

Abstract
Glycerol polymers are attracting increased attention due to the diversity of polymer compositions and architectures available. This article provides a brief chronological review on the current status of these polymers along with representative examples of their use for biomedical applications. First, we describe the underlying chemistry of glycerol, which provides access to a range of monomers for subsequent polymerizations. We then review the various synthetic methodologies to prepare glycerol-based polymers including polyethers, polycarbonates, polyesters, and so forth. Next, we describe several biomedical applications where glycerol polymers are being investigated including carriers for drug delivery, sealants or coatings for tissue repair, and agents possessing antibacterial activity. Fourth, we describe the growing market opportunity for the use of polymers in medicine. Finally we conclude and summarize the findings, as well as discuss potential opportunities for continued research efforts.
Keywords: glycerol, polymer, biomedical application, biodegradable, biocompatible, medicine
1. Introduction
During the past four decades, significant advances in polymer chemistry have been reported including, but not limited to, various controlled polymerization methods (atom transfer radical polymerization, reversible addition-fragmentation polymerization, ring-opening metathesis polymerization, etc.),1–5 polymers of various architectures (linear, branched, hyperbranched, dendritic, etc.),6–9 and self-assembly strategies to prepare supramolecular polymers (hydrogen bonding, metal ligand bonding, ionic interactions).7,10–12 These advances provide new routes to polymer compositions and structures, and consequently new chemical, physical, and mechanical properties or properties with finer control than could be attained earlier. The role of polymers in commercial products continues to increase with hundreds of millions of metric tons being produced each year, with polyethylene being the largest, accounting for 37% of global polymer production.13
Synthetic polymers intended for use in biomedical applications require the additional criteria of biocompatibility and sometimes biodegradability included within the design parameters along with mechanical properties, manufacturability, etc. for the specific application in mind. The composition of the monomer and the type of linker within the main chain polymer as well as the chemical reactivity of these chemical entities will define the degradation rates and the conditions under which degradation will or will not occur. For some applications, the degradation rate of the polymer is minimized, as with polymers used in long term implants (e.g., 20+ years for a high density polyethylene coated hip implant),14 while for other applications the polymer degrades relatively rapidly (e.g., less than 10 days for a polyanhydride composed of 1,3-bis-(p-carboxyphenoxy)propane and sebacic acid loaded with carmustine for the treatment of brain cancer).15 However, biocompatibility is usually a built-in characteristic and related to the polymer (and monomer) composition and is not easily engineered into an existing polymer by converting it from being non-biocompatible to biocompatible.16 Consequently, a majority of the biocompatible polymers used in medical devices or evaluated for biomedical uses are composed of substances that are natural metabolites or known to be biocompatible and nontoxic. Using this design principle, a number of successful examples of biocompatible polymers are reported such as poly(lactic acid), poly(glycolic acid), and their copolymers, and nowadays, all of these polymers are used in US and EU approved devices.
Another example of a well-known natural metabolite is glycerol or glycerin / glycerine (IUPAC name, propane-1,2,3-triol). Karl W. Scheele discovered glycerol in 1779 when he mixed and heated olive oil with lead monoxide.17 He refereed to the product as the “sweet principle of fat.” It took more than 100 years before the chemical formula C3H5(OH)3 was established by Berthelot and Lucea in 1883.17 Glycerol serves many roles in the human body, including as the backbone unit of triglycerides and phospholipids, which are the main form of energy storage and a major component of cell membranes, respectively. Glycerol has an oral LD50 of 12600 mg/kg in rats, and is generally considered to be nontoxic. It is listed on the GRAS list (FDA website) as “generally regarded as safe”. Glycerol is widely used in the pharmaceutical and cosmetic industries as an additive (e.g., a plasticizer, thickener, emollient, demulcent, humectant, bodying agent, lubricant) due to its physical properties.
Historically, glycerol was typically synthesized from propylene via a four-step procedure which involved first chlorination of propylene to give allyl chloride, oxidation with hypochlorite to afford dichlorohydrin, reaction with a strong base to give epichlorohydrin, and finally, hydrolysis to yield glycerol. Today, glycerol is becoming a renewable raw material of ample supply due to the increases in biodiesel production.18,19 For example, the global glycerol production soared from 60,000 tons in 2001 to 800,000 tons in 2005 with 400,000 tons from biodiesel production alone.20 This large surplus of glycerol is fueling the production of downstream value-added products and stimulating research and development activities.18,19,21
Glycerol is a high boiling, viscous liquid that is colorless, odorless, hydroscopic, and sweat to the taste. The physiochemical properties of glycerol are summarized in Table 1. From a synthetic perspective, glycerol possesses a wealth of chemical diversity and opportunities for selective chemical reactivity and manipulation for being such a small molecule (C3H8O3; 92.09 g/mol).22–25 As shown in Figure 1, glycerol is a simple polyol containing two primary and one secondary hydroxyl groups with a prostereogenic center at the C2 position. Consequently, glycerol can undergo a large number and variety of chemical transformations such as selective oxidation,26–28 dehydration and hydrogenolysis,29–31 and selective protection and esterification32,33 to afford an array of value-added small molecule building blocks with increased chemical complexity for both small molecule chemical synthesis or polymer synthesis (Figure 1).34
Table 1.
Physical-chemical Properties of Glycerol
| Molecular Weight | 92.09 | Specific Gravity | 1.2620 (25 °C) |
| Specific Heat | 0.5795 cal/g·deg (26 °C) | Refractive Index | (N420)1.47399 |
| Vapor Pressure | 0.0025 mm (50 °C) | Fire Point | 204 °C |
| Boiling Point | 290 °C (760 mm), 152.0 °C (5 mm) | Flash Point | 177 °C |
| Melting Point | 18.17 °C | Viscosity | 1.499 Pa·s (20 °C) |
| Freezing Point | (66.7% glycerol solution) −46.5 °C | Surface Tension | 63.4 dynes/cm (20 °C) |
Figure 1.

(Chemoselective conversion of glycerol into value-added compounds via different catalytic pathways.)
For all of these reasons, glycerol-based polymers are attracting increasingly more attention for both fundamental studies and practical applications. Various glycerol polymer architectures from linear to dendritic are reported for pure polyglycerol ethers35,36 and carbonates37–40 as well as copolymers with hydroxyacids,37,41 for example, to give polyether esters or polycarbonate esters. This article provides a brief chronological review on the current status of polymers synthesized from glycerol and glycerol derivatives. Herein, we describe the underlying chemistry that affords these various glycerol polymers, their structural characteristics, their chemical, physical, and rheological properties, and their applications with a focus on biomedical uses. This article is not a comprehensive review (please see the reviews by Frey and Haag),42–44 but rather a perspective on recent advances of glycerol and glycerol derivative polymers, and highlights representative examples in synthetic methodologies and polymer architectures as well as the evaluation of these polymers in a range of different biomedical applications.
2. Polymer Chemistry
2.1. Linear 1,2-Linked Glycerol
Linear 1,2-linked glycerol polymers represent an important structural class of glycerol polymers. Depending on the linkage, this class of glycerol polymers includes poly(1,2-glycerol ether)s and poly(1,2-glycerol carbonate)s.
2.1.1. Linear Poly(1,2-glycerol ether)s
Linear poly(1,2-glycerol ether)s are also referred to as polyglycerols or polyglycidols and constitute the largest structural class of glycerol polymers. Because of the low price of glycidol (~0.3$/gram), the easy access to the glycidyl ether monomers (usually only require one step protection), and the controlled living polymerization process which leads to well-defined polymer structures, both the chemistry and application of poly(1,2-glycerol ether)s are extensively studied and reported over the past 40 years. For example, a Web of Science search on this topic gives more than 150 references. As the recent review by Frey et al. provides an excellent comprehensive summary of this specific area,44 we will focus on a few representative examples of the polymers synthesized and the polymer chain end and backbone functionalization chemistry reported.
Linear poly(1,2-glycerol ether)s are most frequently accessed by anionic ring-opening polymerization (AROP) of glycidyl ether such as trimethyl glycidyl ether (TMSGE), allyl glycidyl ether (AGE), tert-butyl glycidyl ether (tBGE), and ethoxyethyl glycidyl ether (EECE) with an anionic initiator. Although first attempted in 1968, poly(1,2-glycerol ether)s with a relatively high molecular weight (Mw ~ 30 kg/mol) were not reported until 1994 when EECE, as the monomer, was polymerized with cesium hydroxide as the initiator (Figure 2).45 Since then, a variety of monomers and initiators46–49 have been described for preparing diverse linear poly(1,2-glycerol ether)s with defined structures and narrow polydispersities (<1.20). In addition, due to the living nature of the AROP of glycidyl ethers, direct termination of the living polymer chain with an electrophile allows preparation of a well-defined polymer chain with one desired chain end. For example, in situ termination of a living polymer chain with p-chloromethylstyrene affords a macroinitiator bearing only one styrene on one chain end. The resultant polymer can be subsequently copolymerized with styrene to give a copolymer.46,50 After an acidic deprotection, a “surfmer” (surfactant and monomer) is obtained, which effectively serves as both emulsifier and monomer in the emulsion copolymerization of styrene. Slomkowski et al. investigated these surfmers and their use to prepare microspheres,51–56 and the results are comprehensively summarized in a review.57
Figure 2.

(Cesium hydroxide initiated polymerization of ethoxyethyl glycidyl ether to afford high molecular weight poly(1,2-glycerol ether).)
Besides p-chloromethylstyrene, various other end caps have been utilized including vinyl sulfonyl chloride,58 propargyl alcohol,59 and 2-halogenopropionyl halides,60 etc., to afford poly(1,2-glycerol ether)s with exact one chain end functionality for coupling with nucleophiles, click reactions with azides, and as initiators for atom transfer radical polymerization (ATRP). Polymers bearing exactly one terminal methacrylate can also be accessed by this end capping method and serve as valuable macromonomers in ATRP with other (macro)monomers. The research groups of both Frey48 and Haag49 report significant findings on this topic (Figure 3). Frey’s work features the direct termination of the living polymer chain with methacrylic anhydride and triethylamine. A well-defined poly(methacrylate-graft-1,2-glycerol ether) is obtained after polymerizing using standard ATRP condition. In contrast, Haag and co-workers use a two-step protocol in which the polymers are first purified and then reacted with methacryloyl chloride to afford poly(1,2-glycerol ether)s with methacrylate chain ends. Both oligomeric linear or dendritic macromonomers with methacrylate chain ends are prepared, and subsequent polymerization of these oligomeric poly(1,2-glycerol ether)s affords a series of polymer brushes with either linear or dendritic oligomeric poly(1,2-glycerol ether)s grafted as the side chain, respectively. These polymer brushes can be used for a number of applications including anchoring them onto a gold surface to form a brush polymer layer to reduce protein adsorption as measured by surface plasma resonance spectroscopy.49
Figure 3.
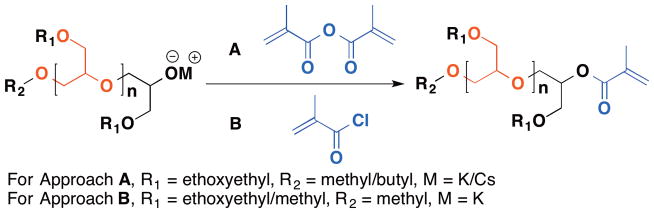
(Different approaches utilized by Frey (A) and Haag (B) to access methacrylate end-capped poly(1,2-glycerol ether)s.)
In contrast to the end cap approach, the initiator approach also allows for the introduction of exactly one chain end by initiating the polymerization with a functional initiator. Both protected amine61,62 and thiol61 initiators are successfully described and give poly(1,2-glycerol ether)s with well-defined chain ends. After polymerization, the protecting groups are removed and the exposed amine or thiol groups are subsequently used for anchoring proteins onto a gold surface. Besides these conventional end groups, other functional initiators bearing catechol,63 cholesterol,64–66 and admantyl67 groups are also reported. For example, catechol-terminated poly(1,2-glycerol ether)s bind to manganese oxide nanoparticles and impart greater water solubility and stability.63
Due to the living nature of the AROP of glycidyl ethers, diblock or triblock block copolymers with AB, ABC, or ABA structures can be readily accessed by sequential addition of different monomers or bi-directional growth from a central block.68–70 Furthermore, a variety of modifications such as hydrophobic / hydrophilic functionalization, grafting-from and hypergrafting using poly(1,2-glycerol ether)s as macroinitiators afford functionalized linear poly(1,2-glycerol ether)s. These topics are discussed at full length by Frey, and the reader is referred to this review for additional details.44
2.1.2. Linear Poly(1,2-glycerol carbonate)s
Although poly(1,2-glycerol ether)s show excellent biocompatibility,61,62,71 the degradation profiles may limit these polymers for some applications since ether linkages are generally non-degradable or degrade at relatively slow rates under physiological conditions. On the other hand, poly(1,2-glycerol carbonate)s, which possess carbonate backbone linkages instead of ether linkages, are readily biodegradable and biocompatible, providing an alternative and complementary polymer structure for investigation.
However, the synthesis of poly(1,2-glycerol carbonate)s is challenging. Traditionally polycarbonates are made either by ring-opening polymerization of cyclic carbonates or polycondensation of diols with phosgene. Both routes are not favored for accessing poly(1,2-glycerol carbonate)s, because unlike poly(1,3-carbonate)s (such as poly(trimethylene carbonate) (PTMC)) which are polymerized from six-membered cyclic carbonates, five-membered cyclic carbonates are remarkably thermodynamically stable72 and ring-opening polymerizations generally do not occur under mild conditions or proceed with extensive decarboxylation under vigorous condition.73–75 However, an exception is the polymerization of a trans-fused bicyclic structure to give trans poly(cyclohexane carbonate) analogous structures.76–78 Attempts to polycondense 1,2-diols with phosgene mainly produce five-membered cyclic carbonates instead of 1,2-linked poly(glycerol carbonate)s.34
Pioneered by Inoue,79 catalytic copolymerization of epoxide with CO2 is receiving increasingly more attention for both environmental and economical reasons, and a number of catalytic systems based on zinc, chromium, and cobalt have been described over the past 15 years.80–85 However, beyond the environmental and economical benefits, catalytic epoxide with CO2 copolymerization provides an exciting platform for synthesizing poly(1,2-carbonate)s that are otherwise not accessible by traditional methods from inexpensive and abundant materials. Recently, the groups of Grinstaff,38 Frey,39 and Luinstra86 successfully described the preparation of poly(1,2-glycerol carbonate)s using this approach. In all cases, poly(1,2-glycerol carbonate)s were synthesized by the copolymerization of benzyl or 2-nitrobenzyl protected glycidyl ether with CO2 using cobalt or zinc catalysts (Figure 4). While both cobalt and zinc catalysts are effective in the copolymerizations and produce polymers with high carbonate linkage selectivity (>99%) and high polymer selectivity over cyclic carbonate (>99%), the salen cobalt system exhibits greater activity (with turnover frequency ~150 h−1), producing polymers of higher molecular weights (as high as 45 k), as well as providing polymers of narrow molecular weight distributions (as low as ~1.1) due to the immortal nature of the polymerization. However, the drawbacks of the cobalt system are the toxicity of the cobalt metal, and the multi-step synthesis required to prepare the bifunctional cobalt complex.87 It is also worth noting that although stereoregularity usually imparts a significant influence on polymer properties, poly(benzyl 1,2-glycerol carbonate) is an amorphous material when it is atactic or isotactic.88
Figure 4.
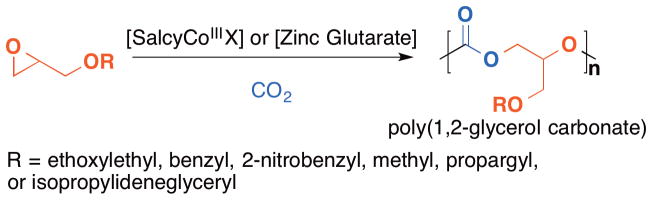
(Catalytic copolymerization of glycidyl ether with carbon dioxide using cobalt or zinc systems.)
After copolymerization, the obtained polymers are deprotected by either hydrogenolysis or photocleavage, depending on the protecting group, to afford poly(1,2-glycerol carbonate)s. One of most unique properties of poly(1,2-glycerol carbonate)s is its degradation.38,39 Poly(1,2-glycerol carbonate)s show a remarkably increased degradation rate as compared to poly(1,3-glycerol carbonate)s with a t1/2 ~ 2–3 days. The accelerated degradation rate is attributed to the intramolecular attack of the pendant primary hydroxyl group onto the carbonate linkage, leading to the formation of the thermally stable five-membered cyclic carbonate (Figure 5).
Figure 5.
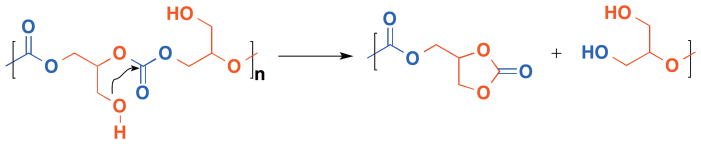
(Proposed mechanism of accelerated degradation of poly(1,2-glycerol carbonate).)
Several follow-up studies are recently described. For example, Frey et al. copolymerized the isopropylidene(glyceryl glycidyl ether) monomer with CO2 to afford a hydrophilic polyglycerol carbonate with two hydroxyl groups on each repeating unit after acidic deprotection.89 Likewise, terpolymers of benzyl glycidyl ether / methyl glycidyl ether / CO2,90 propargyl glycidyl ether / methyl glycidyl ether / CO2,91 and benzyl glycidyl ether / propylene oxide / CO292 are also accessed using cobalt or zinc systems.
However, catalytic epoxide / CO2 systems are particularly sensitive to trace amounts of water acting as a chain transfer agent, resulting in polymers with significantly lower molecular weights comparing to theoretical molecular weights.93 As a result of the chain transfer reaction, a bimodal distribution of molecular weights is observed in most cases,94 which translates to the formation of two different populations of polymers, one population with one initiator on one end and one hydroxyl group on the other end, while the other population is a telechelic species possessing two hydroxyl groups on both ends. This renders poly(1,2-glycerol carbonate)s relatively ill-defined as compared to poly(1,2-glycerol ether)s, and a synthetic challenge that calls for continued efforts.
2.2. Linear 1,3-Linked Glycerol Polymers
Linear 1,3-linked glycerol polymers feature a 1,3-linkage with a protected or free secondary hydroxyl group on the polymer backbone. Due to the significant knowledge on the ring-opening polymerization of six-membered cyclic carbonates and the known higher reactivity of two primary hydroxyl groups compared to the secondary hydroxyl group during polycondensation, linear poly(1,3-glycerol carbonate)s and polyesters synthesized from the polycondensation of glycerol with diacid, e.g., adipic acid, are the two main structural classes of 1,3-linked glycerol polymers synthesized, respectively.
In the early 2000’s, both the Zhuo and Grinstaff research groups described the syntheses of poly(1,3-glycerol carbonate)s via ring-opening polymerization reactions. Zhuo et al. published the first synthesis of poly(1,3-glycerol carbonate)s in 2002.40 The monomer, 5-benzyloxy-trimethylene carbonate (5-BTC), is synthesized from glycerol in four steps and can be polymerized using either Sn(Oct)2 or Al(Oi-Pr)3 as the catalyst. The resultant polymers exhibited molecular weight ranging from 8k to 24k g/mol with molecular weight distribution ranging from 1.49 to 1.96 (Figure 6). As compared to the known and structurally analogous biocompatible PTMC, the DSC results showed poly(benzyl 1,3-glycerol carbonate)s to be soft rubbery materials possessing a glass transition temperature of 0 °C, which is 18 °C higher than PTMC. Follow-up metal-free polymerizations are also reported. For example, both silica gel-immobilized lipase-catalyzed and organocatalytic approaches are reported to polymerize 5-BTC by Zhuo and Guillaume,95,96 respectively. These approaches represent elegant alternatives to the metal-catalyzed routes.
Figure 6.
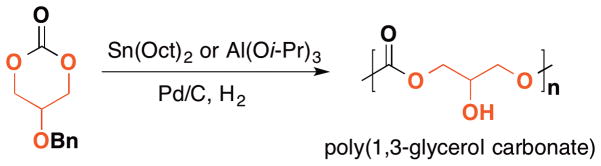
(Ring-opening polymerization of 5-BTC using metal alkoxide followed by catalytic dehydrogenation to afford poly(1,3-glycerol carbonate)s.)
In 2003, Grinstaff et al. reported the synthesis of a copolymer of glycerol and lactic acid,37 which expanded upon an earlier conference proceeding report in 2001 on the synthesis of a polycarbonate of glycerol.97 The resulting polycarbonates and poly(carbonate-ester)s exhibited molecular weights from 11k to 30k g/mol with molecular weight distributions ranging from 1.21 to 1.76. The glass transition temperatures of the poly(carbonate-ester)s are dependent on lactic acid content and vary from 18 °C with 25% lactic acid to 53 °C with 90% lactic acid content. Building off these results, Grinstaff et al. copolymerize 5-BTC with ε-caprolactone or lactide to obtain a copolymer with improved thermal and mechanical properties.41,98 Moreover, after deprotection, the free secondary hydroxyl group is available for functionalization to introduce specific nucleophiles or fluorescence dyes (Figure 7). These hydroxyl-functionalized polymers can be cast into films or electospun to form meshes and used as drug delivery vehicles.99–103
Figure 7.

(Functionalization of the secondary hydroxyl group of poly(ε-caprolactone-co-1,3-glycerol carbonate).)
Another class of 1,3-linked glycerol polymers is polyesters from glycerol and a diacid prepared via a conventional condensation reaction. Diacids including sebacic acid and adipic acid (as well as other diacids with different chain lengths) can be copolymerized with glycerol to yield poly(glycerol-co-sebacic acid)104 and poly(glycerol-co-adipic acid).105,106 For example, traditional synthetic biodegradable polymers (poly(lactic acid), polyhydroxyalkanoates, etc.) are often limited by inferior mechanical properties. The Langer group reported a tough biodegradable and biocompatible elastomer synthesized by polycondensation of 1:1 molar ratio of glycerol and sebacic acid (Figure 8).104 It should be noted that a 1:1 molar ratio is critical for maintaining a small number of crosslinks, and a cross-linked thermoset polymer is obtained with a 2:3 molar ratio. This slightly cross-linked elastomeric glycerol polymer is biocompatible as determined by in vitro MTT assay and in vivo subcutaneous implantation in rats. The study also demonstrated that subcutaneous implants were completely absorbed after 60 days with the restoration of normal tissue architecture.
Figure 8.
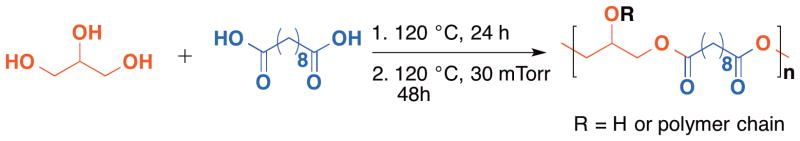
(Polycondensation of glycerol with sebacic acid to afford lightly cross-linked poly(glycerol-co-sebacic acid).)
While glycerol-diacid-based polyesters are synthesized by well-established thermal condensation polymerization, enzyme-catalyzed condensation polymerization reactions are also described by Gross et al. For example, using immobilized lipase B from Candida Antarctica, copolymers of glycerol, octanediol, and adipic acid,107,108 glycerol and oleic diacid,109 and glycerol, linoleic diacid and oleic diacid110 can be successfully synthesized and subsequently characterized (e.g., physical, thermal, structural properties). Compared to conventional thermal condensation polymerizations, which usually involve energy intensive processess (high heat and high vacuum) and a metal catalyst, lipase-catalyzed polymerization represents a mild and environmental benign approach that provides better control over branching while avoiding crosslinking reactions.110 Such crosslinking reactions are common during the polycondensation of a polyol (e.g., glycerol) with a diacid, as exemplified by the synthesis of a slightly crosslinked poly(glycerol-co-sebacic acid) by Langer et al.104
As compared to well-known biocompatible polymers such as poly(lactic acid) or poly(glycolic acid), glycerol-based poly(1,3-carbonate)s and polyesters as well as their copolymers possess the additional benefits of an easily modifiable structure, nonacidic (or a reduced number of acidic) products upon biodegradation, biocompatibility, and controllable degradation rates. These properties are advantageous, and, thus, these polymers are being evaluated for biomedical applications such as nanoparticles for drug delivery105,106 and drug buttressing films for prevention of tumor recurrence after surgical resection.99–102
2.3. Dendritic and Hyperbranched Glycerol Polymers
Dendritic and hyperbranched glycerol polymers constitute an important class of glycerol polymers. Haag et al. reported the first synthesis of dendritic glycerol polymers: dendritic poly(1,2-glycerol ether)s.36 Starting from trimethylolpropane core, a simple and efficient two-step approach involving allylation of hydroxyl group and dihydroxylation of the double bond afforded access to the globular dendritic poly(1,2-glycerol ether) structure (Figure 9). For example, a generation 3 ([G3]) dendrimer with 24 peripheral end hydroxyl groups is obtained in 75% yield after three cycles of allylation-dihydroxylation. It is also worth noting that column chromatography purification is only necessary after the allylation steps, which is important for large-scale synthesis. In a series of follow-up studies, Haag111,112 and Zimmerman113 also used 2-amino-1,3-propane, triglycerol, and TBDPS-protected allyl alcohol to synthesize amine and azido-ended polyglycerol dendrons which can be further coupled to 4,4′-biphenyldicarboxylic acid core to form a dendritic triblock amphiphile or to a porphyrin core to form a polyglycerol dendron coated porphyrin.
Figure 9.

(First synthesis of a globular [G3] polyglycerol dendrimer.)
In contrast to the globular dendritic pure poly(1,2-glycerol ether) structure, Grinstaff et al. synthesized a new type of glycerol dendrimer - poly(glycerol-co-lactic acid),114 poly(glycerol-co-succinic acid),115 and poly(glycerol-co-adipic acid)116 dendrimers of generation zero to four. In these dendrimers, the structures are intricately designed in such a way that the C2 position of glycerol serves as the branching point while lactic acid or succinic acid serve both as the core and the linker between the branching points. These key constituents in the dendrimers are combined in reiterative divergent reactions steps leading to the dendritic structures in good to high yields (Figure 10). A convergent route to poly(glycerol-co-succinic acid) dendrons is also described that utilizes two orthogonal protecting groups; namely, the benzylidene acetal for the protection of the 1,3-diols of glycerol and the tert-butyldiphenyl (TBDPS) for protection of the carboxylic acid of succinic acid.117 This methodology can be extended to the synthesis of dumb-bell dendrimer structures where the dendritic glycerol-succinic acid units are attached to a linear polyetheylene glycol of 3400, 5000, or 20,000 g/mol molecular weight.118 These glycerol based dendrimers, with architectures ranging from globular to dumb-bell, are termed biodendrimers, where the biocompatible building blocks are incorporated into the dendritic structure.119
Figure 10.

(Synthesis of a poly(glycerol-co-lactic acid) dendrimer.)
While the synthesis of dendrimers requires tedious, repetitive protection and deprotection steps, structurally similar hyperbranched glycerol polymers possess comparable properties and can be conveniently synthesized in a one-pot manner.120 Although some monomers used for synthesizing hyperbranched polymers require a few steps to synthesize, this methodology still possesses advantages such as higher yields, reliable scalability as well as the readiness of running the polymerization as compared to the convergent or divergent synthesis of dendrimers, which is critical for their use by non-experts and large scale production. Hyperbranched glycerol polymer are synthesized using glycidol,121 glycerol carbonate,122 6-hydroxymethyl-1,4-dioxan-2-one (6-HDON),123 5-hydroxymethyl-1,4-dioxan-2-one (5-HDON)120 and 5-(3-[(2-hydroxyethyl)thio]propoxy)-1,3-dioxan-2-one (HTPDO)124 (Figure 11). All of these structures represent a latent cyclic AB2 type monomer and can be polymerized to form hyperbranched glycerol-based polyether and poly(ether-ester) by ring-opening multi-branching polymerization (ROMBP). Compared to direct polycondensation of ABm type monomers, ROMBP gives polymers of more well-defined structures and narrow molecular weight distributions. For example, in cases of glycidol and glycerol carbonate, 1,1,1-tris(hydroxymethyl)propane (TMP) is used as the initiator and opens up a glycidol or glycerol carbonate with simultaneous decarboxylation. This process frees the latent hydroxyl group and leads to the exponential increase of both branching points and end groups affording the hyperbranched poly(glycerol ether) structure. Slow addition of the monomer gives polymers with a polydispersity index as low as 1.1–1.6, which is exceptionally low for hyperbranched polymers. Hyperbranched poly(glycerol ether)s are being extensively studied and the reader is referred to insightful reviews by both Frey43 and Haag42,125 for additional information and data.
Figure 11.
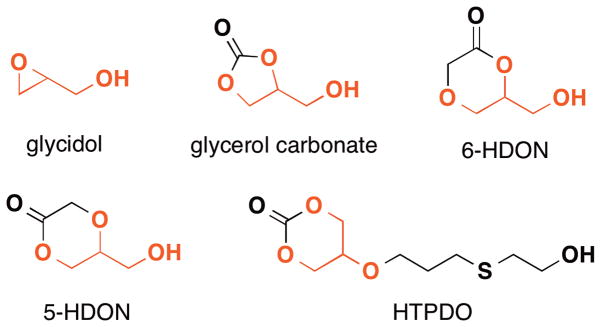
(Various monomers reported for synthesizing glycerol based hyperbranched polymers.)
An elegant extension of hyperbranched poly(glycerol ether)s is the preparation of micro / nanogels, described by Haag et al.126–132 These micro / nanogels span a range of 20 nm – 350 nm and merge the gap between hyperbranched polymers (<10 nm) and macroscopic or bulk hydrogels (>1000 nm).125 Two main approaches are employed to synthesize those micro/ nanogels. The first approach is termed “megamer” and combines partially alkyne or azide-functionalized low molecular weight hyperbranched polyglycerols under click condition to form a giant “megamer” (Figure 12A).130,131 The second approach, namely, acid-catalyzed polyaddition, utilizes the polyaddition of polyol-polyepoxide monomers to form hyperbranched yet cross-linked network under acid catalysis (Figure 12B).126–128 While the “megamer” approach is general, controllable, and conducted under mild conditions, the acid catalyzed polyaddition approach possesses the advantage of being efficient and affords a structure solely derived from ether bonds without the need for further chemical cross-linking.125 Both strategies involve creating a confined micro / nano sphere in a continuous phase which is accomplished by inverse miniemulsion or inverse nanoprecipitation with or without the assistance of surfactant. Being hyperbranched and cross-linked in nature, these micro / nanogels combine the characteristics from both hyperbranched polyglycerol and cross-linked hydrogel such as low viscosity, high solubility, and limited swelling ratio. Moreover, environmental responsive modules such as pH or redox sensitive moieties are being successfully engineered into these micro / nanogels,130–132 and therefore, these compositions are of interest for biomedical applications.
Figure 12.

(Schematic presentation of (A) “Megamer” approach and (B) Acid-catalyzed polyaddition approach).
While glycidol and glycerol carbonate polymerize under anionic or cationic conditions to afford hyperbranched poly(glycerol ether)s in one step, a more intricately designed hyperbranched glycerol system is reported by Zhuo123 and Parzuchowski120. In both reports, two biocompatible components, i.e. glycerol and glycolic acid, are incorporated into one monomer – 5-HDON or 6-HDON. A self-condensing ring-opening polymerization affords hyperbranched poly(glycerol-co-glycolic acid) with a degree of branching of 0.4 and 0.5 for 6-HDON and 5-HDON, respectively (Figure 13).
Figure 13.

(Ring-opening multibranching polymerization of 6-hydroxymethyl-1,4-dioxan-2-one (6-HDON) to afford glycerol-glycolic acid based hyperbranched polymer.)
Compared to linear polymers, these dendritic and hyperbranched polymers possess unique characteristics such as high surface area-to-volume ratio, numerous end groups for functionalization, and nearly monodisperse structures with well-defined interior and exterior regions. These characteristics translate to unique properties such as low viscosity, high solubility, capability for small molecule encapsulation, and adhesiveness.7,133 When coupled with the additional benefit of being built from building blocks that are known to be biocompatible and nontoxic, these structures are of interest for biomedical applications.
2.4. Glycerol Derivative Polymers
Polymers derived from 1,3-dihydroxyacteone represent another important class of glycerol polymers. Dihydroxyacetone is one of the downstream metabolites of glycerol and also an intermediate metabolite in glycolysis. It is readily available (<0.4 $/gram on SigmaAldrich) as a cyclic dimer or by convenient selective catalytic oxidation of glycerol. Putnam et al. reported the first 1,3-dihydroxyacetone-based polymer in 2005.134 In their report, 1,3-dihydroxyacetone is locked in its dimer form by acetal protection. The resultant locked cyclic dimer is polycondensed with triphosgene to afford poly(carbonate-acetal)s (Figure 14A). Due to the cyclic structure on the polymer backbone, the resultant 1,3-dihydroxyacetone-based poly(carbonate-acetal)s possess glass transition temperatures >40 °C and thermal decomposition temperatures >300 °C, which is higher than those typically associated with aliphatic polycarbonates. Subsequently, Putnam,16,135 Waymouth,136 and Guilaume137 reported the ring-opening polymerization of dimethylketal dihydroxyacetonecarbonate using either Sn(Oct)2-catalyzed or organo-catalytic ring-opening polymerization to afford poly(dihydroxyacetone carbonate)s (Figure 14B) and copolymers with ethylene glycol,135 polylactic acid,138 and glycerol.139 After (co)polymerization, the dimethylketal is removed by either trifluoroacetic acid or trityl tetrafluoroborate with water to give the desired polymer. Poly(dihydroxyacetone carbonate) is a brittle semicrystalline polymer with a melting temperature (Tm) of 245 °C, which is close to the Tm of the structurally analogous commercial copolymer from ethylene and carbon monoxide (Carilon™) (257 °C).136 Interestingly, although poly(dihydroxyacetone carbonate) is insoluble in water and most organic solvents, when cast on a surface it appears hydrophilic as determined by contact angle measurement,16 presumably due to the dipole moment of the carbonyl group on the polymer backbone. This carbonyl group is critical for some applications because of its ability to react with the amine residues on a tissue surface to give a polymer coating.140
Figure 14.

(Synthesis of poly(dihydroxyacetone carbonate) by ring-opening polymerization of a six-membered cyclic carbonate derived from dihydroxyacetone.)
3. Biomedical Applications
With efficient synthetic routes reported, diverse glycerol polymer architectures available, tunable physical and mechanical properties, biodegradability, and biocompatibility, glycerol polymers are actively investigated for a number of biomedical applications, and several representative examples are described below.
3.1. Drug Delivery
A number of reports describe glycerol polymers as drug delivery vehicles for prolonged release. For example, Murphy and Shabir et al. reported a microsphere drug delivery system based on poly(glycerol adipate-co-ω-pentadecalactone).106 The polymer microparticles are produced by spray drying from a double emulsion with L-leucine as a dispersibility enhancer. Incorporation of L-leucine significantly reduces the burst release (24.04±3.87%) of a doped model hydrophilic drug (e.g., sodium fluorescein) compared to unmodified formulation (41.87±2.46%). Another glycerol polymer-based drug delivery system is reported by Garnett et al in 2005.105 They prepared a library of linear poly(glycerol-co-adipate) via a condensation reaction, and these polymers possess a pendant secondary hydroxyl group on each repeating glycerol unit. The hydroxyl groups are further functionalized with different degrees of fatty acid (C8 or C18) substitution. The fatty acid-functionalized polymers self-assembled into nanoparticles of ~200 nm in diameter, depending on the degree of fatty acid substitution and the length the fatty acid chain. The release profile of a hydrophilic radiolabelled 125I-Deoxy-uridine, incorporated in the nanoparticle, is delayed without an initial burst release even in serum, which is in contrast to the drug release profiles observed with poly(lactic-co-glycolic acid) (PLGA) or poly(lactic acid-co-ethylene glycol) (PLA-PEG) systems.141,142
Dendritic glycerol-based polymers are also reported for delivery of therapeutic agents. For example, Grinstaff et al. discovered that a dendritic [G4] poly(glycerol-co-succinic acid) forms supramolecular assemblies with hydrophobic compounds in aqueous solution, and can be used for the delivery of hydrophobic chemotherapeutic such as 10-hydroxycamptothecin (10-HCPT). Enhanced in vitro anticancer activity is observed as a consequence of increased drug solubility, cell uptake, and cellular retention.143–145 In a recent report, Haag et al. described a dendritic polyglycerol-doxorubicin conjugate possessing an antibody for targeting, a fluorescence tag for labeling, and a PEG for solublization. The conjugates demonstrated specific targeting to cancer cell lines expressing epidermal growth factor receptor (A431, MDA-MB-468, Panc-1) with increased tolerability and enhanced cytotoxic activity.146
The standard of care for patients with early stage non-small-cell lung cancer (NSCLC) is surgical resection. This is a delicate and complicated procedure as the surgeon must remove as much of the cancerous tissue as possible while sparing enough lung tissue for normal function and reduced patient morbidity. To address this unmet clinical need, Grinstaff et al. evaluated chemotherapeutic-loaded polymer films, that can be stapled into the resection margin during surgery, for the prevention of locoregional recurrence.99–101 Building upon their earlier work on the copolymerization of 5-BTC and ε-caprolactone using Sn(Oct)2 as the catalyst followed by hydrogenolysis, they coupled various fatty acids of chain lengths C8, C12, C14, C16, and C18 to the copolymer in order to impart different degrees of hydrophobicity, crystallinity, film forming capacity, and drug release rates.102 Drug-loaded films cast from solutions of polymer and the anticancer agent, 10-hydroxycamptothecin, exhibited prolonged release from four to over seven weeks with the rate of release being fastest for the C12 and slowest for the C18 analog. The 10-hydroxycamptothecin loaded poly(glycerol monostearate-co-caprolactone) films were cytotoxic to Lewis lung carcinoma cells in vitro, and prevented the local growth and establishment of Lewis lung carcinoma tumors in vivo. In contrast, animals treated with a larger intravenous dose of 10-hydroxycamptothecin or unloaded films rapidly developed local tumors. Next, Grinstaff et al. encapsulated paclitaxel (a first line chemothereauptic used in later staged cancer patients) within the poly(glycerol monostearate-co-caprolactone) and evaluated the performance of the films in a lung cancer surgical resection model (Figure 15). The paclitaxel-loaded poly(glycerol monostearate-co-caprolactone) films exhibited prolonged tumor cytotoxicity in vitro against LLC, NCI-H460, and NCI-H292 cells for 50+ days, and prevented locoregional recurrence of tumor in vivo.100 Specifically, an overall freedom from locoregional recurrence of 83% is observed in the paclitaxel-loaded film treatment group, while the freedom from recurrence in mice treated with unloaded films or paclitaxel administered intraperitoneally or locally at the surgical site is significantly lower at 12, 22, 0%, respectively. This drug delivery concept can be translated to other cancers where surgical resection is performed, and similar results are described in a nude murine model of human sarcoma tumors.99
Figure 15.
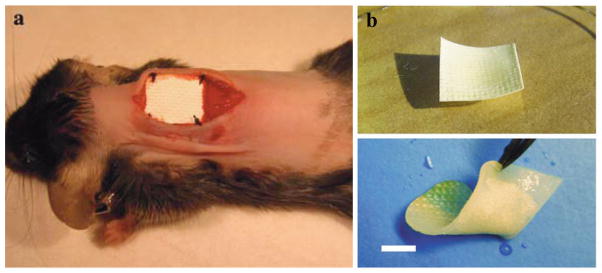
((a) Implantation of PGC-C18 film on a collagen scaffold. (b) PGC-C18 film on surgical collagen scaffold. The upper panel shows a dry film; the lower panel shows flexibility of a wet film. Scale bar, 5 mm.)
Reproduced with permission.[100] Copyright © 2010, Weily.
Given the above dependence of drug release rates on the choice of fatty acid-functionalized to the poly(glycerol-co-caprolactone) backbone, Grinstaff et al. explored a more intricately designed drug delivery system to take advantage of the superhydrophobic effect by fabricating drug loaded nano-fiber meshes.103 In particular, poly(glycerol monostearate-co-caprolactone) was blended with polycaprolactone, the model camptothecin drug, SN-38, and electrospun to give a non-woven polymeric mesh (Figure 17). This resultant mesh exhibits a high apparent contact angle, which arises from the hydrophobicity of C18 chain, the rough surface of the polymeric nanofibers, and the 3D porous network. The release of SN-38 showed a striking dependence on the apparent contact angle of the mesh, with higher apparent contact angles slowing drug release relatively to lower apparent contact angles. Release of drug from the meshes is controlled by mesh wetting, which occurs as the air is slowly displaced from the mesh. In fact, wetting the mesh (with an ethanol treatment) or displacing the entrapped air using an ultrasound treatment accelerated drug release.103 The entrapped air layer within the superhydrophobic meshes is stable even in the presence of serum, and drug-loaded meshes are efficacious against several cancer cells in vitro for >60 days, thus demonstrating their applicability for long-term drug delivery. Importantly, this work highlights a new concept for drug delivery, that is, using air as a barrier component to control the rate at which drug is released.
Figure 17.

Schematic representation of the nanoprecipitation process. (A) Injection of aqueous solution of alkyne functionalized polyglycerol macromonomers (red spheres), azide functionalized polyglycerol macromonomers (blue spheres) and the proteins asparaginase, IgG, Lysozyme and BSA (3D structure of α-helixes and β-turns). (B) Particle templation by diffusion of aqueous phase (blue arrow) into acetone phase (violet). (C) Particle gelation by azide alkyne 2+3 cycloaddition.
Reproduced with permission.[131] Copyright © 2013, Elsevier.
Besides linear glycerol polymers, polyglycerol micro / nanogels are also being investigated for drug delivery applications. In particular, environment responsive modules are engineered into the micro / nanogel structures to control the release of therapeutic agents by an external stimuli. An excellent example is presented by Haag et al. who develop a pH responsive nanogel system based on acid sensitive benzylidene acetal moiety.131 The nanogel is synthesized by inverse precipitation of two low molecular hyperbranched polyglycerols with alkyne and azide moieties, respectively (Figure 17). The benzylidene acetal protected polyglycerol nanogel is stable under physiological pH while rapidly hydrolyzes at acidic pH, leading to the disintegration of the nanogel and release of the incorporated biomacromolecule, such as Asparaginase. The enzyme is encapsulated with 100% efficiency, and the structural integrity and enzyme activity is fully retained after release. A similar dual responsive system, based on the incorporation of both a pH sensitive hydrazone and redox responsive disulfide bond, is also reported.132
3.2. Ophthalmic Sealant for Corneal Wounds
Dendritic polyglycerol are also used as ophthalmic adhesives for repairing corneal lacerations, securing penetrating keratoplasties, and sealing laser in situ keratoplasties flaps.118,147–149 As a result of the globular branched structure of dendrimers, which translates to low intrinsic viscosity and highly reactive surface, dendrimers are particularly suitable for preparing highly crosslinked hydrogels. Grinstaff et al. first synthesized a series of ABA triblock dendritic polymers (dumb-bell or bola-type architecture) of differing generation (G0, 1, 2, and 3) consisting of two methacrylate end-capped poly(glycerol-co-succinic acid) dendrons on the two chain ends of a nonimmunogenic poly(ethylene glycol) (PEG) (([Gn]-polyglycerol-succinic acid-methacrylate)2-PEG (([Gn]-PGLSA-MA)2-PEG)), Figure 18).118 Application of these photocrosslinkable hybrid linear-dendritic copolymers to a 4.1 mm linear laceration followed by photolysis in the presence of a photoinitiator results in closure of the wound (Figure 18). The G1 formulation performed the best, and withstood a pressure of about 171 mm Hg, well in excess of normal the intraocular pressure (15 and 20 mmHg). Next, they synthesized a series of first generation (G1) dendritic polymers, ([G1]-polyglycerol-succinic acid-methacrylate)2-PEG) with three different central PEG block lengths (i.e., molecular weight = 3400, 10000, and 20000). As before, aqueous solutions of these polymers when applied to an ex vivo 4.1 mm cornea laceration, in an enucleated porcine eye, sealed the wound upon in situ polymerization with light. For example, the hydrogel sealant formed from the 20 wt% solution of ([G1]-PGLSA-MA)2-PEG10,000 maintained a leak proof seal until > 300 mm Hg. Besides providing a leaking pressure well above the normal intraocular pressure, the formed hydrogel also acts as a barrier for post-operational microbial infection. Based on these encouraging ex vivo results, they conducted an in vivo study.149 Full-thickness 4.1-mm lacerations were made in the right eyes of 60 white leghorn chickens, and half of the wounds were treated with the ([G1]-PGLSA-MA)2-PEG3,400 sealant and half were closed with three interrupted 10-0 nylon sutures (Figure 18). All the wounds are sealed by postoperative day one with no leaks observed. Although scarring is more prominent at day 7 in sealant treated corneas; by day 28, the sutured corneas exhibited more inflammation, scarring, and a more irregular anterior corneal surface. Clinically, all sealant treated corneas remained clear while nearly all sutured corneas displayed some degree of corneal scarring persisting through day 28. This hybrid linear-dendritic sealant represents a safe, effective, and technically easier alternative to traditional suture repair of corneal wounds.
Figure 18.

(Top: Chemical structure of the photo cross-linkable biodendrimer. Middle: Photographs of a secured autograft in an enucleated porcine eye after placement of (A) 8 or (B) 16 interrupted 10-0 nylon sutures followed by application and photo cross-linking of the hydrogel adhesive. Bottom: Corneal laceration repair in an in vivo chicken model, postoperative day 5 cornea. (C) Adhesive, (D) Sutured.)
Reproduced with permission.[118] Copyright © 2007, Investigative Ophthalmology & Visual Science and ARVO.
3.3. Anti-bacterial and Anti-inflammatory Agent
e the various known agents that show anti-bacterial activity, amphiphiles are an important class of anti-bacterial agents, which act through the perturbation and disruption of the prokaryotic membrane. Grinstaff et al. reported an anti-bacterial agent based on an amphiphilic dendritic poly(glycerol-co-succinic acid).150 The dendritic glycerol polymers are designed in such a way that they contain one hydrophilic dendritic terminal end group with four sodium carboxylate and one hydrophobic terminal with either one (1) or two (2) fatty acid chains connecting to the core of the dendrimer (Figure 19). Dendrimer 1, sodium dodecyl sulfate (3), and Triton X-100 (4) are cytotoxic to both prokaryotes and eukaryotes at similar concentrations with the compounds exhibiting a ratio of eukaryotic to prokaryotic EC50 less than a factor of 3.8, which is not ideal for an antibacterial compound. Dendrimer 2, however, exhibits a ≥36-fold eukaryotic to prokaryotic EC50 ratio. These results are of interest, as few reports describe linear polyanionic polymers with anti-bacterial activity.151,152 Importantly, this work showed that glycerol polymers, with their excellent built-in biocompatibility, can not only be used as structural and functional materials, but also the polymer entity itself with specific structural motif can be used as an agent that exhibits therapeutic efficacy.
Figure 19.

(1 and 2: bola-shaped [G1] dendrimers investigated for anti-bacterial activity; 3: sodium dodecyl sulfate; 4: non-ionic surfactant Triton X-100.)
Another exciting example in this category is the anti-inflammatory dendritic compound - polyglycerol sulfate (dPGS). Recruitment of the leukocytes to the inflammation site is a critical step of the development of various acute or chronic inflammatory diseases, and is mediated by the interaction between selectin and carbohydrate ligand. Therefore, specific inhibitors of selectin may provide a potent treatment of inflammatory disease. Haag et al. designed a sulfated dendritic polyglycerol which features a dendritic polyether core with sulfated hydroxyl groups on the surface (Figure 20).153 dPGS exhibits high binding affinity to L-selectin and P-selectin. Interestingly, the binding affinity data show both dependence on the core size and the degree of sulfation: increasing the core size from 240 Da to 2500 Da and 6000 Da results in increasing binding affinity from 2 mM, 90 nM, to 8 nM, respectively. Surprisingly, however, for the dendrimer with 6000 Da core size, the degree of sulfation shows an inverse relationship with binding affinity, indicating that ligand spacing also plays a critical role in binding affinity.
Figure 20.

(Chemical structure of dendritic polyglycerol sulfate.)
3.4. Coatings for Prevention of Seroma
While glycerol polymers are used as structural or functional materials in drug delivery and ophthalmic surgery, as well as chemical entities with specific motifs that exhibit therapeutic efficacy itself, the glycerol polymer discussed in this example captures both of these features. Seroma formation is a common postoperative complication particularly following ablative and reconstructive surgeries, where the fluid collects in the void space remaining after extensive tissue removal.140,154,155 Putnam et al. reported a biocompatible and biodegradable poly(ethylene glycol-co-dihydroxyacetone carbonate) (MPEG-pDHA) that is effective for prevention of seroma. They designed and synthesized MPEG-pDHA with varying pDHA block molecular weights (5000-3000, 5000-5000, 5000–7000 g/mol) (Figure 21).140 MPEG-pDHA forms a physically cross-linked hydrogel upon hydration and shows thixotropic behavior, which allows remote delivery of the hydrogel through extrusion from a needle (Figure 21). Importantly, MPEG-pDHA 5000-3000 g/mol treated rats display a significant decrease in seroma volume while surprisingly, both 5000-5000 g/mol and 5000–7000 g/mol show no statistical difference from untreated group. Addition of lysine, into the hydrogel which competitively reacts with the carbonyl on the polymer backbone, rendered the polymer ineffective suggesting that formation of the Schiff-base may be a critical step for the observed performance. MPEG-pDHA serves as the structural material that fills in the space and the carbonyl groups on the MPEG-pDHA react with amine on the tissue surface to provide an optimal outcome.
Figure 21.

(Top: Synthesis of poly(ethylene glycol-block-dihydroxyacetone carbonate); Bottom: (A) Viscosity readings show the MPEG-pDHA hydrogel thixotropic behavior (1% strain, 28 °C). (B) An example of MPEG-pDHA 5,000–5,000 extruded from a 26-gauge needle.)
Reproduced with permission.[140] Copyright © 2010, National Academy of Sciences.
Besides the above-mentioned applications, glycerol and glycerol derivative polymers are also investigated in other applications such as a biorepellent coating for surfaces,61,71,156 an alternative to heparin as an injectable anticoagulant,157 and as a vehicle for gene delivery,158–160 which are comprehensively summarized in a review by Haag.42 All these applications add to the diversity of the biomedical applications that glycerol polymers can be engineered into, provide insights into the structure-property relationship of glycerol polymers, and further justify continued efforts in this area.
4. Medical Polymers Market
Medical polymers such as biodegradable polymers (e.g. poly(lactic acid) (PLA) and poly(glycolic acid) (PGA)), polymethylmethacrylate, polypropylene, polyvinyl chloride, nylons, polyurethanes, and poly(tetrafluoroethylene) (Teflon) are used in a variety of medical device products. These products include wound dressings, catheters, vascular grafts, implantable defibrillator, blood filters, contact lenses, automated analyzers, diagnostic and imaging devices, surgical screws, nails and plates, etc. The global biomaterials market is projected to increase at a CAGR (compound annual growth rate) of 14% (2007–2017) according to Industry Experts.161 Consequently, medical polymers represent a major commercial market, and this market is growing and estimated to exceed $3.5 billion by 2018.162 Polymeric biomaterials used in medical applications are driven by the clinical and market needs. Once the design requirements for performance are identified, the task of manufacturing, pre-clinical testing, and regulatory approval must be completed. Glycerol and glycerol derivative polymers, with their builtin biocompatibility, possesses benefits such as: 1) a variety of accessible architectures and compositions to afford linear, hyperbranched, dendritic polyether or polycarbonate, as well as copolymers with hydroxyacids; 2) an easily modifiable structure for introducing specific moieties or functional groups or preparation of block polymers via backbone and chain end conjugation. These benefits translate to efficient routes for the synthesis of specific polymers and to unique properties including, but not limited to, degradation, gel-forming ability, and thixotropic behavior. In many ways, glycerol polymers are the next generation biodegradable polymers like the well-known PLA, first discovered in the 1960’s, but with all of its advantages and more. Coupled with the low cost, ample supply, bioavailability and renewability of glycerol, we envision that glycerol and glycerol polymers represent a major opportunity for replacing or complementing the current polymers used in the biomedical market, and making a difference in patient care.
5. Summary and Future Perspectives
Glycerol polymers are actively investigated and these efforts will continue, given the successes that are reported to date. Various polymer architectures from linear to dendritic are reported for pure polyglycerol ethers and carbonates as well as copolymers with hydroxyacids, for example, to give polyether esters or polycarbonate esters. In many ways, these polymers provide users the capabilities of well-known polymers like PLA with the additional benefits of an easily modifiable structure and nonacidic products upon biodegradation. Interdisciplinary, collaborative, and translational efforts are affording a number of successful applications of glycerol polymers in the biomedical field as either structural or functional materials or both.
This area is still in its infancy and there is a wealth of opportunities for basic research. Designing and synthesizing polymers of novel architectures and compositions with desirable (i.e., controllable) properties still remains a challenge. The synthesis of poly(1,2-glycerol carbonate)s using the cobalt or zinc-catalyzed copolymerization of epoxides with CO2 provides a recent example where robust and efficient polymerization methods afforded access to structures that are not otherwise attainable by conventional methods. Continued development of new polymerization methods involving monomers and catalysts will provide novel polymer compositions and architectures, which will further translate to unique properties. The structure of glycerol with its three hydroxyl functionalities enables facile and selective functional group manipulation to afford a range of different monomers for subsequent polymerization. However, there is also a demand and an emerging trend to focus our attention towards other natural metabolite building blocks for the synthesis of glycerol-based polymers, such as the upstream or downstream metabolites of glycerol. The polycarbonates based on 1,3-dihydroxyacetone represent an excellent example that illustrates this trend, and the success of this approach. While developing and utilizing novel robust polymerization methods to access new polymer architectures and compositions, as well as investigating into novel biocompatible building blocks remain critical for the development of this area, the further evaluation and continued use of known glycerol polymers is key to growth in this area. There are opportunities for chemists and engineers to use existing polymers, polymerization methods, or fabrication processes to make tailored-made materials with a specific property(ies).
With regards to identifying biomedical applications appropriate for glycerol-based polymers, this is and remains a challenge. We find that discussing with oncologists, surgeons, radiologists, and pathologists about their work, and asking them questions about the current standard of care, is a good first step. Communication is key and by learning their language and understanding their challenges we will gain insight into the needs of patients, and, hopefully, motivation for a potential new project in our laboratories. We also need to be aware of the advances occurring in medicine, for example, understanding the underlying cause(s) of a disease or new imaging modalities being developed. To encourage these interactions, my students and I meet more than once a month with a MD or MD / PhD to discuss their day-to-day activities and what they would like to do better in the clinic. This is one example way to foster interactions between individuals, but other approaches are equally as good. Through concerted and collaborative efforts, glycerol-based polymers will continue to play a critical role in the chemical and engineering disciplines, and find uses in medicine.
This feature article is intended to briefly summarize the advances in the glycerol polymer field, stimulate critical discussions, and pique scientific (and clinical) interest. It is not a comprehensive review, and we apologize upfront for not referencing all who have made important contributions to this area. It is an invitation to the community to examine this exciting area of polyglycerols broadly defined.
Figure 16.

(Top: (A) PCL was used as the base polymer for fabrication of electrospun meshes and melted electrospun meshes. (B) PGC-C18 was used as the hydrophobic dopant in electrospun PCL meshes to decrease their wettability. (C) Electrospun PCL mesh with an average fiber size of 7.7 ± 1.2 μm. (D) 10% PGC-C18-doped electrospun PCL mesh with an average fiber size of 7.2 ± 1.4 μm. (E) A melted PCL mesh. (F) A melted 10% PGC-C18-doped electrospun PCL mesh. Bottom: Graphic illustration of the principle of using entrapped air layer as barrier to control drug release rate.)
Reproduced with permission.[103] Copyright © 2012, American Chemical Society.
Acknowledgments
The authors would like to thank BU, the Nanomedicine Program and Cross-Disciplinary Training in Nanotechnology for Cancer (NIH R25 CA153955), BU Nanotheranostics ARC, National Institute of Health R01CA149561 and R01EY13881, and the National Science Foundation DMR-1410450 for support. MWG also thanks his past and current students and postdoctoral fellows for their hard work and dedication to synthesizing and evaluating new glycerol polymers.
Biographies

Mr. Heng Zhang is a Ph.D. candidate in the Department of Chemistry at Boston University. He received his B.S. in Pharmaceutics from Nanjing University of Technology in 2009 and M.S. in Pharmacy from University of Iowa in 2011. Since joining Prof. Grinstaff’s group, his research interests have focused on the design and synthesis of novel biocompatible and biodegradable polymeric materials for biomedical applications, as well as organometallic chemistry and polymerization methods. He recently reported the synthesis of poly(1,2-glycerol carbonate)s via the copolymerization of benzyl protected glycidyl ether with CO2 using a cobalt salen complex (H. Zhang, M. W. Grinstaff J. Am. Chem. Soc., 2013, 135, 6806).

Mark W. Grinstaff is the Distinguished Professor of Translational Research and Professor of Biomedical Engineering, Chemistry, and Materials Science and Engineering, at Boston University. He is also the Director of the NIH T32 Program in Translational Research in Biomaterials. Mark’s awards include the ACS Nobel Laureate Signature Award, NSF Career Award, Pew Scholar in the Biomedical Sciences, Camille Dreyfus Teacher-Scholar, Alfred P. Sloan Research Fellowship, the Edward M. Kennedy Award for Health Care Innovation, and a Fellow of the National Academy of Inventors. He has published more than 200 peer-reviewed manuscripts. He is a co-founder of four companies that are commercializing his ideas, and he has three products being sold and used in the clinic. His current research activities involve the synthesis of new macromolecules and amphiphiles, self-assembly chemistry, drug delivery, wound repair, and contrast agents for medical imaging (http://people.bu.edu/mgrin/).
References
- 1.Matyjaszewski K, Tsarevsky NV. J Am Chem Soc. 2014;136:6513. doi: 10.1021/ja408069v. [DOI] [PubMed] [Google Scholar]
- 2.McCormack CL, Lowe AB. Acc Chem Res. 2004;37:312. doi: 10.1021/ar0302484. [DOI] [PubMed] [Google Scholar]
- 3.Sutthasupa S, Shiotsuki M, Sanda F. Polym J. 2010;42:905. [Google Scholar]
- 4.Bielawski CW, Grubbs RH. Prog Polym Sci. 2007;32:1. [Google Scholar]
- 5.Schrock RR. Acc Chem Res. 1990;23:158. [Google Scholar]
- 6.Frechet JMJ. Science. 1994;263:1710. doi: 10.1126/science.8134834. [DOI] [PubMed] [Google Scholar]
- 7.Zeng FW, Zimmerman SC. Chem Rev. 1997;97:1681. doi: 10.1021/cr9603892. [DOI] [PubMed] [Google Scholar]
- 8.Frechet JM, Tomalia DA. Dendrimers and Other Dendritic Polymers. John Wiley & Sons; New York, NY: 2002. [Google Scholar]
- 9.Newkome GR, Shreiner C. Chem Rev. 2010;110:6338. doi: 10.1021/cr900341m. [DOI] [PubMed] [Google Scholar]
- 10.Lin XR, Grinstaff MW. Isr J Chem. 2013;53:498. [Google Scholar]
- 11.Brunsveld L, Folmer BJB, Meijer EW, Sijbesma RP. Chem Rev. 2001;101:4071. doi: 10.1021/cr990125q. [DOI] [PubMed] [Google Scholar]
- 12.Badjic JD, Nelson A, Cantrill SJ, Turnbull WB, Stoddart JF. Acc Chem Res. 2005;38:723. doi: 10.1021/ar040223k. [DOI] [PubMed] [Google Scholar]
- 13.Polymer Solutions. One Word for 2014: Plastics! 2014. [Google Scholar]
- 14.Kim WY, Muddu BN. Int Orthop. 2006;30:182. doi: 10.1007/s00264-006-0075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ewend MG, Brem S, Gilbert M, Goodkin R, Penar PL, Varia M, Cush S, Carey LA. Clin Cancer Res. 2007;13:3637. doi: 10.1158/1078-0432.CCR-06-2095. [DOI] [PubMed] [Google Scholar]
- 16.Zelikin AN, Zawaneh PN, Putnam D. Biomacromolecules. 2006;7:3239. doi: 10.1021/bm060544e. [DOI] [PubMed] [Google Scholar]
- 17.Miner CS, Dalton NN. Glycerol, American Chemical Society Monograph Series. Reinhold Publishing Company; New York, NY: 1953. p. 1. [Google Scholar]
- 18.Ayoub M, Abdullah AZ. Renew Sust Energ Rev. 2012;16:2671. [Google Scholar]
- 19.Yang FX, Hanna MA, Sun RC. Biotechnol Biofuels. 2012:5. doi: 10.1186/1754-6834-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pagliaro M, Ciriminna R, Kimura H, Rossi M, Della Pina C. Angew Chem Int Ed. 2007;46:4434. doi: 10.1002/anie.200604694. [DOI] [PubMed] [Google Scholar]
- 21.Rahmat N, Abdullah AZ, Mohamed AR. Renew Sust Energ Rev. 2010;14:987. [Google Scholar]
- 22.Li MY, Zhou CH, Beltramini JN, Yu WH, Fan YX. Prog Chem. 2008;20:1474. [Google Scholar]
- 23.Katryniok B, Kimura H, Skrzynska E, Girardon JS, Fongarland P, Capron M, Ducoulombier R, Mimura N, Paul S, Dumeignil F. Green Chemistry. 2011;13:1960. [Google Scholar]
- 24.Tran NH, Kannangara GSK. Chem Soc Rev. 2013;42:9454. doi: 10.1039/c3cs60227c. [DOI] [PubMed] [Google Scholar]
- 25.Mizugaki T, Arundhathi R, Mitsudome T, Jitsukawa K, Kaneda K. Acs Sustain Chem Eng. 2014;2:574. [Google Scholar]
- 26.Kimura H, Tsuto K, Wakisaka T, Kazumi Y, Inaya Y. Appl Catal a-Gen. 1993;96:217. [Google Scholar]
- 27.Porta F, Prati L. J Catal. 2004;224:397. [Google Scholar]
- 28.Dimitratos N, Porta F, Prati L. Appl Catal a-Gen. 2005;291:210. [Google Scholar]
- 29.Montassier C, Dumas JM, Granger P, Barbier J. Appl Catal a-Gen. 1995;121:231. [Google Scholar]
- 30.Buhler W, Dinjus E, Ederer HJ, Kruse A, Mas C. J Supercrit Fluids. 2002;22:37. [Google Scholar]
- 31.Ott L, Bicker M, Vogel H. Green Chemistry. 2006;8:214. [Google Scholar]
- 32.Vieville C, Yoo JW, Pelet S, Mouloungui Z. Catal Lett. 1998;56:245. [Google Scholar]
- 33.Cassel S, Debaig C, Benvegnu T, Chaimbault P, Lafosse M, Plusquellec D, Rollin P. Eur J Org Chem. 2001:875. doi: 10.1016/s0021-9673(01)00801-9. [DOI] [PubMed] [Google Scholar]
- 34.Zhou CHC, Beltramini JN, Fan YX, Lu GQM. Chem Soc Rev. 2008;37:527. doi: 10.1039/b707343g. [DOI] [PubMed] [Google Scholar]
- 35.Walach W, Kowalczuk A, Trzebicka B, Dworak A. Macromol Rapid Commun. 2001;22:1272. [Google Scholar]
- 36.Haag R, Sunder A, Stumbe JF. J Am Chem Soc. 2000;122:2954. [Google Scholar]
- 37.Ray WC, Grinstaff MW. Macromolecules. 2003;36:3557. [Google Scholar]
- 38.Zhang H, Grinstaff MW. J Am Chem Soc. 2013;135:6806. doi: 10.1021/ja402558m. [DOI] [PubMed] [Google Scholar]
- 39.Geschwind J, Frey H. Macromolecules. 2013;46:3280. [Google Scholar]
- 40.Wang XL, Zhuo RX, Liu LJ, He F, Liu G. J Polym Sci Pol Chem. 2002;40:70. [Google Scholar]
- 41.Wolinsky JB, Ray WC, Colson YL, Grinstaff MW. Macromolecules. 2007;40:7065. [Google Scholar]
- 42.Calderon M, Quadir MA, Sharma SK, Haag R. Adv Mater. 2010;22:190. doi: 10.1002/adma.200902144. [DOI] [PubMed] [Google Scholar]
- 43.Wilms D, Stiriba SE, Frey H. Acc Chem Res. 2010;43:129. doi: 10.1021/ar900158p. [DOI] [PubMed] [Google Scholar]
- 44.Thomas A, Müller SS, Frey H. Biomacromolecules. 2014;15:1935. doi: 10.1021/bm5002608. [DOI] [PubMed] [Google Scholar]
- 45.Taton D, Leborgne A, Sepulchre M, Spassky N. Macromol Chem Phys. 1994;195:139. [Google Scholar]
- 46.Dworak A, Panchev I, Trzebicka B, Walach W. Macromolecular Symposia. 2000;153:233. [Google Scholar]
- 47.Hans M, Gasteier P, Keul H, Moeller M. Macromolecules. 2006;39:3184. [Google Scholar]
- 48.Thomas A, Wolf FK, Frey H. Macromol Rapid Commun. 2011;32:1910. doi: 10.1002/marc.201100432. [DOI] [PubMed] [Google Scholar]
- 49.Gunkel G, Weinhart M, Becherer T, Haag R, Huck WTS. Biomacromolecules. 2011;12:4169. doi: 10.1021/bm200943m. [DOI] [PubMed] [Google Scholar]
- 50.Dworak A, Panchev I, Trzebicka B, Walach W. Polym Bull. 1998;40:461. [Google Scholar]
- 51.Basinska T, Slomkowski S, Dworak A, Panchev I, Chehimi MM. Colloid Polym Sci. 2001;279:916. [Google Scholar]
- 52.Basinska T, Slomkowski S, Kazmierski S, Dworak A, Chehimi MM. J Polym Sci Pol Chem. 2004;42:615. [Google Scholar]
- 53.Griffete N, Dybkowska M, Glebocki B, Basinska T, Connan C, Maitre A, Chehimi MM, Slomkowski S, Mangeney C. Langmuir. 2010;26:11550. doi: 10.1021/la100537v. [DOI] [PubMed] [Google Scholar]
- 54.Gosecka M, Griffete N, Mangeney C, Chehimi MM, Slomkowski S, Basinska T. Colloid Polym Sci. 2011;289:1511. doi: 10.1007/s00396-011-2447-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slomkowski S, Basinska T. New Frontiers in Macromolecular Science. 2010;295:13. [Google Scholar]
- 56.Basinska T, Krolik S, Slomkowski S. Macromolecular Symposia. 2009;281:96. [Google Scholar]
- 57.Basinska T, Slomkowski S. Chem Pap. 2012;66:352. [Google Scholar]
- 58.Haamann D, Keul H, Klee D, Moller M. Macromolecules. 2010;43:6295. [Google Scholar]
- 59.Thomas A, Niederer K, Wurm F, Frey H. Polym Chem-Uk. 2014;5:899. [Google Scholar]
- 60.Mendrek S, Mendrek A, Adler HJ, Walach W, Dworak A, Kuckling D. J Polym Sci Pol Chem. 2008;46:2488. [Google Scholar]
- 61.Weinhart M, Grunwald I, Wyszogrodzka M, Gaetjen L, Hartwig A, Haag R. Chem-Asian J. 2010;5:1992. doi: 10.1002/asia.201000127. [DOI] [PubMed] [Google Scholar]
- 62.Weinhart M, Becherer T, Schnurbusch N, Schwibbert K, Kunte HJ, Haag R. Adv Eng Mater. 2011;13:B501. [Google Scholar]
- 63.Wilms VS, Bauer H, Tonhauser C, Schilmann AM, Muller MC, Tremel W, Frey H. Biomacromolecules. 2013;14:193. doi: 10.1021/bm3015889. [DOI] [PubMed] [Google Scholar]
- 64.Hofmann AM, Wipf R, Stuhn B, Frey H. Macromolecules. 2011;44:6767. [Google Scholar]
- 65.Hofmann AM, Wurin F, Frey H. Macromolecules. 2011;44:4648. [Google Scholar]
- 66.Zhao JP, Schlaad H, Weidner S, Antonietti M. Polym Chem-Uk. 2012;3:1763. [Google Scholar]
- 67.Moers C, Nuhn L, Wissel M, Stangenberg R, Mondeshki M, Berger-Nicoletti E, Thomas A, Schaeffel D, Koynov K, Klapper M, Zentel R, Frey H. Macromolecules. 2013;46:9544. [Google Scholar]
- 68.Siebert M, Henke A, Eckert T, Richtering W, Keul H, Moller M. Langmuir. 2010;26:16791. doi: 10.1021/la102780y. [DOI] [PubMed] [Google Scholar]
- 69.Meyer J, Keul H, Moller M. Macromolecules. 2011;44:4082. [Google Scholar]
- 70.Kwon W, Rho Y, Kamoshida K, Kwon KH, Jeong YC, Kim J, Misaka H, Shin TJ, Kim J, Kim KW, Jin KS, Chang T, Kim H, Satoh T, Kakuchi T, Ree M. Adv Funct Mater. 2012;22:5194. [Google Scholar]
- 71.Wyszogrodzka M, Haag R. Langmuir. 2009;25:5703. doi: 10.1021/la803017b. [DOI] [PubMed] [Google Scholar]
- 72.Rokicki G. Prog Polym Sci. 2000;25:259. [Google Scholar]
- 73.Harris RF. J Appl Polym Sci. 1989;37:183. [Google Scholar]
- 74.Vogdanis L, Martens B, Uchtmann H, Hensel F, Heitz W. Makromol Chem. 1990;191:465. [Google Scholar]
- 75.Lee JC, Litt MH. Macromolecules. 2000;33:1618. [Google Scholar]
- 76.Azechi M, Matsumoto K, Endo T. J Polym Sci Pol Chem. 2013;51:1651. [Google Scholar]
- 77.Tezuka K, Komatsu K, Haba O. Polym J. 2013;45:1183. [Google Scholar]
- 78.Haba O, Tomizuka H, Endo T. Macromolecules. 2005;38:3562. [Google Scholar]
- 79.Inoue S. Journal of Macromolecular Science-Chemistry. 1979;A13:651. [Google Scholar]
- 80.Allen SD, Moore DR, Lobkovsky EB, Coates GW. J Am Chem Soc. 2002;124:14284. doi: 10.1021/ja028071g. [DOI] [PubMed] [Google Scholar]
- 81.Darensbourg DJ, Yarbrough JC. J Am Chem Soc. 2002;124:6335. doi: 10.1021/ja012714v. [DOI] [PubMed] [Google Scholar]
- 82.Qin ZQ, Thomas CM, Lee S, Coates GW. Angew Chem Int Ed. 2003;42:5484. doi: 10.1002/anie.200352605. [DOI] [PubMed] [Google Scholar]
- 83.Lu XB, Shi L, Wang YM, Zhang R, Zhang YJ, Peng XJ, Zhang ZC, Li B. J Am Chem Soc. 2006;128:1664. doi: 10.1021/ja056383o. [DOI] [PubMed] [Google Scholar]
- 84.Cohen CT, Chu T, Coates GW. J Am Chem Soc. 2005;127:10869. doi: 10.1021/ja051744l. [DOI] [PubMed] [Google Scholar]
- 85.Noh EK, Na SJ, Sujith S, Kim SW, Lee BY. J Am Chem Soc. 2007;129:8082. doi: 10.1021/ja071290n. [DOI] [PubMed] [Google Scholar]
- 86.Wu X, Zhao H, Nornberg B, Theato P, Luinstra GA. Macromolecules. 2014;47:492. [Google Scholar]
- 87.Ren WM, Liu ZW, Wen YQ, Zhang R, Lu XB. J Am Chem Soc. 2009;131:11509. doi: 10.1021/ja9033999. [DOI] [PubMed] [Google Scholar]
- 88.Unpublished results. During the course of our work, we found that even thought we made the poly(benzyl 1,2-glycerol carbonate) isotactic, the polymer was still an amorphous polymer.
- 89.Geschwind J, Frey H. Macromol Rapid Commun. 2013;34:150. doi: 10.1002/marc.201200682. [DOI] [PubMed] [Google Scholar]
- 90.Hilf J, Phillips A, Frey H. Polym Chem-Uk. 2014;5:814. [Google Scholar]
- 91.Hilf J, Frey H. Macromol Rapid Commun. 2013;34:1395. doi: 10.1002/marc.201300425. [DOI] [PubMed] [Google Scholar]
- 92.Zhang H, Grinstaff MW. J Appl Polym Sci. 2014:131. [Google Scholar]
- 93.Shi L, Lu XB, Zhang R, Peng XJ, Zhang CQ, Li JF, Peng XM. Macromolecules. 2006;39:5679. [Google Scholar]
- 94.Lu XB, Ren WM, Wu GP. Acc Chem Res. 2012;45:1721. doi: 10.1021/ar300035z. [DOI] [PubMed] [Google Scholar]
- 95.He F, Wang YX, Feng J, Zhuo RX, Wang XL. Polymer. 2003;44:3215. [Google Scholar]
- 96.Helou M, Miserque O, Brusson JM, Carpentier JF, Guillaume SM. Chem-Eur J. 2010;16:13805. doi: 10.1002/chem.201001111. [DOI] [PubMed] [Google Scholar]
- 97.Ray WC, Grinstaff MW. 222nd American Chemical Society Div Polym Chem. 2001;42:123. doi: 10.1021/ja005726+. [DOI] [PubMed] [Google Scholar]
- 98.Kaplan JA, Lei H, Liu R, Padera R, Colson YL, Grinstaff MW. Biomacromolecules. 2014;15:2548. doi: 10.1021/bm500410h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu R, Wade JE, Wolinsky JB, Winer JH, Catalano PJ, Wagner AJ, Grinstaff MW, Colson YL, Raut CP. Ann Surg Oncol. 2010;17:S31. [Google Scholar]
- 100.Liu R, Wolinsky JB, Walpole J, Southard E, Chirieac LR, Grinstaff MW, Colson YL. Ann Surg Oncol. 2010;17:1203. doi: 10.1245/s10434-009-0856-z. [DOI] [PubMed] [Google Scholar]
- 101.Liu R, Wolinsky JB, Catalano PJ, Chirieac LR, Wagner AJ, Grinstaff MW, Colson YL, Raut CP. Ann Surg Oncol. 2012;19:199. doi: 10.1245/s10434-011-1871-4. [DOI] [PubMed] [Google Scholar]
- 102.Wolinsky JB, Yohe ST, Colson YL, Grinstaff MW. Biomacromolecules. 2012;13:406. doi: 10.1021/bm201443m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yohe ST, Colson YL, Grinstaff MW. J Am Chem Soc. 2012;134:2016. doi: 10.1021/ja211148a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang YD, Ameer GA, Sheppard BJ, Langer R. Nat Biotechnol. 2002;20:602. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 105.Kallinteri P, Higgins S, Hutcheon GA, St Pourcain CB, Garnett MC. Biomacromolecules. 2005;6:1885. doi: 10.1021/bm049200j. [DOI] [PubMed] [Google Scholar]
- 106.Tawfeek H, Khidr S, Samy E, Ahmed S, Murphy M, Mohammed A, Shabir A, Hutcheon G, Saleem I. Pharm Res. 2011;28:2086. doi: 10.1007/s11095-011-0433-6. [DOI] [PubMed] [Google Scholar]
- 107.Fu HY, Kulshrestha AS, Gao W, Gross RA, Baiardo M, Scandola M. Macromolecules. 2003;36:9804. [Google Scholar]
- 108.Kulshrestha AS, Gao W, Gross RA. Macromolecules. 2005;38:3193. [Google Scholar]
- 109.Yang YX, Lu WH, Cai JL, Hou Y, Ouyang SY, Xie WC, Gross RA. Macromolecules. 2011;44:1977. [Google Scholar]
- 110.Zhang YR, Spinella S, Xie WC, Cai JL, Yang YX, Wang YZ, Gross RA. Eur Polym J. 2013;49:793. [Google Scholar]
- 111.Wyszogrodzka M, Haag R. Chem-Eur J. 2008;14:9202. doi: 10.1002/chem.200800892. [DOI] [PubMed] [Google Scholar]
- 112.Wyszogrodzka M, Mows K, Kamlage S, Wodzifiska J, Plietker B, Haag R. Eur J Org Chem. 2008:53. [Google Scholar]
- 113.Elmer SL, Man S, Zimmerman SC. Eur J Org Chem. 2008:3845. doi: 10.1002/ejoc.200800401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Carnahan MA, Grinstaff MW. J Am Chem Soc. 2001;123:2905. doi: 10.1021/ja005726+. [DOI] [PubMed] [Google Scholar]
- 115.Carnahan MA, Grinstaff MW. Macromolecules. 2001;34:7648. [Google Scholar]
- 116.Carnahan MA, Grinstaff MW. Macromolecules. 2006;39:609. [Google Scholar]
- 117.Luman NR, Smeds KA, Grinstaff MW. Chem-Eur J. 2003;9:5618. doi: 10.1002/chem.200305172. [DOI] [PubMed] [Google Scholar]
- 118.Degoricija L, Johnson CS, Wathier M, Kim T, Grinstaff MW. Invest Ophth Vis Sci. 2007;48:2037. doi: 10.1167/iovs.06-0957. [DOI] [PubMed] [Google Scholar]
- 119.Grinstaff MW. Chem-Eur J. 2002;8:2838. [Google Scholar]
- 120.Parzuchowski PG, Grabowska M, Tryznowski M, Rokicki G. Macromolecules. 2006;39:7181. [Google Scholar]
- 121.Sunder A, Hanselmann R, Frey H, Mulhaupt R. Macromolecules. 1999;32:4240. [Google Scholar]
- 122.Rokicki G, Rakoczy P, Parzuchowski P, Sobiecki M. Green Chemistry. 2005;7:529. [Google Scholar]
- 123.Yu XH, Feng J, Zhuo RX. Macromolecules. 2005;38:6244. [Google Scholar]
- 124.Parzuchowski PG, Jaroch M, Tryznowski M, Rokicki G. Macromolecules. 2008;41:3859. [Google Scholar]
- 125.Sisson AL, Haag R. Soft Matter. 2010;6:4968. [Google Scholar]
- 126.Sisson AL, Steinhilber D, Rossow T, Welker P, Licha K, Haag R. Angew Chem Int Ed. 2009;48:7540. doi: 10.1002/anie.200901583. [DOI] [PubMed] [Google Scholar]
- 127.Steinhilber D, Sisson AL, Mangoldt D, Welker P, Licha K, Haag R. Adv Funct Mater. 2010;20:4133. [Google Scholar]
- 128.Zhou HX, Steinhilber D, Schlaad H, Sisson AL, Haag R. Reactive & Functional Polymers. 2011;71:356. [Google Scholar]
- 129.Rossow T, Heyman JA, Ehrlicher AJ, Langhoff A, Weitz DA, Haag R, Seiffert S. J Am Chem Soc. 2012;134:4983. doi: 10.1021/ja300460p. [DOI] [PubMed] [Google Scholar]
- 130.Steinhilber D, Rossow T, Wedepohl S, Paulus F, Seiffert S, Haag R. Angew Chem Int Ed. 2013;52:13538. doi: 10.1002/anie.201308005. [DOI] [PubMed] [Google Scholar]
- 131.Steinhilber D, Witting M, Zhang XJ, Staegemann M, Paulus F, Friess W, Kuchler S, Haag R. J Controlled Release. 2013;169:289. doi: 10.1016/j.jconrel.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 132.Zhang XJ, Achazi K, Steinhilber D, Kratz F, Dernedde J, Haag R. J Controlled Release. 2014;174:209. doi: 10.1016/j.jconrel.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 133.Matthews OA, Shipway AN, Stoddart JF. Prog Polym Sci. 1998;23:1. [Google Scholar]
- 134.Zelikin AN, Putnam D. Macromolecules. 2005;38:5532. [Google Scholar]
- 135.Zawaneh PN, Doody AM, Zelikin AN, Putnam D. Biomacromolecules. 2006;7:3245. doi: 10.1021/bm0605457. [DOI] [PubMed] [Google Scholar]
- 136.Simon J, Olsson JV, Kim H, Tenney IF, Waymouth RM. Macromolecules. 2012;45:9275. [Google Scholar]
- 137.Helou M, Brusson JM, Carpentier JF, Guillaume SM. Polym Chem-Uk. 2011;2:2789. [Google Scholar]
- 138.Weiser JR, Zawaneh PN, Putnam D. Biomacromolecules. 2011;12:977. doi: 10.1021/bm101342p. [DOI] [PubMed] [Google Scholar]
- 139.Guerin W, Helou M, Slawinski M, Brusson JM, Carpentier JF, Guillaume SM. Polym Chem-Uk. 2014;5:1229. [Google Scholar]
- 140.Zawaneh PN, Singh SP, Padera RF, Henderson PW, Spector JA, Putnam D. P Natl Acad Sci USA. 2010;107:11014. doi: 10.1073/pnas.0811529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. J Controlled Release. 2001;70:1. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 142.Vandervoort J, Yoncheva K, Ludwig A. Chemical & Pharmaceutical Bulletin. 2004;52:1273. doi: 10.1248/cpb.52.1273. [DOI] [PubMed] [Google Scholar]
- 143.Morgan MT, Carnahan MA, Immoos CE, Ribeiro AA, Finkelstein S, Lee SJ, Grinstaff MW. J Am Chem Soc. 2003;125:15485. doi: 10.1021/ja0347383. [DOI] [PubMed] [Google Scholar]
- 144.Morgan MT, Carnahan MA, Finkelstein S, Prata CAH, Degoricija L, Lee SJ, Grinstaff MW. Chem Commun. 2005:4309. doi: 10.1039/b502411k. [DOI] [PubMed] [Google Scholar]
- 145.Morgan MT, Nakanishi Y, Kroll DJ, Griset AP, Carnahan MA, Wathier M, Oberlies NH, Manikumar G, Wani MC, Grinstaff MW. Cancer Res. 2006;66:11913. doi: 10.1158/0008-5472.CAN-06-2066. [DOI] [PubMed] [Google Scholar]
- 146.Hussain AF, Kruger HR, Kampmeier F, Weissbach T, Licha K, Kratz F, Haag R, Calderon M, Barth S. Biomacromolecules. 2013;14:2510. doi: 10.1021/bm400410e. [DOI] [PubMed] [Google Scholar]
- 147.Velazquez AJ, Carnahan MA, Kristinsson J, Stinnett S, Grinstaff MW, Kim T. Arch Ophthalmol-Chic. 2004;122:867. doi: 10.1001/archopht.122.6.867. [DOI] [PubMed] [Google Scholar]
- 148.Kang PC, Carnahan MA, Wathier M, Grinstaff MW, Kim T. J Cataract Refr Surg. 2005;31:1208. doi: 10.1016/j.jcrs.2004.10.067. [DOI] [PubMed] [Google Scholar]
- 149.Berdahl JP, Johnson CS, Proia AD, Grinstaff MW, Kim T. Arch Ophthalmol-Chic. 2009;127:442. doi: 10.1001/archophthalmol.2008.582. [DOI] [PubMed] [Google Scholar]
- 150.Meyers SR, Juhn FS, Griset AP, Luman NR, Grinstaff MW. J Am Chem Soc. 2008;130:14444. doi: 10.1021/ja806912a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Anderson RA, Feathergill KA, Diao XH, Cooper MD, Kirkpatrick R, Herold BC, Doncel GF, Chany CJ, Waller DP, Rencher WF, Zaneveld LJD. J Androl. 2002;23:426. [PubMed] [Google Scholar]
- 152.Herold BC, Bourne N, Marcellino D, Kirkpatrick R, Strauss DM, Zaneveld LJD, Waller DP, Anderson RA, Chany CJ, Barham BJ, Stanberry LR, Cooper MD. J Infect Dis. 2000;181:770. doi: 10.1086/315228. [DOI] [PubMed] [Google Scholar]
- 153.Dernedde J, Rausch A, Weinhart M, Enders S, Tauber R, Licha K, Schirner M, Zugel U, von Bonin A, Haag R. P Natl Acad Sci USA. 2010;107:19679. doi: 10.1073/pnas.1003103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Agrawal A, Ayantunde AA, Cheung KL. Anz J Surg. 2006;76:1088. doi: 10.1111/j.1445-2197.2006.03949.x. [DOI] [PubMed] [Google Scholar]
- 155.Kuroi K, Shimozuma K, Taguchi T, Imai H, Yamashiro H, Ohsumi S, Saito S. Jpn J Clin Oncol. 2006;36:197. doi: 10.1093/jjco/hyl019. [DOI] [PubMed] [Google Scholar]
- 156.Weinhart M, Becherer T, Haag R. Chem Commun. 2011;47:1553. doi: 10.1039/c0cc04002a. [DOI] [PubMed] [Google Scholar]
- 157.Turk H, Haag R, Alban S. Bioconjugate Chem. 2004;15:162. doi: 10.1021/bc034044j. [DOI] [PubMed] [Google Scholar]
- 158.Ganguli M, Jayachandran KN, Maiti S. J Am Chem Soc. 2004;126:26. doi: 10.1021/ja037534v. [DOI] [PubMed] [Google Scholar]
- 159.Kainthan RK, Gnanamani M, Ganguli M, Ghosh T, Brooks DE, Maiti S, Kizhakkedathu JN. Biomaterials. 2006;27:5377. doi: 10.1016/j.biomaterials.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 160.Fischer W, Calderon M, Schulz A, Andreou I, Weber M, Haag R. Bioconjugate Chem. 2010;21:1744. doi: 10.1021/bc900459n. [DOI] [PubMed] [Google Scholar]
- 161.Industry Experts. Biomaterials – A Global Market Overview. 2011. [Google Scholar]
- 162.NanoMarkets. Worldwide Medical Polymer Markets 2013–2020. 2013. [Google Scholar]