2 (InnaPhase Corp., Philadelphia, Pennsylvania, USA). The area under the plasma concentration–time curve from time zero to the last quantifiable concentration [AUC(0,tlast)] and extrapolated to infinity [AUC(0,∞)] were calculated using the log-linear trapezoidal method. The t1/2 was estimated as ln2/λz, in which the slope of the terminal phase of the plasma concentration–time profile (λz) was determined by the least-squares method (log linear regression of at least three data points) with a weighting factor of 1.
Safety determinations
Adverse event (AE) data were obtained from information volunteered or solicited from subjects and from investigator review of vital signs, laboratory test results and electrocardiogram (ECG) data. All AEs were reviewed for severity, relation to study drugs and clinical importance. Any marked abnormalities in clinical laboratory test results were reviewed. Twelve-lead ECGs and physical examinations were performed at screening, baseline and before study discharge.
Statistical analyses
Statistical analyses were carried out using SAS/STAT® version 8.2 (SAS Institute, Cary, North Carolina, USA). Geometric means and CVs were presented for Cmax, AUC(0,tlast) and AUC(0,∞), medians, minima and maxima were reported for tmax and means and SDs were reported for t1/2. To assess the effect of concomitant administration of ketoconazole or diltiazem on the single dose PK parameters of apixaban, a general linear mixed model analysis was performed on ln(Cmax), ln(AUC(0,tlast)) and ln(AUC(0,∞)) to obtain point estimates and 90% confidence intervals (CIs) on ratios of apixaban Cmax, AUC(0,tlast) and AUC(0,∞) geometric means with and without concomitant medication. A general linear mixed effects model was fitted to ln(Cmax), ln(AUC(0,tlast)) and ln(AUC(0,∞)). Factors in the model were treatment as fixed effect and measurements within subjects as repeated measures. Means and differences between means on a logarithmic scale were exponentiated to obtain point estimates and 90% CIs for the geometric ratios of apixaban Cmax, AUC(0,tlast) and AUC(0,∞) with and without ketoconazole or diltiazem. No adjustments were made for multiplicity. Trough plasma concentrations of both ketoconazole and diltiazem were summarized by study day.
An absence of effect of ketoconazole or diltiazem on apixaban PK parameters would be concluded if the 90% CIs for the ratios of geometric means for apixaban Cmax and AUC(0,∞) were contained within an 80–125% interval. On the basis of the observed variation between individual subjects in previous apixaban PK studies 14 and assuming log normal distributions of AUC(0,∞) and Cmax, a sample size of 18 subjects was expected to provide greater than 90% power to conclude absence of an interaction. Twenty subjects were treated in the ketoconazole DDI study to allow for dropouts. Eighteen subjects were treated in the diltiazem study.
Results
Study populations
Eighteen of 20 subjects completed the ketoconazole DDI study. Two subjects discontinued the study prior to receiving all treatments. One subject discontinued the study due to an AE (rash) on day 1 after receiving a 10 mg dose of apixaban and one subject withdrew consent because of a family emergency. All 18 subjects in the diltiazem DDI study completed the study. Demographic characteristics of subjects are shown in Table 1.
Table 1.
Subject demographics for the ketoconazole and diltiazem DDI studies
| Characteristics | Ketoconazole (n = 20*) | Diltiazem (n = 18) | |
|---|---|---|---|
| Gender, n (%) | Male | 20 (100) | 13 (72) |
| Age (years) | Mean (SD) | 29 (6) | 29 (7) |
| Range | 21–45 | 22–45 | |
| Race, n (%) | White | 7 (35) | 7 (39) |
| Black | 10 (50) | 10 (56) | |
| Asian | 3 (15) | 1 (6) | |
| Body weight (kg) | Mean (SD) | 79 (11) | 78 (16) |
| Range | 64–107 | 53–109 | |
| BMI (kg m−2) | Mean (SD) | 25 (3) | 26 (3) |
| Range | 20–30 | 21–30 | |
Eighteen subjects completed the study: one subject discontinued the study due to an adverse event (rash) and one subject withdrew consent because of a family emergency. BMI, body mass index; DDI, drug–drug interaction; SD, standard deviation.
Pharmacokinetic results
Mean plasma concentration–time profiles with and without co-administration of ketoconazole and diltiazem are shown in Figure 2A and B, respectively, and PK parameters are summarized in Table 2. Ketoconazole increased apixaban Cmax by 62% and increased AUC(0,tlast) and AUC(0,∞) by 97% and 99%, respectively. The 90% CIs for Cmax and AUC(0,∞) were not contained within the 80–125% interval and excluded the value of 100%, indicating the presence of a drug interaction. Although apixaban exposure increased with co-administration of ketoconazole, there was no change in median tmax. The mean t1/2 of apixaban increased by 2.5 h (22%) following co-administration of ketoconazole. The geometric mean trough plasma concentrations of ketoconazole ranged from 173 ng ml−1 to 295 ng ml−1 on days 7–10.
Figure 2.
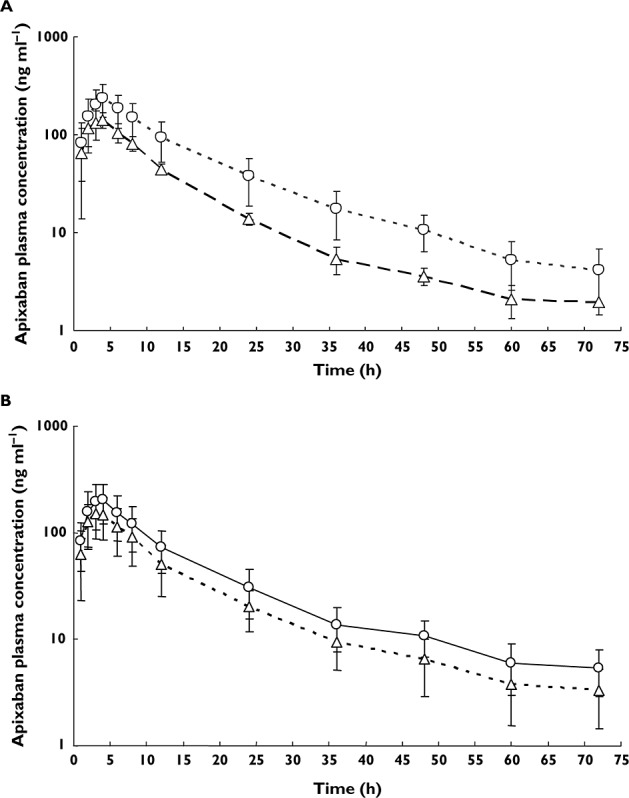
Mean plasma concentration–time profiles of apixaban (error bars show one standard deviation) in healthy subjects following a single 10 mg dose of apixaban alone or in the presence of multiple doses of (A) ketoconazole,  , apixaban 10 mg (n = 19);
, apixaban 10 mg (n = 19);  , apixaban + ketoconazole 400 mg once daily (n = 18) and (B) diltiazem,
, apixaban + ketoconazole 400 mg once daily (n = 18) and (B) diltiazem,  , apixaban 10 mg (n = 18);
, apixaban 10 mg (n = 18);  , apixaban 10 mg + diltiazem 360 mg once daily (n = 18)
, apixaban 10 mg + diltiazem 360 mg once daily (n = 18)
Table 2.
Summary statistics for apixaban pharmacokinetics
| Pharmacokinetic parameter* | Ketoconazole DDI study | Diltiazem DDI study | ||||
|---|---|---|---|---|---|---|
| (n = 18) | (n = 18) | |||||
| Apixaban | Apixaban + ketoconazole | Point estimate of RGM (90% CI) | Apixaban | Apixaban + diltiazem | Point estimate of RGM (90% CI) | |
| Cmax (ng ml−1) | 139.5 (32) | 225.3 (36) | 1.62 (1.47, 1.78) | 148.1 (38) | 194.6 (41) | 1.31 (1.16, 1.49) |
| [80, 250] | [103, 391] | [76, 304] | [119, 463] | |||
| AUC(0,tlast) (ng ml−1 h) | 1490 (28) | 2939 (38) | 1.97 (1.80, 2.16) | 1779 (40) | 2475 (40) | 1.39 (1.24, 1.56) |
| [818, 2165] | [1288, 5612] | [892, 3574] | [1228, 5144] | |||
| AUC(0,∞) (ng ml−1 h) | 1523 (28) | 3027 (37) | 1.99 (1.81, 2.18) | 1897† (38) | 2606 (39) | 1.40 (1.23, 1.59) |
| [846, 2229] | [1306, 5859] | [941, 3589] | [1242, 5229] | |||
| tmax (h) | 4 (1, 4) | 4 (3, 4) | – | 3 (2, 8) | 4 (2, 4) | – |
| t1/2 (h) | 11.3 (5.8) | 13.8 (6.3) | – | 17.2† (7.4) | 16.3 (7.8) | – |
| [4.6, 25] | [5.6, 34] | [8.6, 30] | [5.6, 37] | |||
Geometric mean (%CV) [min, max] for Cmax, AUC(0,tlast) and AUC(0,∞); median (min,max) for tmax; arithmetic mean (SD) [min, max] for t1/2.
n = 17; in one subject AUC(0,∞) and t1/2 could not be calculated. AUC(0,tlast), area under concentration–time curve from time zero to last quantifiable concentration; AUC(0,∞), area under concentration–time curve from time zero to infinity; CI, confidence interval; Cmax, observed peak plasma concentration; CV, coefficient of variation; DDI, drug–drug interaction; RGM, ratio of geometric means estimated as concomitant treatment vs. apixaban alone; SD, standard deviation; t1/2, elimination half-life; tmax, time taken to reach Cmax.
Comparison of plasma concentration–time profiles and PK parameters showed that diltiazem also increased apixaban exposure (Figure 2B, Table 2). Concomitantly administered diltiazem increased apixaban Cmax by 31% and AUC(0,tlast) and AUC(0,∞) by 39% and 40%, respectively, but had no effect on the tmax or t1/2 of apixaban. The 90% CIs for Cmax and AUC(0,∞) were not contained within the 80–125% interval and excluded the value of 100%, indicating the presence of a drug interaction. The geometric mean trough concentrations of diltiazem ranged from 125.0 ng ml−1 to 177.9 ng ml−1 on days 8–11.
Safety results
Adverse events reported in the studies were mild or moderate in intensity and all resolved without treatment. None was rated as serious. One subject discontinued the study due to an AE of a papular rash that lasted for 20 days beginning on day 8 in the ketoconazole DDI study. A total of 14 AEs occurred in six subjects during the ketoconazole DDI study and 34 AEs occurred in 10 subjects during the diltiazem DDI study. Headache was the most frequently reported AE in the ketoconazole DDI study (four subjects) and in the diltiazem DDI study (seven subjects). In addition, four subjects reported dizziness after taking diltiazem alone. Bleeding-related AEs occurred in one subject after receiving apixaban and ketoconazole (epistaxis) and in two subjects after receiving apixaban and diltiazem (subconjunctival haemorrhage and petechiae/contusions). All three events were considered mild and resolved without treatment.
Discussion
The data from these studies indicate that co-administration of apixaban with modulators of CYP3A4 and P-gp activity affect apixaban PK and that the extent of interaction is dependent on the strength of the modulator. Co-administration of apixaban with ketoconazole resulted in a 2-fold increase in apixaban AUC and a 1.6-fold increase in apixaban Cmax. Diltiazem led to a 1.4- and 1.3-fold increase in apixaban AUC and Cmax, respectively. Neither ketoconazole nor diltiazem impacted apixaban tmax. Apixaban t1/2 was numerically longer by 2.5 h when administered in the presence of ketoconazole. This apparent small increase in apixaban t1/2 is not expected to have a meaningful impact on the time to reach apixaban steady-state. Assuming that the volume of distribution of apixaban was not altered by ketoconazole or diltiazem, these data suggest that changes in systemic clearance are not solely responsible for the observed effects on apixaban exposure. The combination of increased Cmax and AUC, and unchanged or slightly prolonged t1/2, suggests that increased bioavailability also contributes to higher apixaban exposure, as would result from inhibition of intestinal/hepatic CYP3A4 metabolism and P-gp–mediated intestinal efflux.
Ketoconazole and diltiazem were administered to steady-state to ensure the maximum inhibition of CYP3A4 was achieved for each agent 46,47. The steady-state trough concentrations of ketoconazole and diltiazem observed in these studies were consistent with those previously reported in the literature 48,49. The effects of co-administered ketoconazole and diltiazem on the PK parameters of apixaban are likely to be representative of other drugs that inhibit CYP3A4 activity and/or P-gp 41. The effect of ketoconazole on apixaban exposure in this study (a 2-fold increase in apixaban AUC) was much less than that reported in the literature for sensitive CYP3A4 substrates such as midazolam (15-fold increase) and simvastatin (12.5-fold increase) 48. The results of the ketoconazole and diltiazem DDI studies demonstrate that moderate-to-strong CYP3A4/P-gp inhibitors increased apixaban exposure and they do not differentiate between the effects on CYP3A4 and P-gp. The doubling of apixaban exposure seen with concomitant ketoconazole likely represents the maximum potential for a DDI via combined inhibition of CYP3A4 metabolism and P-gp–mediated drug efflux (i.e. the increase in apixaban exposure with concomitant use of other strong inhibitors of CYP3A4 and/or P-gp is expected to be less than or generally comparable with that seen with ketoconazole) 50,51.
The clinical relevance of an increase in apixaban exposure depends on the assessment of the benefit–risk profile within the patient population. Since apixaban is a direct reversible inhibitor of factor Xa, the primary concern with increases in exposure would be an increased risk of bleeding. The use of strong CYP3A4 inhibitors was prohibited in apixaban phase III clinical trials. Given the limited clinical experience with concomitant administration of apixaban with strong inhibitors of CYP3A4 and P-gp, co-administration is generally not recommended 52,53.
The use of moderate inhibitors of CYP3A4 and/or P-gp was allowed in phase III trials 4–8 and dose reduction is not necessary when apixaban is administered with moderate inhibitors of CYP3A4 and/or P-gp. However, caution is warranted in the presence of additional factors that increase apixaban exposure or that may pose an inherent risk of bleeding, such as significant renal impairment 52,53.
In conclusion, a 2-fold and 1.4-fold increase in apixaban exposure was observed with co-administration of ketoconazole and diltiazem, respectively. The decision to administer apixaban in the presence of inhibitors of CYP3A4 and P-gp should follow apixaban approved product labelling.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: C.E. Frost, Y. Song, J. Wang, A.E. Schuster, D. Zhang, Z. Yu, C. Dias, A. Shenker and F. LaCreta were all employees of Bristol-Myers Squibb Company at time of the research and received salaries and benefits commensurate with employment; W. Byon and R.A. Boyd are employees of Pfizer Inc and receive salaries and benefits commensurate with employment.
This study was funded by Bristol-Myers Squibb and Pfizer Inc. Medical writing and editorial assistance were provided by Andy Shepherd, PhD and Dana Fox, PhD, CMPP, at Caudex Medical and funded by Bristol-Myers Squibb Company and Pfizer Inc.
Contributors
C.E. Frost: Wrote manuscript, designed research, analyzed data; J. Wang: Designed research, analyzed data; A.E. Schuster: Analyzed data; Y. Song: Wrote manuscript; W. Byon: Wrote manuscript; R.A. Boyd: Wrote manuscript; D. Zhang: Performed research; Z. Yu: Wrote manuscript, designed research, analyzed data; C. Dias: Wrote manuscript, designed research, analyzed data; A. Shenker: Wrote manuscript, designed research, analyzed data; F. LaCreta: Wrote manuscript, designed research, analyzed data.
References
- Kaiser B. Factor Xa – a promising target for drug development. Cell Mol Life Sci. 2002;59:189–192. doi: 10.1007/s00018-002-8415-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto DJ, Orwat MJ, Koch S, Rossi KA, Alexander RS, Smallwood A, Wong PC, Rendina AR, Luettgen JM, Knabb RM, He K, Xin B, Wexler RR, Lam PY. Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem. 2007;50:5339–5356. doi: 10.1021/jm070245n. [DOI] [PubMed] [Google Scholar]
- Wong PC, Crain EJ, Xin B, Wexler RR, Lam PY, Pinto DJ, Luettgen JM, Knabb RM. Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies. J Thromb Haemost. 2008;6:820–829. doi: 10.1111/j.1538-7836.2008.02939.x. [DOI] [PubMed] [Google Scholar]
- Connolly SJ, Eikelboom J, Joyner C, Diener HC, Hart R, Golitsyn S, Flaker G, Avezum A, Hohnloser SH, Diaz R, Talajic M, Zhu J, Pais P, Budaj A, Parkhomenko A, Jansky P, Commerford P, Tan RS, Sim KH, Lewis BS, Van Mieghem W, Lip GY, Kim JH, Lanas-Zanetti F, Gonzalez-Hermosillo A, Dans AL, Munawar M, O'Donnell M, Lawrence J, Lewis G, Afzal R, Yusuf S. Apixaban in patients with atrial fibrillation. N Engl J Med. 2011;364:806–817. doi: 10.1056/NEJMoa1007432. [DOI] [PubMed] [Google Scholar]
- Granger CB, Alexander JH, McMurray JJ, Lopes RD, Hylek E, Hanna M, Al-Khalidi HR, Ansell J, Atar D, Avezum A, Bahit MC, Diaz R, Easton JD, Ezekowitz JA, Flaker G, Garcia D, Geraldes M, Gersh BJ, Golitsyn S, Goto S, Hermosillo AG, Hohnloser S, Horowitz J, Mohan P, Jansky P, Lewis BS, Lopez-Sendon J, Pais P, Parkhomenko A, Verheugt F, Zhu J, Wallentin L. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365:981–992. doi: 10.1056/NEJMoa1107039. [DOI] [PubMed] [Google Scholar]
- Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Portman RJ. Apixaban or enoxaparin for thromboprophylaxis after knee replacement. N Engl J Med. 2009;361:594–604. doi: 10.1056/NEJMoa0810773. [DOI] [PubMed] [Google Scholar]
- Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Hornick P. Apixaban versus enoxaparin for thromboprophylaxis after knee replacement (ADVANCE-2): a randomised double-blind trial. Lancet. 2010;375:807–815. doi: 10.1016/S0140-6736(09)62125-5. [DOI] [PubMed] [Google Scholar]
- Lassen MR, Gallus A, Raskob GE, Pineo G, Chen D, Ramirez LM. Apixaban versus enoxaparin for thromboprophylaxis after hip replacement. N Engl J Med. 2010;363:2487–2498. doi: 10.1056/NEJMoa1006885. [DOI] [PubMed] [Google Scholar]
- Agnelli G, Buller HR, Cohen A, Curto M, Gallus AS, Johnson M, Masiukiewicz U, Pak R, Thompson J, Raskob GE, Weitz JI. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369:799–808. doi: 10.1056/NEJMoa1302507. [DOI] [PubMed] [Google Scholar]
- Agnelli G, Buller HR, Cohen A, Curto M, Gallus AS, Johnson M, Porcari A, Raskob GE, Weitz J. Apixaban for extended treatment of venous thromboembolism. N Engl J Med. 2013;368:699–708. doi: 10.1056/NEJMoa1207541. [DOI] [PubMed] [Google Scholar]
- Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e44S–88S. doi: 10.1378/chest.11-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition) Chest. 2008;133:160S–198. doi: 10.1378/chest.08-0670. [DOI] [PubMed] [Google Scholar]
- Frost C, Nepal S, Wang J, Schuster A, Byon W, Boyd RA, Yu Z, Shenker A, Barrett YC, Mosqueda-Garcia R, LaCreta F. Safety, pharmacokinetics and pharmacodynamics of multiple oral doses of apixaban, a factor Xa inhibitor, in healthy subjects. Br J Clin Pharmacol. 2013;76:776–786. doi: 10.1111/bcp.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost C, Wang J, Nepal S, Schuster A, Barrett YC, Mosqueda-Garcia R, Reeves RA, LaCreta F. Apixaban, an oral, direct factor Xa inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects. Br J Clin Pharmacol. 2013;75:476–487. doi: 10.1111/j.1365-2125.2012.04369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost C, Yu Z, Nepal S, Bragat A, Moore K, Shenker A, Barrett YC, LaCreta F. Apixaban, a direct factor Xa inhibitor: single-dose pharmacokinetics and pharmacodynamics of an intravenous formulation [abstract] J Clin Pharmacol. 2008;48:1132. [Google Scholar]
- Vakkalagadda B, Frost C, Wang J, Nepal S, Schuster A, Zhang D, Dias C, Yu Z, Shenker A, LaCreta F. Effect of rifampin on the pharmacokinetics of apixaban, an oral direct inhibitor of factor Xa [abstract] J Clin Pharmacol. 2009;49:1091–1130. [Google Scholar]
- Zhang D, He K, Herbst JJ, Kolb J, Shou W, Wang L, Balimane P, Han Y-H, Gan J, Frost CE. Characterization of transporters involved in distribution and disposition of apixaban. Drug Metab Dispos. 2013;41:827–835. doi: 10.1124/dmd.112.050260. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan N, Frost CE, Yu Z, He K, Zhang H, Humphreys WG, Pinto D, Chen S, Bonacorsi S, Wong PC, Zhang D. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos. 2009;37:74–81. doi: 10.1124/dmd.108.023143. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang D, Raghavan N, Yao M, Ma L, Frost CE, Maxwell BD, Chen SY, He K, Goosen TC, Humphreys WG, Grossman SJ. In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos. 2010;38:448–458. doi: 10.1124/dmd.109.029694. [DOI] [PubMed] [Google Scholar]
- Wang L, He K, Maxwell B, Grossman SJ, Tremaine LM, Humphreys WG, Zhang D. Tissue distribution and elimination of [14C]apixaban in rats. Drug Metab Dispos. 2011;39:256–264. doi: 10.1124/dmd.110.036442. [DOI] [PubMed] [Google Scholar]
- Wang X, Tirucherai G, Pannacciulli N, Wang J, Elsrougy A, Teslenko V, Chang M, Zhang D, Frost C. Effect of activated charcoal on the pharmacokinetics of apixaban in healthy subjects. 2012. ASCPT. 15-3-2012.
- Zhang D, He K, Raghavan N, Wang L, Mitroka J, Maxwell BD, Knabb RM, Frost C, Schuster A, Hao F, Gu Z, Humphreys WG, Grossman SJ. Comparative metabolism of 14C-labeled apixaban in mice, rats, rabbits, dogs, and humans. Drug Metab Dispos. 2009;37:1738–1748. doi: 10.1124/dmd.108.025981. [DOI] [PubMed] [Google Scholar]
- Wang L, Raghavan N, He K, Luettgen JM, Humphreys WG, Knabb RM, Pinto DJ, Zhang D. Sulfation of o-demethyl apixaban: enzyme identification and species comparison. Drug Metab Dispos. 2009;37:802–808. doi: 10.1124/dmd.108.025593. [DOI] [PubMed] [Google Scholar]
- Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. The human intestinal cytochrome P450 ‘pie’. Drug Metab Dispos. 2006;34:880–886. doi: 10.1124/dmd.105.008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostami-Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6:140–148. doi: 10.1038/nrd2173. [DOI] [PubMed] [Google Scholar]
- Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–423. [PubMed] [Google Scholar]
- Aithal GP, Day CP, Kesteven PJ, Daly AK. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet. 1999;353:717–719. doi: 10.1016/S0140-6736(98)04474-2. [DOI] [PubMed] [Google Scholar]
- Crewe HK, Notley LM, Wunsch RM, Lennard MS, Gillam EM. Metabolism of tamoxifen by recombinant human cytochrome P450 enzymes: formation of the 4-hydroxy, 4′-hydroxy and N-desmethyl metabolites and isomerization of trans-4-hydroxytamoxifen. Drug Metab Dispos. 2002;30:869–874. doi: 10.1124/dmd.30.8.869. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Russell DW. Clinical importance of the cytochromes P450. Lancet. 2002;360:1155–1162. doi: 10.1016/S0140-6736(02)11203-7. [DOI] [PubMed] [Google Scholar]
- Plastaras JP, Guengerich FP, Nebert DW, Marnett LJ. Xenobiotic-metabolizing cytochromes P450 convert prostaglandin endoperoxide to hydroxyheptadecatrienoic acid and the mutagen, malondialdehyde. J Biol Chem. 2000;275:11784–11790. doi: 10.1074/jbc.275.16.11784. [DOI] [PubMed] [Google Scholar]
- Bailey DG, Malcolm J, Arnold O, Spence JD. Grapefruit juice–drug interactions. Br J Clin Pharmacol. 1998;46:101–110. doi: 10.1046/j.1365-2125.1998.00764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durr D, Stieger B, Kullak-Ublick GA, Rentsch KM, Steinert HC, Meier PJ, Fattinger K. St John's Wort induces intestinal P-glycoprotein/MDR1 and intestinal and hepatic CYP3A4. Clin Pharmacol Ther. 2000;68:598–604. doi: 10.1067/mcp.2000.112240. [DOI] [PubMed] [Google Scholar]
- Roby CA, Anderson GD, Kantor E, Dryer DA, Burstein AH. St John's Wort: effect on CYP3A4 activity. Clin Pharmacol Ther. 2000;67:451–457. doi: 10.1067/mcp.2000.106793. [DOI] [PubMed] [Google Scholar]
- Edgar B, Bailey D, Bergstrand R, Johnsson G, Regardh CG. Acute effects of drinking grapefruit juice on the pharmacokinetics and dynamics of felodipine – and its potential clinical relevance. Eur J Clin Pharmacol. 1992;42:313–317. doi: 10.1007/BF00266354. [DOI] [PubMed] [Google Scholar]
- Nappi JM. Warfarin and phenytoin interaction. Ann Intern Med. 1979;90:852. doi: 10.7326/0003-4819-90-5-852_1. [DOI] [PubMed] [Google Scholar]
- Shenfield GM, Page M. Potentiation of warfarin action by miconazole oral gel. Aust N Z J Med. 1991;21:928. doi: 10.1111/j.1445-5994.1991.tb01422.x. [DOI] [PubMed] [Google Scholar]
- Jiang X, Williams KM, Liauw WS, Ammit AJ, Roufogalis BD, Duke CC, Day RO, McLachlan AJ. Effect of St John's wort and ginseng on the pharmacokinetics and pharmacodynamics of warfarin in healthy subjects. Br J Clin Pharmacol. 2004;57:592–599. doi: 10.1111/j.1365-2125.2003.02051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benet LZ, Cummins CL. The drug efflux-metabolism alliance: biochemical aspects. Adv Drug Deliv Rev. 2001;50(Suppl. 1):S3–11. doi: 10.1016/s0169-409x(01)00178-8. [DOI] [PubMed] [Google Scholar]
- University of Washington. Metabolism and transport drug interaction database. Available at http://www.druginteractioninfo.org (last accessed February 2012)
- Food and Drug Administration. Guidance for industry. Drug interaction studies – study design, data analysis and implications for dosing and labeling. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf (last accessed February 2013)
- Rowland YK, Jamei M, Yang J, Tucker GT, Rostami-Hodjegan A. Physiologically based mechanistic modelling to predict complex drug-drug interactions involving simultaneous competitive and time-dependent enzyme inhibition by parent compound and its metabolite in both liver and gut – the effect of diltiazem on the time-course of exposure to triazolam. Eur J Pharm Sci. 2010;39:298–309. doi: 10.1016/j.ejps.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Cornwell MM, Pastan I, Gottesman MM. Certain calcium channel blockers bind specifically to multidrug-resistant human KB carcinoma membrane vesicles and inhibit drug binding to P-glycoprotein. J Biol Chem. 1987;262:2166–2170. [PubMed] [Google Scholar]
- Jones DR, Gorski JC, Hamman MA, Mayhew BS, Rider S, Hall SD. Diltiazem inhibition of cytochrome P-450 3A activity is due to metabolite intermediate complex formation. J Pharmacol Exp Ther. 1999;290:1116–1125. [PubMed] [Google Scholar]
- Pursley J, Shen JX, Schuster A, Dang OT, Lehman J, Buonarati MH, Song Y, Aubry A-F, Arnold ME. LC-MS/MS determination of apixaban (BMS562247) and its major metabolite in human plasma – an application of polarity switching and monolithic HPLC column. Bioanalysis. 2014;6:2071–2082. doi: 10.4155/bio.14.66. [DOI] [PubMed] [Google Scholar]
- Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, Kao J, King SP, Miwa G, Ni L, Kumar G, McLeod J, Obach RS, Roberts S, Roe A, Shah A, Snikeris F, Sullivan JT, Tweedie D, Vega JM, Walsh J, Wrighton SA. The conduct of in vitro and in vivo drug–drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos. 2003;31:815–832. doi: 10.1124/dmd.31.7.815. [DOI] [PubMed] [Google Scholar]
- Stoch SA, Friedman E, Maes A, Yee K, Xu Y, Larson P, Fitzgerald M, Chodakewitz J, Wagner JA. Effect of different durations of ketoconazole dosing on the single-dose pharmacokinetics of midazolam: shortening the paradigm. J Clin Pharmacol. 2009;49:398–406. doi: 10.1177/0091270008331133. [DOI] [PubMed] [Google Scholar]
- Daneshmend TK, Warnock DW. Clinical pharmacokinetics of ketoconazole. Clin Pharmacokinet. 1988;14:13–34. doi: 10.2165/00003088-198814010-00002. [DOI] [PubMed] [Google Scholar]
- Sista S, Lai JC, Eradiri O, Albert KS. Pharmacokinetics of a novel diltiazem HCl extended-release tablet formulation for evening administration. J Clin Pharmacol. 2003;43:1149–1157. doi: 10.1177/0091270003257214. [DOI] [PubMed] [Google Scholar]
- Chung E, Nafziger AN, Kazierad DJ, Bertino JS., Jr Comparison of midazolam and simvastatin as cytochrome P450 3A probes. Clin Pharmacol Ther. 2006;79:350–361. doi: 10.1016/j.clpt.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Olkkola KT, Backman JT, Neuvonen PJ. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;55:481–485. doi: 10.1038/clpt.1994.60. [DOI] [PubMed] [Google Scholar]
- Bristol-Myers Squibb and Pfizer EEIG. Eliquis® (apixaban tablets) Summary of product characteristics. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002148/WC500107728.pdf (last accessed January 2013)
- Bristol-Myers Squibb Company. Eliquis® (apixaban tablets) Prescribing Information. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202155s000lbl.pdf (last accessed January 2013)