

Abstract
Membrane proteins are an important class of proteins in biology and therapeutics. Understanding the dynamic nature of the molecular interactions that stabilize membrane protein structure is critical to dissect the mechanism of action and dysfunction of these proteins. Single-molecule force spectroscopy (SMFS) and dynamic SMFS (DFS) are emerging nanotechniques that allow the study of membrane proteins under the physiologically relevant conditions of a lipid bilayer and buffer conditions. These techniques directly probe the molecular interactions underlying protein structure and reveal unique insights about their properties. Outlined in this report will be procedures on how to conduct SMFS and DFS on rhodopsin in native retinal membranes. Rhodopsin is a membrane protein belonging to the G protein-coupled receptor family of proteins, one of the largest families of proteins in nature.
Keywords: Atomic force microscopy, biological membranes, energy landscape, membrane proteins, intramolecular interactions, protein stability, protein unfolding
1. Introduction
The atomic force microscope (AFM) has become a multifaceted instrument providing novel insights about the dynamic structure of membrane proteins (1,2). Molecular interactions formed by membrane proteins underlie their function, fold, and stability. AFM-based single-molecule force spectroscopy (SMFS) is a powerful tool that has allowed the direct probing and detection of molecular interactions in membrane proteins at the single molecule level (3). In contrast to more traditional structural methods, SMFS allows for the study of membrane proteins within the physiological context of a lipid bilayer and physiological buffer conditions. Thus, the dynamic molecular interactions underlying proteins structure can be investigated under near-native conditions.
SMFS has been successfully applied to rhodopsin to investigate the influence of various factors on the molecular interactions that stabilize this membrane protein. Rhodopsin is a prototypical G protein-coupled receptor (GPCR) found in the rod outer segment (ROS) disc membranes of photoreceptor cells in the retina. Rhodopsin is highly expressed in ROS disc membranes representing about 90% of all proteins in that compartment (4). Thus, isolation of ROS disc membranes essentially provides a purified preparation of rhodopsin embedded in a native lipid bilayer at high concentrations, an ideal preparation for SMFS. The availability of different animal models has provided a method to obtain native preparations of genetically modified forms of rhodopsin.
Initial SMFS studies carried out on rhodopsin from native retinal tissue revealed that the structure of rhodopsin is organized into distinct regions that exhibit intrinsic stability and require mechanical force to unfold (5), which are referred to as stable structural segments (3). SMFS allowed for the determination of the relative stability of each stable structural segment in rhodopsin. The ability to determine the relative stability of different regions of rhodopsin allowed for the determination of the effect of zinc ions and post-translational modification on the structure of rhodopsin. Zinc ions appear to bind specifically to rhodopsin and exert a stabilizing effect since most stable structural segments exhibited a dose-dependent increase in relative stability (6). The absence of normal palmitylation of rhodopsin in its C-terminal region has been shown by SMFS to destabilize this region while leaving the rest of the rhodopsin structure unperturbed (7). The functional effect of this destabilization appears to impact the efficiency of signaling to transducin.
Dynamic SMFS (DFS) is a related method whereby SMFS is conducted at different pulling velocities (8). DFS allows the determination of parameters that describe the underlying energy landscape of protein unfolding, thereby providing information about the kinetic, energetic, and mechanical properties of stable structural segments of rhodopsin (9). DFS has been applied to rhodopsin to determine the effect of amino acid substitutions and the binding of the chromophore 11-cis retinal on the molecular interactions formed in stable structural segments. Investigation of bovine and murine rhodopsin, where there are 23 amino acid residue differences between the receptor homologues, revealed that the kinetic, energetic, and mechanical properties of stable structural segments are largely conserved despite differences in amino acid sequence (10). Thus, although there are over 100 point mutations in rhodopsin that cause retinal disease (11), the structure of rhodopsin can accommodate a certain level of amino acid residue substitutions without displaying significant perturbations in structure.
DFS was able to uncover the perturbations resulting from an amino acid substitution observed in patients with retinal disease, a G90D mutation that causes constitutive activity in rhodopsin and leads to congenital night blindness (12). DFS revealed that the G90D mutant exhibited lower conformational variability, increased unfolding rates, decreased activation energy, and increased mechanical rigidity in most stable structural segments compared to wild-type rhodopsin (13). These changes may underlie the disease-causing constitutive activity promoted by the mutation and may be characteristic of constitutive activity in general since similar changes were also detected by DFS on the apoprotein opsin (14), which exhibits a low level of constitutive activity. Moreover, DFS studies on the apoprotein opsin revealed that the impact of bound 11-cis retinal on stable structural segments in rhodopsin shares some similarities with the impact of bound ligands on stable structural segments of the β2 adrenergic receptor (15), another member of the GPCR family. Thus, DFS studies support the notion that some mechanistic and structural features of GPCRs are conserved.
The SMFS and DFS studies on rhodopsin highlighted here point to the incredible promise the methodology holds to reveal pertinent molecular information required to understand the mechanism underlying the function, folding, and stability of rhodopsin and other GPCRs. Reported in the following sections are detailed procedures for conducting SMFS and DFS on rhodopsin embedded in native ROS disc membranes. The reader is also directed to several excellent reports detailing general methods for the study of membrane proteins by an AFM and for conducting SMFS (16-18).
2. Materials
ROS disc membranes. ROS disc membranes prepared from fresh bovine and murine retina have been used previously in SMFS/DFS applications (5,19).
Mica supports for sample adsorption. Detailed methods for preparing mica supports have been reported previously (16-18).
Scotch tape.
AFM. We have used the Multimode AFM (Bruker Corporation, Santa Barbara, CA) and the NanoWizard AFM (JPK Instruments, Berlin, Germany) in our studies. Both AFMs give similar results. Other commercial AFMs should be suitable as well. AFMs equipped with red or infrared lasers will not bleach rhodopsin and are therefore suitable. A 16-bit data acquisition card (NI PCI-6221, National Instruments, Austin, TX) is required to collect data at high pulling speeds (>1,500 nm/s).
Si3N4 AFM probes (NPS, Bruker Corporation, Camarillo, CA) with nominal spring constants of 0.06-0.08 N/m. Spring constants for AFM probes should be experimentally determined in the buffer that will be used for experiments. Spring constants can be determined using a thermal noise method (20).
Ringer’s buffer: 10 mM HEPES, 130 mM NaCl, 3.6 mM KCl, 2.4 mM MgCl2, 1.2 mM CaCl2, 0.02 mM EDTA, pH 7.4.
IGOR Pro (WaveMetrics, Inc., Lake Oswego, OR) graphing and data analysis software.
3. Methods
All procedures involving rhodopsin should be carried out in the dark under dim red light conditions. All light sources should be covered with red filters.
3.1 Preparation of samples for SMFS
Dilute the ROS disc membrane sample (0.5-1 mg/mL) in Ringer’s buffer. A 100-fold dilution is usually suitable, but may be modified as required (Note 1).
Cleave mica with tape. The adhesive of the tape will remove a layer of the mica to expose an atomically flat surface on which the sample will adsorb.
Add 30 μL of the diluted ROS disc membrane sample (5-10 μg/mL) onto a freshly cleaved mica support. Incubate for 10-20 minutes to allow ROS disc membranes to adsorb onto the mica.
Remove the liquid from the mica surface. Wash the mica surface at least 5 times with 30 μL of Ringer’s buffer to remove unadsorbed debris.
After the last wash, add 30 μL of Ringer’s buffer to the mica support (Note 2). Mount the mica support onto the AFM. Using an appropriate fluid cell, conduct experiments at ambient temperatures in buffer solution.
3.2 Collecting Force-Distance (F-D) curves in SMFS (Note 3)
Set the AFM for contact mode imaging.
Engage the AFM with the scan area set to 0 μm in order to avoid contamination of the AFM probe.
Once the AFM is engaged, retract the AFM probe from the mica surface and set the scan area to the maximum.
Slowly extend the AFM probe to the mica surface until contact is made. Adjust settings so that sufficient force is applied by the probe to maintain contact with the sample surface but minimized so that the sample is not deformed or disrupted. Typically, about 100 pN or less is sufficient. Scan rates and gains should also be optimized. Scan rates of 4-6 Hz usually are satisfactory.
Zoom in on a very small area of exposed mica that does not have any adsorbed material. Obtain a F-D curve to check cleanliness of the AFM probe and determine the deflection sensitivity of the cantilever. The AFM probe must be clean to obtain good quality F-D curves.
Zoom out to the maximum scan area and survey the adsorbed membranes on the surface of the mica.
Zoom in to the central region of an adsorbed ROS disc membrane (Fig. 1A) (Note 4). An area of the ROS disc membrane filling a frame size of 40-60 nm is suitable for manual F-D curve collection.
Collect F-D curves from the zoomed in area. F-D curve collection begins by extending the AFM probe so that it pushes into the membrane with a force of 0.5-1 nN for about 1 s. This contact allows the probe to form non-specific interactions with rhodopsin that is sufficiently strong to allow for protein unfolding. Retract the AFM probe at a constant pulling velocity to unfold a rhodopsin molecule (Fig. 1B). F-D curves record unfolding events that occur during this retraction (Figs. 2A and 2B) (Notes 5-7).
For DFS analysis, F-D curves must be collected at different pulling velocities. Pulling velocities in the range of 100-6000 nm/s have been previously used.
Fig. 1.
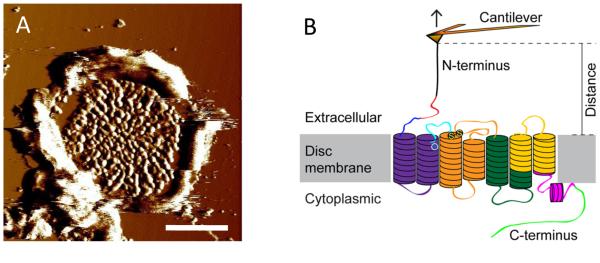
(A) An AFM deflection image of a ROS disc membrane adsorbed on mica. Scale bar, 500 nm. (B) Illustration of a SMFS experiment. The AFM probe attached to a flexible cantilever is non-specifically bound to the N-terminal end of rhodopsin embedded in ROS disc membranes. As the AFM probe is retracted from the sample surface, rhodopsin is mechanically unfolded in a stepwise manner. The stable structural segments of rhodopsin are highlighted in different colors. This figure is reproduced from (13) with permission from the American Society for Biochemistry and Molecular Biology.
Fig. 2.

(A) A set of F-D curves obtained from ROS disc membranes from mice expressing wild-type (black) or G90D mutant rhodopsin (blue). Each F-D curve records the unfolding events of a single rhodopsin molecule. (B) A single F-D curve analyzed by the WLC model. Each peak in the F-D curve has been fit with the WLC model (colored curves). Contour lengths obtained from the fit are denoted. (C) A histogram of contour lengths obtained from WLC model analyses of F-D curves obtained from ROS disc membranes containing wild-type (black) or G90D mutant (blue) rhodopsin. The histogram shows the fitting of peaks to a Guassian function to determine the mean contour length of major force peaks occurring with high frequency. (D) Stable structural segments are highlighted on the secondary structure of rhodopsin by different colors. The boundary amino acid residues are denoted with the corresponding contour length determined from contour length histogram analysis in parentheses. Figures were reproduced from (10) with permission from the American Chemical Society and from (13) with permission from the American Society for Biochemistry and Molecular Biology.
3.3 Analysis of SMFS data
Collected F-D curves must be exported and analyzed in appropriate graphing software. Analysis procedures outlined here are based on those conducted in IGOR Pro (Notes 8-9).
Select F-D curves that correspond to the unfolding of a single rhodopsin molecule from the N-terminal end (Figs. 2A and 2B) (Notes 10-11).
Overlay F-D curves. Exclude F-D curves that do not exhibit similar patterns of force peaks. F-D curves that correspond to the unfolding of rhodopsin will exhibit similar patterns of force peaks. Curves are aligned using frequently observed peaks as a reference (Note 12).
Once F-D curves are aligned, individual curves are analyzed using the worm-like-chain (WLC) model (21). Each force peak is fit with the WLC model to determine the contour length (L) (Fig. 2B) (Note 13).
A histogram of contour lengths is generated from fits of each force peak in all F-D curves analyzed for a single condition (Fig. 2C). The histogram is fit by a Gaussian function to determine the most frequently observed populations of contour lengths (Note 14). Force peaks are classified according to these populations of contour lengths determined from histograms.
Using the contour length information, map the location of stable structural segments on the secondary structure of rhodopsin (Fig. 2D) (Note 15).
For each class of force peaks, determine the rupture forces for the corresponding stable structural segment (Note 16). Generate a histogram of rupture forces and fit the histogram with a Gaussian function to determine the most probable rupture force for a given stable structral segment. Separate histograms should be generated for each data set obtained from different pulling velocities.
3.4 Analysis of DFS data
For each class of force peaks, determine the loading rate. The loading rate is the product of the pulling velocity and the effective spring constant. The effective spring constant can be determined by taking the slope of the force peak prior to rupture. Generate a histogram of loading rates and fit the histrogram with a Gaussian function to determine the most probable loading rate for a given class of force peaks. Separate histograms should be generated for each data set obtained from different pulling velocities.
For each class of force peaks, generate a DFS plot by plotting the most probable rupture force versus the logarithm of the most probable loading rate (Fig. 3).
Fit data in the DFS plot with the Bell-Evans model to compute parameters that describe the underlying energy landscape of unfolding a stable structural segment (Fig. 4)(22). Energy landscape parameters computed from this fitting include xu, the distance from the free energy minimum to the transition-state barrier, and ku, the rate of unfolding in the absence of applied force (Notes 17 and 18). These parameters provide insights about the conformational variability (xu) and kinetic stability (ku) of a stable structural segment.
Parameters determined from fits of the DFS data to Equation 2 are used to determine the height of the energy barrier, ΔG, and mechanical spring constant, κ (Notes 19 and 20). These parameters provide insights about the activation energy (ΔG) and mechanical rigidity (κ) of a stable structural segment.
Fig. 3.

DFS plots for wild-type (black) and G90D mutant (blue) rhodopsin. DFS plots for each stable structural segment of rhodopsin is shown. Data were fit using the Bell-Evans model (Equation 2). This figure is reproduced from (13) with permission from the American Society for Biochemistry and Molecular Biology.
Fig. 4.
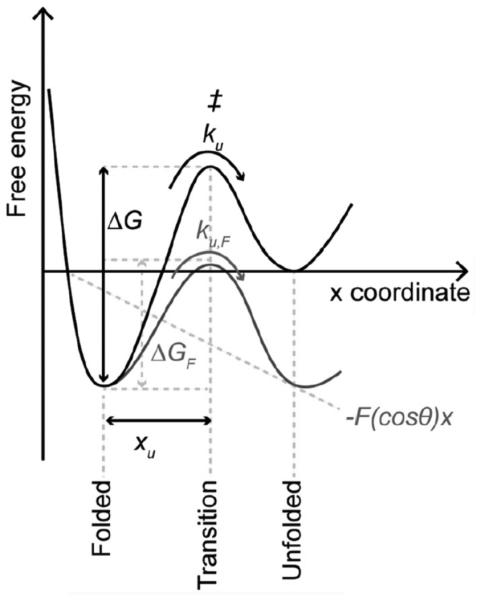
An unfolding energy landscape for a stable structural segment. The folded state of a stable structure segment is separated from the unfolded state by a free energy barrier. In the absence of an externally applied force (i.e., at equilibrium) the folded structure unfolds with a certain rate (ku) by overcoming the transition state (‡). The distance from the folded state to the transition state is characterized by xu and the height of the unfolding free energy barrier is characterized by ΔG. According to the Bell-Evans model, an externally applied force (F) tilts the energy landscape due to the mechanical energy (-F(cosθ)x), thereby lowering the free energy barrier (ΔGF) that separates the folded state from the unfolded state and increasing the rate of unfolding (kuF). Lowering of the free energy barrier is dependent on the pulling direction (x) and the angle (θ) of the externally applied force (F). This figure is reproduced from (13) with permission from the American Society for Biochemistry and Molecular Biology.
Acknowledgements
This work was funded by the European Community's Seventh Framework Programme [FP7/2007-2013] under grant agreement n° [211800], Swiss National Science Foundation, National Institutes of Health (R01EY021731), and Research to Prevent Blindness (Career Development Award).
Footnotes
4. Notes
Procedures outlined here are based on ROS disc membrane preparations obtained from the eyes of 15 mice. Both frozen and freshly prepared membranes have been investigated and shown to give similar results.
Buffer composition can have a significant effect on SMFS results. Thus, only results from data obtained in the same buffer should be compared.
For general considerations pertaining to AFM imaging and SMFS of membrane proteins, the reader can refer to detailed reports published previously (16-18).
ROS disc membranes have a distinct morphology and are easily recognizable (Fig. 1A). About 90% of all proteins in the ROS disc membrane are rhodopsin (4). Rhodopsin is found throughout the ROS disc membrane except in the rim region, where there is a high concentration of structural proteins (23). Thus, SFMS studies are conducted in the central region of disc membranes.
When F-D curves are collected manually, the AFM probe is set to continuously undergo extension/retraction cycles. The drift of the piezoelectric scanner allows for sampling of different regions in the membrane. Only 1-10% of the extension/retraction cycles will result in an interaction between the AFM probe and polypeptide chain of rhodopsin sufficiently strong for protein unfolding (16). The z-position of the piezoelectric scanner must be monitored and adjusted to maintain the appropriate force with which the AFM probe pushes into the membrane. Automation of F-D curve collection is possible with AFMs that have an x-y-z closed-loop piezoelectric scanner (e.g., (24)).
When the rate of F-D curve collection begins to decrease, a different region of the same ROS disc membrane or a different ROS disc membrane should be selected. Adsorbed ROS disc membranes are typically investigated by SMFS for 30-60 min.
The sidedness of ROS disc membranes adsorbed on mica was previously determined utilizing a strategy that involved the digestion of membranes with endoproteinase Glu-C (5). The majority of ROS disc membranes adsorb onto mica exposing the extracellular surface of rhodopsin. Thus, F-D curves obtained from disc membranes represent unfolding of rhodopsin from the N-terminus.
Homemade macros can be created in IGOR Pro to help speed up the manual analysis process. Automated algorithms for analysis of F-D curves have been attempted (e.g., (25,26,24)), however, at the moment these procedures are not suitable for analysis of rhodopsin data.
F-D curves should plot the retraction curve with the tip-sample distance on the x-axis and force on the y-axis (Figs. 2A and 2B). Accurate values for tip-sample distance and force require proper determination of the deflection sensitivity and spring constant of the AFM probe.
F-D curves that represent pulling of a shorter length of polypeptide chain are ambiguous as they can arise from multiple sources. Since attachment of the AFM probe to rhodopsin occurs in a non-specific manner, the probe can make contact at any point of the polypeptide chain of rhodopsin that is exposed to the probe. Pulling from these regions will result in shorter F-D curves. Shorter curves can also arise from a loss of contact between the AFM probe and N-terminal region of rhodopsin prior to the unfolding of the entire polypeptide chain.
Rhodopsin contains a conserved disulfide bond between Cys110 and Cys187. As a result, two major populations of curves are observed (5). One corresponding to the unfolding of the receptor where the disulfide bond remains intact (curve length about 65 nm, Figs. 2A and 2B) and another population corresponding to the unfolding of the receptor in the absence of this disulfide bond (curve length about 95 nm). Since the native receptor contains a disulfide bond, the shorter length curves have been the focus of most of the studies to date.
Unfolding of rhodopsin results in a sequence of force peaks in F-D curves (Figs. 2A and 2B). Each force peak represents the sequential unfolding of a stable structural segment of the membrane protein (3). The last force peak in rhodopsin F-D curves is presumed to arise from palmitate groups that anchor two cysteine residues in the C-terminal region (19).
| (1) |
Frequently observed populations of contour lengths were originally determined by manually grouping force peaks into classes (e.g., (5,6,19)). To more accurately determine frequently observed populations of contour lengths, contour length histograms were utilized in subsequent studies. Individual peaks in histograms were initially fit manually by a Gaussian function to reveal the mean contour length of major force peaks occurring with high frequency (e.g., (10,13)) (Fig. 2C). More recently, peaks in histograms were fit simultaneously using a single Gaussian mixture model (14). This most recent method avoids bias introduced by previous methods and was able to resolve an additional population of frequently observed contour lengths. To improve the resolution of the histograms, data collected at all pulling velocities can be combined. The frequently observed populations of contour lengths appear to be independent of the pulling velocity (10).
The contour length provides an estimate for the number of amino acid residues stretched above the sample surface (Fig. 1B). This information, along with location of secondary structural elements and thickness of the lipid bilayer (≈4 nm), is used to determine the location of unfolding barriers in rhodopsin and to map the location of stable structural segments in rhodopsin (28) (Fig. 2D).
The height of force peaks in F-D curves represents the rupture force for a stable structural segment. The most probable rupture force for a class of force peaks is an indicator of the relative stability of the corresponding stable structural segment.
A linear relationship observed in DFS plots indicates that a single energy barrier separates the folded and unfolded states of a stable structural segment (Figs. 3 and 4).
| (2) |
| (3) |
References
- 1.Muller DJ. AFM: a nanotool in membrane biology. Biochemistry. 2008;47:7986–7998. doi: 10.1021/bi800753x. [DOI] [PubMed] [Google Scholar]
- 2.Whited AM, Park PS. Atomic force microscopy: a multifaceted tool to study membrane proteins and their interactions with ligands. Biochim Biophys Acta. 2014;1838:56–68. doi: 10.1016/j.bbamem.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kedrov A, Janovjak H, Sapra KT, et al. Deciphering molecular interactions of native membrane proteins by single-molecule force spectroscopy. Annu Rev Biophys Biomol Struct. 2007;36:233–260. doi: 10.1146/annurev.biophys.36.040306.132640. [DOI] [PubMed] [Google Scholar]
- 4.Papermaster DS, Dreyer WJ. Rhodopsin content in the outer segment membranes of bovine and frog retinal rods. Biochemistry. 1974;13:2438–2444. doi: 10.1021/bi00708a031. [DOI] [PubMed] [Google Scholar]
- 5.Sapra KT, Park PS, Filipek S, et al. Detecting molecular interactions that stabilize native bovine rhodopsin. J Mol Biol. 2006;358:255–269. doi: 10.1016/j.jmb.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Park PS, Sapra KT, Kolinski M, et al. Stabilizing effect of Zn2+ in native bovine rhodopsin. J Biol Chem. 2007;282:11377–11385. doi: 10.1074/jbc.M610341200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park PS, Sapra KT, Jastrzebska B, et al. Modulation of molecular interactions and function by rhodopsin palmitylation. Biochemistry. 2009;48:4294–4304. doi: 10.1021/bi900417b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans E. Probing the relation between force--lifetime--and chemistry in single molecular bonds. Annu Rev Biophys Biomol Struct. 2001;30:105–128. doi: 10.1146/annurev.biophys.30.1.105. [DOI] [PubMed] [Google Scholar]
- 9.Sapra KT, Park PS, Palczewski K, et al. Mechanical properties of bovine rhodopsin and bacteriorhodopsin: possible roles in folding and function. Langmuir. 2008;24:1330–1337. doi: 10.1021/la702299z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawamura S, Colozo AT, Muller DJ, et al. Conservation of molecular interactions stabilizing bovine and mouse rhodopsin. Biochemistry. 2010;49:10412–10420. doi: 10.1021/bi101345x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendes HF, van der Spuy J, Chapple JP, et al. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11:177–185. doi: 10.1016/j.molmed.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Sieving PA, Richards JE, Naarendorp F, et al. Dark-light: model for nightblindness from the human rhodopsin Gly-90-->Asp mutation. Proc Natl Acad Sci U S A. 1995;92:880–884. doi: 10.1073/pnas.92.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawamura S, Colozo AT, Ge L, et al. Structural, energetic, and mechanical perturbations in rhodopsin mutant that causes congenital stationary night blindness. J Biol Chem. 2012;287:21826–21835. doi: 10.1074/jbc.M112.340182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawamura S, Gerstung M, Colozo AT, et al. Kinetic, energetic, and mechanical differences between dark-state rhodopsin and opsin. Structure. 2013;21:426–437. doi: 10.1016/j.str.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zocher M, Fung JJ, Kobilka BK, et al. Ligand-specific interactions modulate kinetic, energetic, and mechanical properties of the human beta2 adrenergic receptor. Structure. 2012;20:1391–1402. doi: 10.1016/j.str.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muller DJ, Engel A. Atomic force microscopy and spectroscopy of native membrane proteins. Nat Protoc. 2007;2:2191–2197. doi: 10.1038/nprot.2007.309. [DOI] [PubMed] [Google Scholar]
- 17.Fotiadis D, Muller DJ. High-resolution imaging of 2D outer membrane protein F crystals by atomic force microscopy. Methods Mol Biol. 2013;955:461–474. doi: 10.1007/978-1-62703-176-9_24. [DOI] [PubMed] [Google Scholar]
- 18.Sapra KT. Atomic force microscopy and spectroscopy to probe single membrane proteins in lipid bilayers. Methods Mol Biol. 2013;974:73–110. doi: 10.1007/978-1-62703-275-9_5. [DOI] [PubMed] [Google Scholar]
- 19.Park PS, Sapra KT, Jastrzebska B, et al. Modulation of molecular interactions and function by rhodopsin palmitylation. Biochemistry. 2009;48:4294–4304. doi: 10.1021/bi900417b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Butt H-J, Jaschke M. Calculation of thermal noise in atomic force microscopy. Nanotechnology. 1995;6:1–7. [Google Scholar]
- 21.Bustamante C, Marko JF, Siggia ED, et al. Entropic elasticity of lambda-phage DNA. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 22.Evans E. Looking inside molecular bonds at biological interfaces with dynamic force spectroscopy. Biophys Chem. 1999;82:83–97. doi: 10.1016/s0301-4622(99)00108-8. [DOI] [PubMed] [Google Scholar]
- 23.Molday RS, Hicks D, Molday L. Peripherin. A rim-specific membrane protein of rod outer segment discs. Invest Ophthalmol Vis Sci. 1987;28:50–61. [PubMed] [Google Scholar]
- 24.Bosshart PD, Casagrande F, Frederix PL, et al. High-throughput single-molecule force spectroscopy for membrane proteins. Nanotechnology. 2008;19:384014. doi: 10.1088/0957-4484/19/38/384014. [DOI] [PubMed] [Google Scholar]
- 25.Bosshart PD, Frederix PL, Engel A. Reference-free alignment and sorting of single-molecule force spectroscopy data. Biophys J. 2012;102:2202–2211. doi: 10.1016/j.bpj.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Struckmeier J, Wahl R, Leuschner M, et al. Fully automated single-molecule force spectroscopy for screening applications. Nanotechnology. 2008;19:384020. doi: 10.1088/0957-4484/19/38/384020. [DOI] [PubMed] [Google Scholar]
- 27.Rief M, Gautel M, Oesterhelt F, et al. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 28.Müller DJ, Kessler M, Oesterhelt F, et al. Stability of bacteriorhodopsin alpha-helices and loops analyzed by single-molecule force spectroscopy. Biophys J. 2002;83:3578–3588. doi: 10.1016/S0006-3495(02)75358-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dietz H, Rief M. Exploring the energy landscape of GFP by single-molecule mechanical experiments. Proc Natl Acad Sci U S A. 2004;101:16192–16197. doi: 10.1073/pnas.0404549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bieri O, Wirz J, Hellrung B, et al. The speed limit for protein folding measured by triplet-triplet energy transfer. Proc Natl Acad Sci U S A. 1999;96:9597–9601. doi: 10.1073/pnas.96.17.9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howard J. Mechanics of Motor Proteins and the Cytoskeleton. Sinauer Associates, Inc.; Sunderland, Massachusetts: 2001. [Google Scholar]