

Abstract
Sensory transduction in the cochlea and vestibular labyrinth depends on fluid movements that deflect the hair bundles of mechanosensitive hair cells. Mechanosensitive transducer channels at the tip of the hair cell stereocilia allow K+ to flow into cells. This unusual process relies on ionic gradients unique to the inner ear. Linking genes to deafness in humans and mice has been instrumental in identifying the ion transport machinery important for hearing and balance. Morphological analysis is difficult in patients, but mouse models have helped to investigate phenotypes at different developmental time points. This review focuses on cellular ion transport mechanisms in the stria vascularis that generate the major electrochemical gradients for sensory transduction.
The ear detects sound waves, which are pressure variations in air (FIGURE 1). Sound enters the outer ear and sets the tympanic membrane in motion. Motions are conducted by middle ear bones to the oval window, from where they enter the fluid-filled cochlea of the inner ear. These “ossicles” translate air pressure variations into fluid movements along the cochlea and ensure that the impedance of the air-filled outer ear matches the impedance of the fluid-filled inner ear. Due to the mechanical properties of the basilar membrane, and owing to an active amplification mechanism mediated by electromotile properties of outer hair cells, the frequency distribution of the sound is tonotopically projected onto the basilar membrane, with the envelope of a travelling wave peaking at dominant frequencies.
FIGURE 1. Overview of the inner ear.
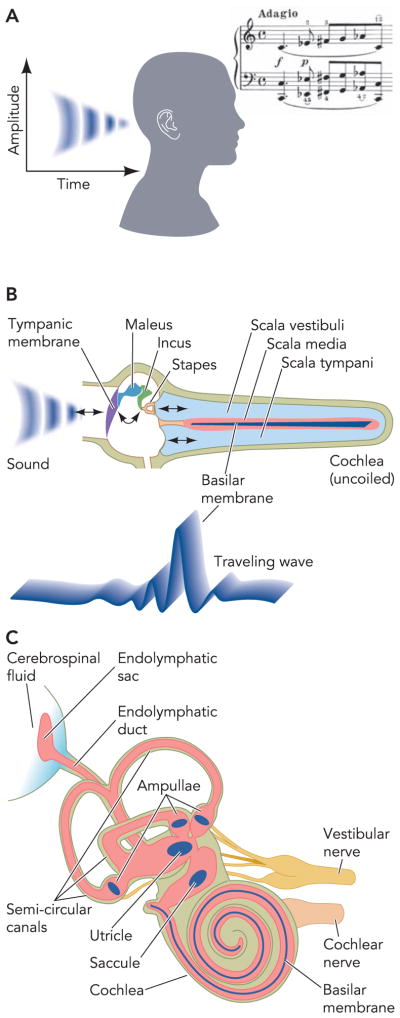
A: sound encodes time and amplitude information for pressure variations in air, which reach the outer ear. The bar of music was taken from “Fantasie in C Moll” by W.A. Mozart, KV475, completed 1785 in Wien. It was reproduced with permission from the publisher (Munchen, Germany: G. Hale Verlag, 1992). B: impedance conversion by the middle ear ossicles from air to the fluid-filled cochlea, depicted uncoiled to reveal the location of frequency detection. Due to passive properties of the basilar membrane and active amplification through the electromotile outer hair cells, a standing wave peaks at the base of the cochlea for high frequencies and at the apex for low frequencies. C: anatomy of the cochlea and vestibular labyrinth. Parts of this figure have been redrawn from Ref. 36a, with permission of the authors, editors, and publisher (Elsevier).
Sensory cells on the basilar membrane in the organ of Corti stretch along the entire length of the cochlea (FIGURE 2). Mechanical stimulation of sensory cells located at the base of the cochlea leads to the sensation of high-frequency sound and stimulation of sensory cells located in the apex of the cochlea lead to the sensation of low-frequency sound (FIGURE 1). This tonotopic organization of the cochlea is at least in part maintained throughout the central processing of auditory stimuli. The sensory process is guarded by intricate feedback mechanisms that include systems of efferent innervation that terminate on the afferent nerves and on the sensory cells.
FIGURE 2. Diagram of a cross section of the coiled cochlea.

The scala media (pink) is filled with endolymph, an unusual extracellular fluid that is high in K+ and low in Na+ and Ca2+ content. The composition of this fluid is maintained by the epithelial cells bounding the cochlear duct lumen that include the stria vascularis in the lateral wall, Reissner’s membrane, and the organ of Corti that contains the sensory inner hair cells and the outer hair cells that provide amplification of the sound-induced mechanical vibrations of the basilar membrane. Parts of this figure have been redrawn from Ref. 36a, with permission of the authors, editors, and publisher (Elsevier).
Hair cell mechanoreceptors rely on ionic gradients with a unique organization in the inner ear: They allow the passive flow of K+ into cells. These electrochemical gradients are achieved by an unusually high K+ concentration and a positive potential of the fluid in the scala media, one of the three major fluid spaces of the cochlea (the other two are the scalae tympani and vestibuli; see FIGURE 2). Both the high potassium concentration and the positive potential are generated by the epithelium of the stria vascularis in the lateral wall of the scala media. The stria vascularis thereby generates the driving force for sound detection by hair cells, which require almost no input of metabolic energy. In the potassium recycling model of the inner ear, K+ ions entering hair cells are brought back to the stria for secretion into scala media using a largely intracellular pathway (FIGURE 3). Several of the molecules involved in this potassium recycling pathway have been identified through mutations in mice and humans that lead to deafness.
FIGURE 3. Overview over the stria marginalis and its K+ transport mechanisms and two alternative K+ pathways removing K+ from the hair cells.

The stria vascularis consists of three distinct cell types: marginal cells, intermediate, and basal cells. Intermediate cells are connected via gap junctions to basal cells, which in turn form connexons with underlying fibrocytes. Model A postulates that K+ released from hair cells cycles back to the stria vascularis through the open perilymph space, whereas model B entails K+ recycling through inner phalangeal cells, marked as supporting cells (SC) here, in the case of inner hair cells (IHC) or Deiters’ cells (DC) in the case of outer hair cells (OHC). These cells act as a K+ buffer in model C. Note that these three models are not mutually exclusive. Parts of this figure have been redrawn from Ref. 36a, with permission of the authors, editors, and publisher (Elsevier).
Sound-induced vibrations of the basilar membrane cause bending of the stereocilia on the apical membrane of the hair cells, which modulates the ionic currents through the transduction channels. The modulated current leads to receptor potentials. Stereocilia in the apical membrane of hair cells are oriented in a pattern resembling organ pipes. Bending of the stereocilia toward the longer stereocilia opens the transduction channel, whereas bending toward the shorter stereocilia closes the channel. The main consequence of receptor potentials in inner hair cells is the modulation of the release of the neurotransmitter glutamate and stimulation of type I afferent dendrites (FIGURE 2). The activity of afferent dendrites is modulated by efferent fibers that terminate on the afferents near the base of the inner hair cells. Type I afferent axons transmit the primary acoustic input to the brain. In contrast to inner hair cells, the main consequences of receptor potentials in outer hair cells are “piezo-electric” length changes of the cell body that lead to an amplification of the sound-induced vibrations of the basilar membrane (FIGURE 2). Outer hair cells express the voltage-sensitive protein prestin at high density in their lateral cell wall. Membrane voltage changes cause this relative of anion transporters to slightly contract or expand, thereby leading to a shortening of the cell body during membrane potential depolarization and a lengthening during membrane potential hyperpolarization. Chicken and zebrafish prestin, which are believed to be non-motile, have been shown to exchange SO42− or oxalate for Cl− with 1:1 stoichiometry (52), but whether the electromotile mammalian orthologs are nonconductive is still controversial (5, 39). The resulting amplification of basilar membrane vibrations is necessary for the high sensitivity and the sharpness of frequency discrimination of the mammalian cochlea. The role protein prestin plays in cochlear sound amplification is subject of excellent recent reviews (4, 16).
Depolarizing K+ entry into sensory hair cells relies on a unique arrangement and composition of fluids surrounding them. The stria vascularis of the inner ear secretes the high K+ containing fluid (endolymph) that bathes the apical poles of the hair cells with their mechanoreceptors. In the vestibular organ, functionally similar vestibular dark cells perform this task. Complementing the high K+ content, endolymph contains much less Na+ and Ca2+ than extracellular fluid found elsewhere in the body. The ion composition of the endolymph resembles intracellular fluid, whereas that of the perilymph corresponds to usual extracellular fluids (with ~5 mM K+ ). The cochlear but not the vestibular endolymph is additionally kept at a strongly positive “endocochlear potential” (EP). It is generated to a large extent by specialized cells of the stria vascularis (intermediate cells) that have no equivalent in the vestibular organs (FIGURE 3). Therefore, the potential of the vestibular endolymph is on the order of only a few mV, whereas the EP is as high as +100 mV. The EP adds to the potential generated at the basolateral membrane of the cochlear hair cells, boosting their sensitivity. Consequently, disrupting the EP results in deafness.
Because the electrochemical potential for K+ is very different across the apical and basolateral membranes of hair cells, K+ can flow passively both into hair cells at the apical pole and out of the cell at the basal side. Since the driving force for K+ exit at the basal side is lower than that for K+ entry at the apical pole, they may need more K+-conducting channels at the basal pole. Given that hair cells have a negative resting membrane potential and experience virtually no K+ concentration gradient across their apical membrane, there is already a large driving force for apical K+ into vestibular hair cells. In the cochlea, where it is augmented by the EP, it is huge. Since K+ is the main cation in the cytosol, the relative change of intracellular ionic concentrations during sensory transduction is minimal.
Why has nature chosen K+ instead of Na+ as carrier ion for the depolarizing current of hair cells? The continuous passive influx of K+ at the apical side facing the endolymph allows detection of hair bundle movement in either direction, reducing or increasing K+ influx. If these currents were carried by Na+, metabolic energy would be required to constantly remove Na+ actively from the cell. The energy required for ATP-driven pumps might require vascularization close to the hair cells, changing the micromechanics of the cochlea. Blood flow would also cause vibration perceived as noise. The hair cells thus use stria vascularis as a remote “power plant” to generate the energy necessary for sound transduction. To perform its task, the stria is one of the most highly vascularized tissues found in the adult mammalian body and is the only epithelium with intraepithelial vessels.
The different K+ concentrations of endolymph and perilymph, which are crucial for the passive flux of K+ through hair cells, require an efficient separation of both fluid spaces. Tight junctions containing claudin-14 and claudin-9 are likely to play this role by controlling paracellular permeability. Mutations in the genes encoding claudin-14 and claudin-9 that are present at tight junctions in the organ of Corti underlie autosomal recessive deafness in humans (DFNB29) (69) and mice (41). A knockout mouse model shows very early degeneration of both outer and inner hair cells (7). Expression of this claudin decreases paracellular permeability by making the tight junctions of model MDCK cells less permeable to Na+ and K+, a process that may be regulated by phosphorylation (71).
Before we turn in detail to the mechanisms involved in the secretion of K+ into the endolymph and the generation of the EP, we shall trace the path of K+ entering the hair cells and recycling through a system of channels, transporters, and gap junctions toward the stria vascularis and discuss the proteins involved. It should be noted that, although many pieces of evidence suggest the presence of K+ recycling in the cochlea and in the vestibular system, this concept is not unchallenged (58). Furthermore, several cycling pathways have been described. Part of the K+ may travel through the open perilymph space of the scala tympani. This model is based on current measurements (74) and measurements of sound-induced increases in the extracellular K+ concentration in the tunnel of Corti (23). These suggested that K+ may reach the scala tympani directly, flowing out from hair cells, as depicted in model A of FIGURE 3 or indirectly from Deiters’ cells that may be engaged to buffer extracellular K+ concentrations (23). Another concept envisages K+ taken up into Deiters’ cells and inner phalangeal cells into which the basal poles of hair cells protrude. They may merely buffer K+ (model C of FIGURE 3) or they may relay it to the stria via a system of gap junctions and transporters (model B of FIGURE 3) (27), and this has gained support from several mouse models described below.
Which are the molecular identities of the channels involved in the conduction of K+ through hair cells? Biophysical studies showed that the mechanosensitive channels, through which K+ enters and depolarizes hair cells, are nonselective cation channels. ENaC and members of the ASIC and Trp channel families have all been candidates for these channels in the past but have been ruled out by subsequent studies (13). NompC was identified as a channel involved in mechanosensation in Drosophila, and a homolog was localized in mechanosensitive organs in C. elegans. Therefore, their homolog TrpA1 was suggested to play this role in mammalian hair cells (14) based on in situ hybridization and immunohistochemistry. However, this candidate has seemingly been ruled out by the generation of a knockout mouse model that is not deaf (31). It is still likely that a member of the Trp family of cation channels mediates mechanosensation in hair cells, but an unexpected protein family could be involved as well.
A major pathway for K+ exit from outer hair cells is probably the KCNQ4 (also known as Kv7.4) K+ channel. Mutations in the gene encoding this channel subunit underlie a slowly progressive dominant form of human deafness (DFNA2). These patients carry dominant negative mutations leading to hearing loss over years and decades (30). In the cochlea, KCNQ4 is present at the basal pole of outer hair cells and to a minor degree also in inner hair cells (25, 26). It is also expressed in type I vestibular hair cells and in tracts and nuclei of the central auditory pathway (26). The basal localization fits well to a role in K+ exit from hair cells. Mouse models with altered Kcnq4 genes have shed light on the patho-physiological mechanisms underlying DFNA2 deafness (25). When KCNQ4 is absent, cochlear outer hair cells degenerate. Inner hair cells and vestibular hair cells appear unaffected, correlating with expression levels of KCNQ4. Outer hair cells degenerated first (over several weeks) at the basal turn of the cochlea, which mediates high-frequency hearing. Another mouse model was analyzed carrying a dominant negative KCNQ4 mutant (which was found in humans with DFNA2) inserted into the mouse genome (25). The mutant channel subunit decreases KCNQ4 currents when present together with wild-type subunits in heteromeric channels. Since Kv-type K+ channels assemble into tetramers, the resulting K+ conductance is expected to be reduced to 6.25% in mice and humans heterozygous for this mutation. Accordingly, outer hair cell degeneration was much slower in mice heterozygous for the dominant negative mutant than in knockout (KO) mice. Patch-clamp recording carried out before onset of degeneration revealed that outer hair cells in such KO mice were depolarized by ~13 mV, whereas inner hair cells were only depolarized by ~7 mV. The depolarization is expected to increase Ca2+ influx through voltage-gated Ca2+ channels into these cells and may thereby underlie their slow degeneration. In both the KO and the KI, the hearing threshold declined by ~50 dB. Whereas this maximal hearing loss was reached after ~6–8 wk in the KO, heterozygous dominant negative KI mice reached this level of deafness only after 42–60 wk. This is compatible with the ~6% of KCNQ4 current remaining in the latter mice and reflects the slow progression of DFNA2-type hearing loss in humans. This extent of hearing loss is compatible with a total and selective loss of outer hair cell function (remember that inner hair cells were unaffected). Indeed, the electromechanical sound amplification by outer hair cells increases hearing sensitivity by 40–60 dB (33).
Even though the depolarization of outer hair cells in the KO mouse model demonstrated a crucial role of KCNQ4 in the maintenance of their resting potential, other K+ channels might contribute to the exit of K+ from hair cells as well. One such channel might be the BK Ca2+-activated K+ channel, as suggested by another mouse model (48). The degeneration of outer hair cells in those mice was attributed to a secondary loss of KCNQ4 expression, the mechanism of which is unclear. As such, a downregulation of outer hair cell KCNQ4 protein levels was also observed in the barttin KO mouse (Ref. 47; see below) and by hypothyroidism (40), and may be an unspecific stress response of these cells.
How is K+ Released From Hair Cells Subsequently Removed?
The basal poles of cochlear hair cells are oriented toward special supporting cells. These are called Deiters’ cells in the case of outer hair cells and inner phalangeal cells in the case of inner hair cells. The membranes of Deiters’ cells facing the hair cells express KCC4 and KCC3, K+-Cl− cotransporters (inner phalangeal cells only show KCC3 expression at these locations) (9, 10). Mice lacking KCC4 show rapidly progressive hearing loss due to a degeneration of outer hair cells (9). The time course of degeneration was comparable to that of KCNQ4 knockout mice. Outer hair cells are also expected to depolarize when K+ removal from the clefts between basal poles and Deiters’ cells is impaired. KCC cotransporters may relay the K+ ions released via KCNQ4 into the underlying Deiters’ cell/epithelial cell/fibrocyte system. Although KCC cotransporters mostly mediate the exit of KCl from cells, they usually operate close to equilibrium (22). It is conceivable that in the tiny cleft between Deiters’ cell and outer hair cell a high K+ concentration ensures transport directed into Deiters’ cells. KCl uptake will be favored by a low intracellular Cl− concentration in Deiters’ cells. This might be achieved by Cl− extrusion through KCC3, which is not only expressed in Deiters’ cells but also in the epithelial cells that are coupled to them via gap junctions. KCl transport may proceed both in and out of this electrically coupled syncythium if the K+ concentration at the Deiters’ cell side is high, whereas it is kept low through active removal at the other side. Usually cells employ the 3Na+/2K+-ATPase or Na+/K+/2Cl− cotransporters for K+ accumulation, but both processes require the input of metabolic energy (Na+/K+/2Cl− cotransport requires Na+ extrusion by the ATPase). Thus a role of KCC4 in K+ removal fits very well into the scheme of largely passive K+ movement in the organ of Corti. It also fits to the observation that outer hair cells degenerate before morphological alterations in Deiters’ cells can be detected (9). Hence, a loss of outer hair cells due to Deiters’ cell degeneration, which might be a consequence of defective cell volume regulation (a known role of KCC4), can be excluded (9).
The importance of KCC cotransporters for hearing is highlighted by a second mouse model. Mice in which KCC3 has been disrupted also develop deafness, again due to hair cell degeneration (10). Compared with KCC4 knockout mice, the degeneration occurs at a much slower time scale. In addition to Deiters’ cells, epithelial cells attached to them, and fibrocytes underlying the stria vascularis express KCC3. These fibrocytes degenerate likewise during adolescence in these mice (10). Interestingly, fibrocytes below the organ of Corti and below the stria vascularis express KCC3, but there is a conspicuous region where KCC3 staining is absent (type II fibrocytes). In gerbil, this particular region shows high NKCC1 expression (15). Interestingly, Deiters’ cells and the epithelial cells sitting on the basilar membrane are connected by gap junctions, as are the fibrocytes in the lateral wall of the cochlea. It is likely that K+ is transported through these gap junctions from cell to cell. However, between these two gap junction systems, K+ has to pass the extracellular space, exiting from the epithelial system and being subsequently taken up by type III fibrocytes. It is attractive to assume that K+ exits the epithelial gap junction system through KCC3, then is taken up by the fibrocyte Na+/K+/2Cl− cotransporter. The cotransporter would create a low K+ concentration in the space between those cells, just like in the space between strial intermediate and marginal cells (see below). This low extracellular K+, in turn, will favor the outward movement of K+ and Cl− through KCC3, resulting in a low Cl− concentration in the epithelial gap junction system that connects these cells with Deiters’ cells. As discussed above, a low Cl− concentration in Deiters’ cells would favor K+ and Cl− entry though KCC4 (and KCC3) based on the reasonable assumption that extracellular K+ is higher close to Deiters’ cells (where it is supplied by K+ efflux from outer hair cells) than close to type II fibrocytes (where it is lowered by uptake through NKCC1).
In this attractive K+ recycling model (model B in FIGURE 3), potassium ions may therefore be transported through many layers of cells efficiently via gap junctions and KCC cotransporters until again metabolic energy [dissipating Na+ gradients using the NKCC1 (Na+/K+/2Cl−) cotransporter] provides new impetus for the rest of the pathway toward the stria vascularis. The idea of K+ recycling is in fact older and was originally based on morphology and the finding that K+ supply to the strial marginal cells originates from perilymph rather than blood (29, 59). The finding that disruption of proteins involved in K+ transport along this recycling pathway leads to a common phenotype, hair cell degeneration, supports this concept. However, alternative K+ pathways have been postulated and may well act in parallel to the pathway outlined above, such as models A and C of FIGURE 3. All K+ pathways are shown as arrows in FIGURE 3.
Although disruption of the ion channels and transporters discussed above mostly results in outer hair cell degeneration, disruption of many of the K+ secretory mechanisms in the stria additionally leads to a physical collapse of the endolymph space. This reflects the loss of fluid secretion associated with impaired KCl transport into the endolymph. Endolymph is enclosed by heterogeneous epithelia that include the stria vascularis, the organ of Corti, and Reissner’s membrane. As its volume decreases, the more compliant Reissner’s membrane approaches the basilar membrane, organ of Corti, and stria vascularis. Likewise, the membraneous semicircular canals of the vestibular organ may collapse and may assume a starlike shape in cross sections.
The stria vascularis does not merely secrete K+-containing fluid. It also generates the lumen-positive EP. Two tight-junction barriers at the marginal cell layer and the intermediate/basal cell layer ensure that neither K+ nor electric potential is dissipated. The K+ secretory mechanism in the marginal cells is depicted in FIGURE 3. Energized by inward movement of one Na+ ion, a basolateral Na+/K+/2Cl− cotransporter (NKCC1) moves 2 Cl− and 1 K+ ion against their electrochemical gradients into the cell. It additionally provides Na+ for the operation of the 3Na+/2K+-ATPase, which ultimately energizes NKCC1 but also contributes to K+ transport. The accompanying Cl− is recycled at the basolateral membrane via Cl− channels. K+ is secreted into the endolymph via K+ channels. The apical membrane potential difference is on the order of few mV because K+ concentrations are similar on both sides. This also ensures that the voltage-activated K+ conductance formed by KCNQ1/KCNE1 heteromeric channels is active (6, 51). However, based on microelectrode studies, the basolateral membrane voltage is very low as well (38). This membrane has a dominant Cl− conductance, which ensures together with a high intracellular Cl− concentration (36) that the EP generated by the preceding cell layer is not dissipated. Since the mechanisms described above are tightly linked, isolated disruption in mice often leads to similar phenotypes based on a loss of KCl secretion into the endolymph space and consecutive loss of fluid secretion.
Knockout models for NKCC1 show a lack of K+ secretion into the endolymphatic space and, as a consequence, exhibit a collapse of Reissner’s membrane and of the vestibular endolymph system (17, 18). Their phenotypes include bidirectional circling, hyperactivity, and head bobbing described as shaker/waltzer behavior, although mice may also exhibit neurological deficits related to the lack of neuronal NKCC1 (46).
The Cl− ions accompanying the K+ are recycled at the basolateral membrane via ClC-Ka and ClC-Kb, as was proposed from whole cell patch-clamp analysis and single-cell PCR (2, 34, 49). Both subunits heteromerize with a β-subunit called barttin. This is illustrated by deafness in human Bartter’s syndrome type IV that results from mutations in BSND, the gene encoding barttin (8), and deafness in a recent mouse model we produced (47). Barttin is present in the basolateral membranes of strial marginal and various cell types of the distal nephron (19).
The absence of functional ClC-K chloride channels from the basolateral membrane in kidney tubule cells due to mutations in either ClC-Kb (Bartter’s syndrome type III) or barttin (type IV) entails salt loss (8, 56). Interestingly, expression of ClC-Ka and -Kb is redundant in the inner ear, as hypothesized from the finding that Bartter type III patients hear normally and that ClC-Ka knockout mice have not been reported to be deaf. This is supported by patients showing Bartter type IV symptoms with mutations in both ClC-Ka and -Kb but not barttin (45, 53). Thus ClC-Kb expression is rate limiting only in the kidney, explaining why its disruption alone leads to Bartter’s syndrome without deafness.
In barttin KO mice, NKCC1 cotransporter function is expected to be impaired in strial marginal cells, since Cl− recycling is strongly reduced at the basolateral membrane. This is similar to impairment of Cl− reabsorption in Bartter Type I patients (with defective luminal ROMK potassium channels) where K+ recycling limits NaCl reabsorption in the thick ascending limb of the kidney tubule. Unexpectedly, fluid secretion into the endolymph was not affected in barttin KO mice. The position of Reissner’s membrane was normal in mice with selective deletion of barttin in the inner ear. Both spurious Na+ and Cl− conductances could help to maintain K+ uptake at the basolateral membrane via a Na+-K+-ATPase or NKCC1. The mechanism of hearing loss turned out to be related to the loss of EP. This potential is believed to be largely generated across the apical membrane of the intermediate cells facing the marginal cells. It expresses KCNJ10 (Kir 4.1), and mice with a deletion of this inwardly rectifying K+ channel are also deaf but show diminished K+ secretion in addition to loss of the EP (37). Mutations in KCNJ10 have been shown to cause deafness, epilepsy, ataxia, and changes in renal calcium and magnesium handling in humans (8a, 54). To generate a large potential difference across this membrane, intrastrial K+ must be low—this has been established by ion-selective microelectrode studies (44, 50). Its low concentration relies on effective removal of K+ by the strial marginal cells. With Cl− recycling at the basolateral membrane impaired, NKCC1 will cease to operate. This will affect K+ removal from the intrastrial space by both NKCC1 and the 3Na+/2K+-ATPase since the former provides the latter with Na+. The K+ concentration in the intrastrial space is expected to rise, collapsing the voltage at the apical membrane of strial intermediate cells. Accordingly, the EP was reduced from about +100 mV to roughly +15 mV, whereas endolymphatic K+ concentration was normal in barttin-deficient mice (47). A loss of EP impairs hair cell function, and a hearing loss of 60 dB was indeed observed. A loss of otoacoustic emissions apparent at hearing onset indicates that outer hair cell dysfunction occurs before degenerative changes appear.
As expected, disruption of the luminal K+ exit channel from strial marginal cells leads to a very severe loss of K+ secretion. This luminal exit is mediated by KCNQ1/KCNE1 heteromeric channels (35, 65). Loss-of-function mutations in either of these subunits cause recessive Jervell-Lange-Nielsen syndrome in humans (60), characterized by deafness and cardiac arrhythmia. Heteromeric KCNQ1/KCNE1 also play an important role in repolarizing cardiac cells. Notably, dominant mutations in either subunit, leading to Romano-Ward-syndrome (70), affect the heart but not the inner ear. The residual function of ~6% homomeric wild-type channels expected to assemble in these heterozygous patients is apparently sufficient to sustain K+ secretion in the stria. In contrast, a KCNE1 knockout mouse model (64) showed a collapse of Reissner’s membrane and a degeneration of hair cells at an early stage in cochlear development. A mouse deficient for KCNQ1 (32) replicates the ear phenotype of KCNE1 knockout mice, providing genetic evidence that other subunits or other potassium channels cannot substitute for this heteromer in the stria vascularis. The latter had already been suggested by the complete absence of K+ secretion in Ussing chamber experiments on stria from KCNE1 knockout mice (64).
Many transport mechanisms found in stria vascularis were first discovered in the vestibular labyrinth where K+ secretion is mediated by vestibular dark cells that are largely equivalent to strial marginal cells (65). Some K+ transport mechanisms are unique to the stria vascularis and are not found in its vestibular equivalent. Based on prominent expression of gastric-type proton ATPase in the stria and the lateral cochlear wall and the effect of inhibitors on the EP, its role in K+ recycling was hypothesized (55), but very high doses of inhibitors were required, and endolymph pH regulation may be impaired under these circumstances as well. It is therefore unlikely that this process directly contributes to the generation of the EP. A prominent difference between cochlear stria and the vestibular epithelium is the lack of intermediate cells, generating the EP in the stria. KCNJ10 expression was found to be restricted to strial intermediate cells, with no expression in the vestibulum (21, 62). Consistently, KCNJ10 KO mice show deafness but lack an apparent vestibular phenotype (37). In this mouse model, vestibular K+ secretion was unaffected, but the K+ content of the cochlear endolymph was reduced. Absence of KCNJ10 expression and thereby loss of the EP is also found in a Pendred syndrome mouse model (20, 67). Pendrin is a member of the SLC transporter family and may exchange chloride for bicarbonate in the inner ear. A series of studies has established a chain of events that ultimately lead to a loss of KCNJ10 in the stria and, consequently, a reduction of the EP. Loss of pendrin causes acidification of the cochlear endolymph (68), which in turn impairs Ca2+ absorption from endolymph (42, 68), causing free oxygen radical stress (57), which ultimately abolishes KCNJ10 expression in intermediate cells (67). Heterozygous mutations in both pendrin and KCNJ10 lead to a hearing loss with enlarged vestibular aquaduct, as in Pendred syndrome. Data from heterozygous pendrin KO mice suggest this is again due to reduced KCNJ10 expression (72). Altered pH affecting ion transporter expression may hint at the mechanism in two other settings, where mechanisms have not been studied in great detail so far. Deafness is also associated with mutations in two H+ ATPase subunits, ATP6B1 (24) and ATP6V0A4 (61), the latter of which was shown to be expressed in the inner ear. Interestingly, a KO model for claudin-11 (28) shows a similar phenotype as in KCNJ10 KO mice where there is an increased hearing threshold, a strongly reduced EP, but normal K+ concentration in the endolymph. In claudin-11-deficient mice, tight junctions are missing between strial basal cells, whereas they are morphologically normal between strial marginal cells. An intact electrical barrier in the basal cell layer is therefore essential for the generation of the EP.
Intermediate cells and basal cells connect to underlying fibrocytes via gap junctions at their basal side. Gap-junction channels or connexons consist of six connexin hemichannels on each opposing cell membrane. The importance of intermediate cell and fibrocyte gap junctions for hearing is illustrated by human mutations affecting several of their isoforms expressed in the inner ear. Connexin-26, -30, -31, and -43 are mutated in hereditary forms of deafness, and KO mouse models replicate this phenotype [connexin-30 (63), connexin-26 (11)], although mechanisms may be diverse. Mutations in connexin-26 underlie DFNB1, the most frequent form of prelingual human deafness (3). A detailed analysis of mice with connexin-26 inactivated specifically in the epithelial gap-junction network revealed that supporting cells for the IHCs are the first to undergo apoptosis, followed by outer hair cells and their supporting cells (11). The epithelial gap-junction network is believed to funnel K+ away from the hair cells, and this mouse model thus supports the notion of K+ recycling via this route. Both EP and endolymph K+ were normal before any morphological change in the organ of Corti but were reduced later, probably as result of damage to the reticular lamina sealing endolymph from perilymph (11).
Interestingly, the endolymph K+ concentration was initially normal in connexin-30 KO mice, but the EP was absent (63). It is tempting to speculate that the arrangement of electrically coupled cells underlying the intermediate cells allows full exploitation of the K+ diffusion potential created across the “apical” membrane of intermediate cells (facing the marginal cells) for the generation of the EP. If their basal membranes also expressed a dominant K+ conductance, the resulting transepithelial voltage would be nullified. These cells must maintain the basolateral membrane potential close to 0 mV, either via a nonselective cation conductance, which would load a single cell layer heavily with Na+, or a Cl− conductance in the presence of high intracellular chloride. The nature of this conductive pathway, however, remains elusive. Another possibility is that loss of the endothelial barrier in connexin-30 KO mice (12) leads to shunting of the EP, but since the endothelial barrier was already affected before development of the EP, more quantitative examination will be necessary to establish this as the cause. Interestingly, overexpression of connexin-26 restored hearing in connexin-30-deficient mice (1).
Gap junctions most probably have additional functions in the cochlea, which are more related to their transfer of signaling molecules rather than to ion transport (73). A recent review has addressed these functions (43).
Summary
Even with a large variety of mouse models available for components of strial electrolyte transport, answers to many open questions will rely on the elimination of proteins in a cell-specific manner to assess in isolation the effects of their disruption on the K+ recycling pathway. It is still not entirely clear, for example, how gap junctions contribute to the generation of the EP. It should also be noted that, although many pieces of evidence support the concept of K+ recycling in the cochlea and vestibulum, there are also alternative models (reviewed in Ref. 66). However, it is reassuring that the components of the stria have been sufficiently characterized for mathematical models of K+ transport, based on some experimental data and assuming K+ recycling, to predict accurately a number of other experimentally confirmed parameters (35, 43).
References
- 1.Ahmad S, Tang W, Chang Q, Qu Y, Hibshman J, Li Y, Sohl G, Willecke K, Chen P, Lin X. Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human connexin30-linked deafness. Proc Natl Acad Sci USA. 2007;104:1337–1341. doi: 10.1073/pnas.0606855104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ando M, Takeuchi S. mRNA encoding ‘ClC-K1, a kidney Cl− channel’ is expressed in marginal cells of the stria vascularis of rat cochlea: its possible contribution to Cl− currents. Neurosci Lett. 2000;284:171–174. doi: 10.1016/s0304-3940(00)01021-1. [DOI] [PubMed] [Google Scholar]
- 3.Angeli S, Utrera R, Dib S, Chiossone E, Naranjo C, Henriquez O, Porta M. GJB2 gene mutations in childhood deafness. Acta Otolaryngol (Stockh) 2000;120:133–136. doi: 10.1080/000164800750000766. [DOI] [PubMed] [Google Scholar]
- 4.Ashmore J. Cochlear outer hair cell motility. Physiol Rev. 2008;88:173–210. doi: 10.1152/physrev.00044.2006. [DOI] [PubMed] [Google Scholar]
- 5.Bai JP, Surguchev A, Montoya S, Aronson PS, Santos-Sacchi J, Navaratnam D. Prestin’s anion transport and voltage-sensing capabilities are independent. Biophys J. 2009;96:3179–3186. doi: 10.1016/j.bpj.2008.12.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 7.Ben-Yosef T, Belyantseva IA, Saunders TL, Hughes ED, Kawamoto K, Van Itallie CM, Beyer LA, Halsey K, Gardner DJ, Wilcox ER, Rasmussen J, Anderson JM, Dolan DF, Forge A, Raphael Y, Camper SA, Friedman TB. Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum Mol Genet. 2003;12:2049–2061. doi: 10.1093/hmg/ddg210. [DOI] [PubMed] [Google Scholar]
- 8.Birkenhäger R, Otto E, Schürmann MJ, Vollmer M, Ruf EM, Maier-Lutz I, Beekmann F, Fekete A, Omran H, Feldmann D, Milford DV, Jeck N, Konrad M, Landau D, Knoers NVAM, Antignac C, Sudbrack R, Kispert A, Hildebrandt F. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. 2001;29:310–314. doi: 10.1038/ng752. [DOI] [PubMed] [Google Scholar]
- 8a.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van’t Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360:1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boettger T, Hübner C, Maier H, Rust MB, Beck FX, Jentsch TJ. Deafness and renal tubular acidosis in mice lacking the K-Cl cotransporter Kcc4. Nature. 2002;416:874–878. doi: 10.1038/416874a. [DOI] [PubMed] [Google Scholar]
- 10.Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, Lemcke B, Horst J, Leuwer R, Pape HC, Volkl H, Hübner CA, Jentsch TJ. Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J. 2003;22:5422–5434. doi: 10.1093/emboj/cdg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen-Salmon M, Ott T, Michel V, Hardelin JP, Perfettini I, Eybalin M, Wu T, Marcus DC, Wangemann P, Willecke K, Petit C. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr Biol. 2002;12:1106–1111. doi: 10.1016/s0960-9822(02)00904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen-Salmon M, Regnault B, Cayet N, Caille D, Demuth K, Hardelin JP, Janel N, Meda P, Petit C. Connexin30 deficiency causes instrastrial fluid-blood barrier disruption within the cochlear stria vascularis. Proc Natl Acad Sci USA. 2007;104:6229–6234. doi: 10.1073/pnas.0605108104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corey DP. What is the hair cell transduction channel? J Physiol. 2006;576:23–28. doi: 10.1113/jphysiol.2006.116582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corey DP, Garcia-Anoveros J, Holt JR, Kwan KY, Lin SY, Vollrath MA, Amalfitano A, Cheung EL, Derfler BH, Duggan A, Geleoc GS, Gray PA, Hoffman MP, Rehm HL, Tamasauskas D, Zhang DS. TRPA1 is a candidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature. 2004;432:723–730. doi: 10.1038/nature03066. [DOI] [PubMed] [Google Scholar]
- 15.Crouch JJ, Sakaguchi N, Lytle C, Schulte BA. Immunohistochemical localization of the Na-K-Cl co-transporter (NKCC1) in the gerbil inner ear. J Histochem Cytochem. 1997;45:773–778. doi: 10.1177/002215549704500601. [DOI] [PubMed] [Google Scholar]
- 16.Dallos P. Cochlear amplification, outer hair cells and prestin. Curr Opin Neurobiol. 2008;18:370–376. doi: 10.1016/j.conb.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet. 1999;22:192–195. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- 18.Dixon MJ, Gazzard J, Chaudhry SS, Sampson N, Schulte BA, Steel KP. Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Hum Mol Genet. 1999;8:1579–1584. doi: 10.1093/hmg/8.8.1579. [DOI] [PubMed] [Google Scholar]
- 19.Estévez R, Boettger T, Stein V, Birkenhäger R, Otto E, Hildebrandt F, Jentsch TJ. Barttin is a Cl− channel beta-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature. 2001;414:558–561. doi: 10.1038/35107099. [DOI] [PubMed] [Google Scholar]
- 20.Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet. 2001;10:153–161. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- 21.Hibino H, Horio Y, Inanobe A, Doi K, Ito M, Yamada M, Gotow T, Uchiyama Y, Kawamura M, Kubo T, Kurachi Y. An ATP-dependent inwardly rectifying potassium channel, KAB-2 (Kir4.1), in cochlear stria vascularis of inner ear: its specific subcellular localization and correlation with the formation of endocochlear potential. J Neurosci. 1997;17:4711–4721. doi: 10.1523/JNEUROSCI.17-12-04711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jentsch TJ. Chloride transport in the kidney: lessons from human disease and knockout mice. J Am Soc Nephrol. 2005;16:1549–1561. doi: 10.1681/ASN.2005020207. [DOI] [PubMed] [Google Scholar]
- 23.Johnstone BM, Patuzzi R, Syka J, Sykova E. Stimulus-related potassium changes in the organ of Corti of guinea-pig. J Physiol. 1989;408:77–92. doi: 10.1113/jphysiol.1989.sp017448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- 25.Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ. Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J. 2006;25:642–652. doi: 10.1038/sj.emboj.7600951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El-Amraoui A, Petit C, Jentsch TJ. KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci USA. 2000;97:4333–4338. doi: 10.1073/pnas.97.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kikuchi T, Adams JC, Miyabe Y, So E, Kobayashi T. Potassium ion recycling pathway via gap junction systems in the mammalian cochlea and its interruption in hereditary nonsyndromic deafness. Med Electron Microsc. 2000;33:51–56. doi: 10.1007/s007950070001. [DOI] [PubMed] [Google Scholar]
- 28.Kitajiri S, Miyamoto T, Mineharu A, Sonoda N, Furuse K, Hata M, Sasaki H, Mori Y, Kubota T, Ito J, Furuse M, Tsukita S. Compartmentalization established by claudin-11-based tight junctions in stria vascularis is required for hearing through generation of endocochlear potential. J Cell Sci. 2004;117:5087–5096. doi: 10.1242/jcs.01393. [DOI] [PubMed] [Google Scholar]
- 29.Konishi T, Hamrick PE, Walsh PJ. Ion transport in guinea pig cochlea. I. Potassium and sodium transport. Acta Otolaryngol (Stockh) 1978;86:22–34. doi: 10.3109/00016487809124717. [DOI] [PubMed] [Google Scholar]
- 30.Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- 31.Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, Corey DP. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron. 2006;50:277–289. doi: 10.1016/j.neuron.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 32.Lee MP, Ravenel JD, Hu RJ, Lustig LR, Tomaselli G, Berger RD, Brandenburg SA, Litzi TJ, Bunton TE, Limb C, Francis H, Gorelikow M, Gu H, Washington K, Argani P, Goldenring JR, Coffey RJ, Feinberg AP. Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J Clin Invest. 2000;106:1447–1455. doi: 10.1172/JCI10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liberman MC, Gao J, He DZ, Wu X, Jia S, Zuo J. Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature. 2002;419:300–304. doi: 10.1038/nature01059. [DOI] [PubMed] [Google Scholar]
- 34.Maehara H, Okamura HO, Kobayashi K, Uchida S, Sasaki S, Kitamura K. Expression of CLC-KB gene promoter in the mouse cochlea. Neuroreport. 2003;14:1571–1573. doi: 10.1097/00001756-200308260-00006. [DOI] [PubMed] [Google Scholar]
- 35.Marcus DC, Shen Z. Slowly activating voltage-dependent K+ conductance is apical pathway for K+ secretion in vestibular dark cells. Am J Physiol Cell Physiol. 1994;267:C857–C864. doi: 10.1152/ajpcell.1994.267.3.C857. [DOI] [PubMed] [Google Scholar]
- 36.Marcus DC, Takeuchi S, Wangemann P. Two types of chloride channel in the basolateral membrane of vestibular dark cells. Hear Res. 1993;69:124–132. doi: 10.1016/0378-5955(93)90100-f. [DOI] [PubMed] [Google Scholar]
- 36a.Marcus DC, Wangemann P. Cochlear and vestibular function and dysfunction. In: Alvarez-Leefmans FJ, Delpier E, editors. Physiology and Pathology of Chloride Transporters and Channels in the Nervous System—From Molecules to Diseases. Oxford, UK: Elsevier; 2009. In press. [Google Scholar]
- 37.Marcus DC, Wu T, Wangemann P, Kofuji P. KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am J Physiol Cell Physiol. 2002;282:C403–C407. doi: 10.1152/ajpcell.00312.2001. [DOI] [PubMed] [Google Scholar]
- 38.Melichar I, Syka J. Electrophysiological measurements of the stria vascularis potentials in vivo. Hear Res. 1987;25:35–43. doi: 10.1016/0378-5955(87)90077-3. [DOI] [PubMed] [Google Scholar]
- 39.Muallem D, Ashmore J. An anion antiporter model of prestin, the outer hair cell motor protein. Biophys J. 2006;90:4035–4045. doi: 10.1529/biophysj.105.073254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mustapha M, Fang Q, Gong TW, Dolan DF, Raphael Y, Camper SA, Duncan RK. Deafness and permanently reduced potassium channel gene expression and function in hypothyroid Pit1dw mutants. J Neurosci. 2009;29:1212–1223. doi: 10.1523/JNEUROSCI.4957-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakano Y, Kim SH, Kim HM, Sannemann JD, Zhang Y, Smith RJH, Marcus DC, Wangemann Nessler P, Banfi RAB. A Claudin-9-based permeability barrier is essential for hearing. PLoS Genetics. doi: 10.1371/journal.pgen.1000610. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakaya K, Harbidge DG, Wangemann P, Schultz BD, Green ED, Wall SM, Marcus DC. Lack of pendrin HCO3− transport elevates vestibular endolymphatic [Ca2+] by inhibition of acid-sensitive TRPV5 and TRPV6 channels. Am J Physiol Renal Physiol. 2007;292:F1314–F1321. doi: 10.1152/ajprenal.00432.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nickel R, Forge A. Gap junctions and connexins in the inner ear: their roles in homeostasis and deafness. Curr Opin Otolaryngol Head Neck Surg. 2008;16:452–457. doi: 10.1097/MOO.0b013e32830e20b0. [DOI] [PubMed] [Google Scholar]
- 44.Nin F, Hibino H, Doi K, Suzuki T, Hisa Y, Kurachi Y. The endocochlear potential depends on two K+ diffusion potentials and an electrical barrier in the stria vascularis of the inner ear. Proc Natl Acad Sci USA. 2008;105:1751–1756. doi: 10.1073/pnas.0711463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nozu K, Inagaki T, Fu XJ, Nozu Y, Kaito H, Kanda K, Sekine T, Igarashi T, Nakanishi K, Yoshikawa N, Iijima K, Matsuo M. Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J Med Genet. 2008;45:182–186. doi: 10.1136/jmg.2007.052944. [DOI] [PubMed] [Google Scholar]
- 46.Pfeffer CK, Stein V, Keating DJ, Maier H, Rinke I, Rudhard Y, Hentschke M, Rune GM, Jentsch TJ, Hübner CA. NKCC1-dependent GABAergic excitation drives synaptic network maturation during early hippocampal development. J Neurosci. 2009;29:3419–3430. doi: 10.1523/JNEUROSCI.1377-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rickheit G, Maier H, Strenzke N, Andreescu CE, De Zeeuw CI, Muenscher A, Zdebik AA, Jentsch TJ. Endocochlear potential depends on Cl− channels: mechanism underlying deafness in Bartter syndrome IV. EMBO J. 2008;27:2907–2917. doi: 10.1038/emboj.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rüttiger L, Sausbier M, Zimmermann U, Winter H, Braig C, Engel J, Knirsch M, Arntz C, Langer P, Hirt B, Müller M, Kopschall I, Pfister M, Munkner S, Rohbock K, Pfaff I, Rusch A, Ruth P, Knipper M. Deletion of the Ca2+-activated potassium (BK) alpha-subunit but not the BKbeta1-subunit leads to progressive hearing loss. Proc Natl Acad Sci USA. 2004;101:12922–12927. doi: 10.1073/pnas.0402660101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sage CL, Marcus DC. Immunolocalization of ClC-K chloride channel in strial marginal cells and vestibular dark cells. Hear Res. 2001;160:1–9. doi: 10.1016/s0378-5955(01)00308-2. [DOI] [PubMed] [Google Scholar]
- 50.Salt AN, Melichar I, Thalmann R. Mechanisms of endocochlear potential generation by stria vascularis. Laryngoscope. 1987;97:984–991. [PubMed] [Google Scholar]
- 51.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 52.Schaechinger TJ, Oliver D. Nonmammalian orthologs of prestin (SLC26A5) are electrogenic divalent/chloride anion exchangers. Proc Natl Acad Sci USA. 2007;104:7693–7698. doi: 10.1073/pnas.0608583104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schlingmann KP, Konrad M, Jeck N, Waldegger P, Reinalter SC, Holder M, Seyberth HW, Waldegger S. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med. 2004;350:1314–1319. doi: 10.1056/NEJMoa032843. [DOI] [PubMed] [Google Scholar]
- 54.Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA. 2009;106:5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shibata T, Hibino H, Doi K, Suzuki T, Hisa Y, Kurachi Y. Gastric type H+,K+-ATPase in the cochlear lateral wall is critically involved in formation of the endocochlear potential. Am J Physiol Cell Physiol. 2006;291:C1038–C1048. doi: 10.1152/ajpcell.00266.2006. [DOI] [PubMed] [Google Scholar]
- 56.Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 57.Singh R, Wangemann P. Free radical stress-mediated loss of Kcnj10 protein expression in stria vascularis contributes to deafness in Pendred syndrome mouse model. Am J Physiol Renal Physiol. 2008;294:F139–F148. doi: 10.1152/ajprenal.00433.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spicer SS, Schulte BA. Evidence for a medial K+ recycling pathway from inner hair cells. Hear Res. 1998;118:1–12. doi: 10.1016/s0378-5955(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 59.Spicer SS, Schulte BA. The fine structure of spiral ligament cells relates to ion return to the stria and varies with place-frequency. Hear Res. 1996;100:80–100. doi: 10.1016/0378-5955(96)00106-2. [DOI] [PubMed] [Google Scholar]
- 60.Splawski I, Timothy KW, Vincent GM, Atkinson DL, Cober Keating George M, Lecturer, Mark T, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. Proc Assoc Am Physicians. 1997;109:504–511. [PubMed] [Google Scholar]
- 61.Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N, Giersch AB, Morton CC, Axon PR, Akil I, Al-Sabban EA, Baguley DM, Bianca S, Bakkaloglu A, Bircan Z, Chauveau D, Clermont MJ, Guala A, Hulton SA, Kroes H, Li Volti G, Mir S, Mocan H, Nayir A, Ozen S, Rodriguez Soriano J, Sanjad SA, Tasic V, Taylor CM, Topaloglu R, Smith AN, Karet FE. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet. 2002;39:796–803. doi: 10.1136/jmg.39.11.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takeuchi S, Ando M. Inwardly rectifying K+ currents in intermediate cells in the cochlea of gerbils: a possible contribution to the endocochlear potential. Neurosci Lett. 1998;247:175–178. doi: 10.1016/s0304-3940(98)00318-8. [DOI] [PubMed] [Google Scholar]
- 63.Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Sohl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP, Petit C, Willecke K. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet. 2003;12:13–21. doi: 10.1093/hmg/ddg001. [DOI] [PubMed] [Google Scholar]
- 64.Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J. Inner ear defects induced by null mutation of the isk gene. Neuron. 1996;17:1251–1264. doi: 10.1016/s0896-6273(00)80255-x. [DOI] [PubMed] [Google Scholar]
- 65.Wangemann P. Comparison of ion transport mechanisms between vestibular dark cells and strial marginal cells. Hear Res. 1995;90:149–157. doi: 10.1016/0378-5955(95)00157-2. [DOI] [PubMed] [Google Scholar]
- 66.Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol. 2006;576:11–21. doi: 10.1113/jphysiol.2006.112888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wangemann P, Itza EM, Albrecht B, Wu T, Jabba SV, Maganti RJ, Lee JH, Everett LA, Wall SM, Royaux IE, Green ED, Marcus DC. Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2004;2:30. doi: 10.1186/1741-7015-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wangemann P, Nakaya K, Wu T, Maganti RJ, Itza EM, Sanneman JD, Harbidge DG, Billings S, Marcus DC. Loss of cochlear HCO3− secretion causes deafness via endolymphatic acidification and inhibition of Ca2+ reabsorption in a Pendred syndrome mouse model. Am J Physiol Renal Physiol. 2007;292:F1345–F1353. doi: 10.1152/ajprenal.00487.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilcox ER, Burton QL, Naz S, Riazuddin S, Smith TN, Ploplis B, Belyantseva I, Ben-Yosef T, Liburd NA, Morell RJ, Kachar B, Wu DK, Griffith AJ, Friedman TB. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell. 2001;104:165–172. doi: 10.1016/s0092-8674(01)00200-8. [DOI] [PubMed] [Google Scholar]
- 70.Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Pathophysiological mechanisms of dominant and recessive KVLQT1 K+ channel mutations found in inherited cardiac arrhythmias. Hum Mol Genet. 1997;6:1943–1949. doi: 10.1093/hmg/6.11.1943. [DOI] [PubMed] [Google Scholar]
- 71.Yamauchi K, Rai T, Kobayashi K, Sohara E, Suzuki T, Itoh T, Suda S, Hayama A, Sasaki S, Uchida S. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc Natl Acad Sci USA. 2004;101:4690–4694. doi: 10.1073/pnas.0306924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang T, Gurrola JG, 2nd, Wu H, Chiu SM, Wangemann P, Snyder PM, Smith RJ. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet. 2009;84:651–657. doi: 10.1016/j.ajhg.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y, Tang W, Ahmad S, Sipp JA, Chen P, Lin X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc Natl Acad Sci USA. 2005;102:15201–15206. doi: 10.1073/pnas.0501859102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zidanic M, Brownell WE. Fine structure of the intracochlear potential field. I. The silent current. Biophys J. 1990;57:1253–1268. doi: 10.1016/S0006-3495(90)82644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]