

Abstract
This Minireview summarizes a variety of intriguing catalytic studies accomplished by employing unsupported, either solubilized or freely mobilized, and small organic ligand-capped palladium nanoparticles as catalysts. Small organic ligands are gaining more attention as nanoparticle stabilizers and alternates to larger organic supports, such as polymers and dendrimers, owing to their tremendous potential for a well-defined system with spatial control in surrounding environments of reactive surfaces. The nanoparticle catalysts are grouped depending on the type of surface stabilizers with reactive head groups, which include thiolate, phosphine, amine, and alkyl azide. Applications for the reactions such as hydrogenation, alkene isomerization, oxidation, and carbon-carbon cross coupling reactions are extensively discussed. The systems defined as “ligandless” Pd nanoparticle catalysts and solvent (e.g. ionic liquid)-stabilized Pd nanoparticle catalysts are not discussed in this review.
Keywords: nanocatalysis, nanoparticle, organic ligands, palldium, unsupported
Introduction
Interests in metallic nanoparticles stand from a wide array of potential applications in fields such as electronics,[1,2] spectroscopy,[3,4] hydrogen storage,[5–8] drug delivery.[9,10] medicine,[11] biology,[12–14] and catalysis.[5–22] Especially, their possession of highly reactive surfaces, which arise from the high surface to volume ratio along with a low number of atomic neighbors, brands nanoparticles as a practical candidate for many scientific efforts especially in the area of catalysis.[23]
Presently, research endeavors on nanoparticle synthesis have been focused on dimensionally controllable synthetic methods,[24–26] optimal application approaches,[27] and the development of efficient large scale manufacturing.[28] The development of nanoparticles, which have been defined restrictively to sizes ranging from 1 nm to no larger than 100 nm, have even made once inert or less reactive bulk materials into highly efficient catalytic systems.[20] For example, yellow bulk gold has been historically employed in pursuits such as coinage, jewelry, and sculpture which required the metal to display anything other than a low chemical reactivity.[19] The progress of research involving gold nanoclusters has led to the discovery of its copious catalytic activities.[20] Reactions such as the oxidation of alcohols,[29–31] epoxidations,[32–35] reduction of nitrophenol,[36,37] and carbon monoxide oxidation[35,38,39] are commonly achieved by gold-cored nanoparticles.
Unlike gold, palladium has enjoyed a compelling reputation as a chemically active material. In fact, many palladium compounds including palladium complexes are currently being utilized as catalysts in asymmetric syntheses,[40,41] cross coupling,[42–46] and alkylation reactions.[47,48] Additionally, palladium is known for its uniquely high hydrogen gas absorption capacity, which has been documented to occur even at room temperature and pressure.[7,8] Hence, palladium nanoparticles (PdNPs) are fairly well studied regarding their potential as novel catalysts and the basis of highly efficient hydrogen storage materials. Furthermore, the cost of palladium as a starring material is much lower than some other catalytically active metals such as rhodium and platinum, which is an advantage itself in the design of large scale production.[49]
Frequently, the use of transition-metal nanoparticles in catalysis is performed with the aid of superficial solid support.[50–55] Nanoparticles are bound either mechanically[50–52] or chemically[53–55] to a surface, while catalysis is undertaken in a heterogeneous system. The benefits in this set up involve a facile separation of products from the catalyst, an ease of the recyclability in the system, and a protection from degradation of the nano-particle catalyst.[56] However, as nanoparticles are jammed to a solid substrate, turnover rates and selectivity can be affected negatively due primarily to the principles of diffusion and the involvement of two different phases in catalytic reactions.[57] This review mainly focuses on the reactions performed with PdNP catalysts not bound to any solid support including metal oxide, silica, or polymer based materials. Even without a solid support, PdNP catalysts can be stabilized by organic ligands and fully mobilized in a heterogeneous condition. They can also be dissolved or suspended in organic or aqueous solvents depending on the structure and functionality of surface-immobilized ligands and placed in a homogeneous condition during the catalytic reactions. When organic ligand-capped nanoparticles are permanently suspended or dissolved in a homogenous condition, it can promote surface chemistry evenly throughout the reaction mixture and enjoy higher reaction selectivity.[57] Even in the case of organic ligand-capped nanoparticles in an unsupported heterogeneous catalysis systems, the catalytic reactions are not limited to a single site like the organometallic chemistry and the catalysts can be effortlessly separated by the use of gravity.[58]
Understanding and controlling the effects of small organic ligands on the catalytic properties of nanoparticles is important due to the tremendous potential of a well-defined system, especially at near-surface active sites, in providing a spatial control in reactivity and selectivity. In particular, functionalized ligand-capped nanoparticles are ideal candidates for selective catalytic reactions because they can be devised to enhance or resist the adsorption of particular substrates similar to enzymes. This review will present a variety of studies in which organic ligand-capped and unsupported PdNPs behave as effective catalysts with a focus on small organic ligands including up to 3rd generation dendrons and supramolecules, such as β-cyclodextrin, but not polymers and dendrimers. Furthermore, the methods of synthesis and some characterization data of these PdNP catalysts are also articulated and evaluated. Here, each study is partitioned by the type of stabilizing ligands used in the nanoparticle synthesis. Larger organic systems such as polymers and dendrimers as stabilizing supports have been extensively covered by other review articles[59,60] and are outside of the scope of this review (smaller supramolecules such as cyclodextrin and a few generation dendrons are included in this review). “Ligandless” PdNP and solvent (e.g. ionic liquid)-stabilized PdNP catalysts are also not discussed in this review, because the organic ligands have either no or only minimum influence over reactivity.[61–63] These topics have also been extensively covered by other review articles.[61,62]
Small Organic-ligand Stabilization
Challenges associated with the synthesis of PdNPs include uncontrollable growth leading to particle agglomeration, otherwise known as Ostwald ripening. Indeed, small metal nanoparticles tend to be attracted to each other growing continuously until the particles eventually become too big to be solubilized or suspended in solution.[63] A popular remedy for this conundrum is the introduction of organic-ligand stabilizers, such as thiols,[64–70] phosphines,[71] amines,[72] ammonium salts,[73] selenolates,[74] and isocyanides,[75] prior to or during the nanoparticle nucleation-growth stage. These organic-ligand stabilizers are capable of providing control over the core size of the nanoparticles during the synthesis, while providing stability and solubility to the nanoparticles.[76] As organic ligands can alter the surrounding environments of nanoparticles, they have a great influence over the activity and selectivity of nano-particle catalysts.
Thiolate-capped Pd nanoparticle catalysts generated from thiol ligands
Among various small organic capping ligands, thiols have been the most popular choice of protecting monolayers, owing to the good stability of thiolate-capped metal nanoparticles that can be isolated, re-dissolved, and handled as molecular reagents.[76–78] The superior control over the structure and composition of thiol assembly on metal surfaces would be especially beneficial for controlling the catalytic properties of metal nanoparticles. Recently, the controlling activity of Al2O3-supported Pd and Pt catalysts functionalized with well-defined and well-ordered alkanethiol ligands has been introduced by Medlin et al.[79–81] They have shown that the catalytic property of supported Pd catalysts was largely influenced by altering the density, structure, and functionality of surface alkanethiol ligands.
Previously, however, alkanethiols have been denoted as fairly aggressive poisons to metal nanoparticle catalysts.[82] Although there are somewhat limited insights in to the nanoparticle surface atom-sulfur bond, the strong affinity of thiols to noble metals is regarded as the main reason why thiolate-stabilized nanoparticles possess densely covered surfaces and, hence, low catalytic activities.[66] However, the Astruc group was able to synthesize dodecanethiolate-stabilized PdNPs capable of catalyzing Suzuki–Miyaura coupling reactions at room temperature.[83] The synthetic procedure involved the reduction of the palladium precursor, (PdCl2(CH3CN)2), by LiBEt3H, superhydride, in THE in the presence of tetra-n-octylammonium bromide. This reaction produced PdNPs with an average core size of 2.3 nm, which showed the relatively good catalytic activity towards the Suzuki–Miyaura coupling reaction. The reaction in a biphasic system with 1 mol% Pd catalyst loading at room temperature (Scheme 1) was completed in 24 h. Moreover, with the increased reaction temperature, the C—C coupling reactions could be completed in 1 h. From this work, Astruc and his co-workers were able to successfully demonstrate for the first time that straight-chain alkanethiolate-capped PdNPs could be utilized to catalyze a simple organic reaction under mild conditions. Similary, Pd nanoshells coated with alkanethiols were used for the Suzuki coupling of phenylboronic acid with iodobenzene in organic solvents.[84] Pd nano-shells were prepared by coating ≈100 nm silica core particles with a thin layer of Pd (≈20 nm in thickness). The mechanistic studies of the C–C coupling reactions involving ligand-capped PdNPs, however, eventually identified that the C–C coupling reactions were indeed catalyzed by the Pd ions leached from PdNP instead of PdNP surface itself.[61]
Scheme 1.

Reaction for the Suzuki–Miyaura coupling by dodecanethiolate-capped PdNPs.[83]
Although the synthesis of monodispersed alkanethiolate-capped PdNPs has been fairly addressed, the ligand varieties of resulting PdNPs are somewhat limited, as methodologies using reducing agents tend to exclude the use of ligands with reactive functionalized groups.[85] It has been shown that ligand place-exchange reactions can be used to add functionalized ligands to these inert alkanethiolate-capped PdNPs. However, a high functional-group density is almost impossible to achieve using the ligand-exchange reaction because of the equilibrium nature of the metal-nonmetal boncl.[86] Recent studies by the Fornasiero group have devised an approach to prepare functionalized nanoparticles by directly employing 11-mercaptoundecanoic acid (MUA) as a stabilizer for PdNPs.[78] Additionally, their synthetic method was capable of synthesizing PdNPs with mixed ligand monolayers: MUA-MN (9-mercapto-1-nonaol), MUA-DT (1-dodecanethiol), and MN-DT by introducing the mixture of thiols prior to nanoparticle formation which overall led to PdNPs with small core sizes (≈2 nm), narrow distributions, and a large range of solubility.
The interesting aspect regarding this one-phase synthetic system involved sizeable addition of phosphoric acid prior to the addition of ligand. The reduction of the K2PdCl4 precursor required only a slight excess of NaBH4, as the reduction of PdII was faster than hydride decomposition. In the case of using purely MUA stabilizers, the resulting PdNPs possessed ≈2 nm metallic cores and were soluble in polar organic solvents (both protic and aprotic) (Scheme 2). Their poor solubility in water was attributed to the highly favored protonation of the COOH group in the presence of phosphoric acid during the synthesis. Conversely, the absence of phosphoric acid during the synthesis yielded carboxylate (COO−)-functionalized, water soluble PdNPs after the deprotonation by NaBH4. The function of the MUA-PdNP as a catalyst was successfully applied to Suzuki cross-coupling reactions. The MUA-PdNP was converted to its carboxylate form under basic conditions. The carboxylate groups made the MUA-PdNP insoluble in organic solvent and the catalytic reaction was done as the unsupported heterogeneous system. GC-MS results confirmed complete reactions under 12 h with aryl halides with more electronegative halogens. No real loss of catalytic performance was seen throughout a total of five consecutive cycles. Suzuki cross-coupling for iodobenzene and 4-bromobenzonitrile yielded >95% in each cycle.
Scheme 2.
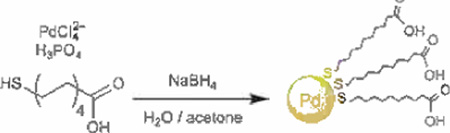
Synthesis of MUA-Pd nanoparticles. Adapted from Ref. [78] with permission from The American Chemical Society.
The surface attachment of thiolated cyclodextrins (CDs) to PdNPs could generate nanocatalysts with good activity towards the hydrogenation of allylamine in aqueous solution.[87] For the synthesis of CD-capped PdNP catalysts, PdCl42− sodium salts were reduced by borohydride in DMSO-H20 in the presence of thiolated CD.[87] Addition of cationic ferrocene derivatives resulted in the inhibition of the catalytic activity of these CD-capped PdNPs for 3-butenyltrimethylammonium bromide.[67] This inhibition is attributed to the ability of ferrocene derivatives to create Coulombic barriers for the approach of cationic olefin substrates to the active sites of PdNPs, which are created by surface bound thiolated CD. This thiolated CD-capped PdNPs were also tested for the Suzuki C–C coupling reactions in a mixed solvent system (acetonitrile/H20).[88] The binding of iodoferrocene to the CD cavities on the surface of PdNPs contributed for the increased product yield compared to the reaction involving free ferrocene inhibitors. This work was reported in 2002 and was the first example of “tunable PdNP catalyst” where the catalytic activity was controlled by manipulating the surface of Pd nanoparticle catalysts using small organic ligands.
In contrast to small organic ligand-stabilized metallic nano-particles, the surface coverage of dendron-capped nanoparticles was limited because of the high steric hindrance of the dendritic branches. Therefore, a substantial amount of surface atoms were uninhibited and available for catalysis. Thiolated dendron-capped PdNPs were prepared by the modified Brust–Schiffrin reaction using toluene-water biphasic system and tested for both Heck and Suzuki coupling reactions.[89] The presence of bulky dendron ligands on the surface PdNPs created more surface-accessible sites and made PdNPs active for both C–C coupling reactions, albeit with somewhat moderate to low yields (38–75%). However, these dendron-capped PdNPs could not be employed as a catalyst for hydrogenation reactions, because the absorption of hydrogen gas induced the decreased stability and precipitation of PdNPs. This work implied such homogeneous PdNPs with ligands-created active sites and less surface-bound thiolate groups can be developed into active catalysts for organic reactions, but it requires a careful control in maintaining the stability while enhancing the efficiency of catalysts.
Thiolate-capped Pd nanoparticle catalysts generated from thiosulfate ligand precursors
In some cases, such as Lindlar catalyst, moderate poisoning to metal catalysts have been found beneficial for selective organic reactions. By decreasing the overall reactivity of a catalyst, higher selectivity is achieved as long as the poisoning can be fully controlled.[82] As mentioned before, thiols are very popular catalyst poisons. Historically, this property of thiols has previously limited thiolate-stabilized PdNPs as catalysts only for cross-coupling reactions and hydrogenation of olefins, as shown above. Shon and his coworkers were able to successfully control the synthesis of dodecanethiolate-capped PdNPs by employing a Bunte Salt precursor, sodium S-dodecylthiosulfate.[90,91] In a two phase catalytic system composed of toluene and water, Bunte Salts were introduced to a solution of PdCl42− phase-transferred to the organic layer by tetra-n-octylammonium bromide prior to reduction by sodium borohydride. At the nanoparticle nucleation-growth stage, the growth of the PdNP is inhibited by the Bunte Salts as they absorb onto the surface and form a Pd-S thiolate bond. After the expunge of the sulfite moiety upon the adsorption on metal surfaces, an easily accessible reaction site is generated as the results of slightly lower density of alkanethiolate ligands on Pd surfaces (Scheme 3). Ultimately, this synthesis yielded small alkanethiolate-capped palladium nanoparticles (2–3 nm) with a lower surface coverage than the method directly employing alkanethiolate ligands prior to reduction.
Scheme 3.

Reaction for synthesis of palladium nanoparticles using sodium S-dodecyl thiosulfate. Adapted from Ref. [92] with permission from The American Chemical Society.
Shon and his coworkers evaluated the catalytic properties of these nanoparticles by monitoring the conversion of allyl alcohol under the atmospheric pressure of H2 gas. The homogenous reaction involving 5 mol% Pd catalyst loading in nonpolar organic solvent yielded a high and selective conversion of allyl alcohol to propanal, the isomerization product. Further analysis of the catalytic properties involved studying the effects of different core sizes and surface ligand density of PdNPs, which revealed a better catalytic activity and selectivity for ≈3.4 nm PdNPs with an overall lower surface coverage of 0.38 ligands per surface Pd atom.[92] In addition, they found that the catalytic selectivity of these PdNPs is greatly influenced by the conformational changes of ligands in different solvents. In polar protic solvent, the reaction of allyl alcohol produced almost exclusively 1-propanol, a hydrogenation product.[93]
Shon et al. further extended their research to the design and synthesis of water-soluble w-carboxylate-functionalized alkanethiolate-capped PdNPs.[94] These PdNPs had the unique and stable structure of exterior ionic functional groups and internal hydrophobic alkane groups, resembling the overall structure of micellar nanostructures in water. Two-phase synthesis was employed for the formation of catalytically active water-soluble PdNPs, because one-phase aqueous synthesis caused the base-catalyzed hydrolysis of thiosulfate ligand precursor and the subsequent formation of densely packed thiolate monolayers on the PdNP surface. The catalytic conversion of allyl alcohol using these water-soluble PdNPs dissolved homogeneously in water greatly favored the hydrogenation of allyl alcohol to 1-propanol. In comparison, PdNPs heterogeneously dispersed in chloroform produced propanal, the isomerization product, in a relatively high yield. The overall results clearly demonstrated that the catalytic activity and selectivity of PdNPs were affected by the thiolate monolayer conformation and controlled by the interaction between monolayers and surrounding solvent environments as shown in Figure 1.
Figure 1.

Proposed conformation of the ligands on Pd nanoparticles and the structure of Pd-alkyl intermediates: a) MUA-PdNP in water, b) MUA-PdNP in chloroform, c) MHA-PdNP in water, and d) MHA-PdNP in chloroform. Reproduced from Ref. [94] with permission from The American Chemical Society.
Alkanethiolate-capped PdNPs synthesized by Shon et al. could also be used for one-pot conversion of simple propargyl alcohols to saturated carbonyl analogues as shown in Scheme 4.[95] The results demonstrated the significance of homogeneous nanoparticle catalysts capable of providing a high selectivity towards atypical products. The direct one-pot conversion of simple propargyl alcohols to saturated carbonyls has not been reported prior to this work. Analysis of the kinetic profile revealed that propargyl alcohol was first converted to the semi-hydrogenation product, allyl alcohol, without producing any full hydrogenation (1-propanol) or the final product (propanal) in the early stage of the reaction. After this first stage, allyl alcohol began its conversion to either 1-propanol or propanal with a higher priority towards the latter.
Scheme 4.

The conversion of propargyl alcohols (R1=H or CH3; R2= H, CH3, or Ph) to their carbonyl analogues using PdNP catalysts. Adapted from Ref. [95] with permission from The Royal Society of Chemistry.
Sulfide-capped Pd nanoparticle catalysts
Thioethers have also been utilized as stabilizing ligands in the one-step procedure producing monodisperse PdNPs with core sizes ranging from 1.7 to 3.5 nm.[96] The size of nanoparticles depended on the reaction temperature, time, and solvent and the carbon chain length of the thioethers. These thioether-stabilized PdNPs turned out to be active catalysts for hydrogenation of acetylene and ethylene. However, owing to the low stability of these PdNPs, the recovery of the nanoparticles after subsequent catalytic reactions was rather challenging. This required a preferential use of SiO2 support for immobilization of thioether-stabilized PdNPs, which increased overall efficiency of PdNP catalysts for hydrogenation.
Phosphine-capped Pd nanoparticle catalysts
Although ligand-stabilized metal nanoparticles have been popularized over the last 20 years, bisphosphines have received little attention as fruitful ligands. Fujihara et al. first demonstrated advantages of employing bisphosphines as effective stabilizers for palladium nanoparticles.[97] The PdNP synthesis involved the reduction of K2PdCl4 in THF by sodium borohydride in the presence of the C8-BINAP ligand as shown in Scheme 5. The resulting nanoparticles possessed ≈1.2 nm core sizes, which was confirmed by small angle X-ray scattering (SAXS) and 25 ligands per assumed 55-atom palladium core proved by TGA analysis. Interestingly, these nanoparticles were shown to be catalytically active in both Suzuki and Stille coupling reactions at room temperature with only 0.06 and 0.02 mol% Pd catalyst, respectively. The Stille coupling experiment involved the reaction between methyl 2-iodobenzene with (tributylstannyl)thiophene, which resulted in a 91 % conversion. As for the Suzuki reaction, the coupling of 4-bromo-toluene with phenylboronic acid afforded 83% isolation of final product. The C8BINAP-Pd nanoparticles were readily soluble in organic solvents granting homogenous catalytic conditions. Moreover, the recycling of the catalysts for the second time showed no loss in activity.
Scheme 5.

Reaction for the Stille coupling by C8BINAP-Pd nanoparticle catalysts and the structure of C8-BINAP. Reproduced from Ref. [97] with permission from The Royal Society of Chemistry.
Fan and his coworkers showed an example of a dendron-capped PdNP coupled by P-Pd bonds.[98] The synthesis of these phosphine dendron-stabilized PdNPs involved the reduction of Pd(acac)2 in THF with H2 gas in the presence of the Fréchetpolyaryl ether Gndendrimer (Figure 2). Examination by TEM showed the nanoparticles have core size from 5.0 nm to 3.2 nm with increasing ratio of dendron ligands. Fan tested the catalytic activity of these PdNPs for both the Suzuki coupling reaction and hydrogenation. The homogeneous reaction afforded high isolated yields of coupling products, even with electron deficient phenylboronic acids, and sterically hindered aryl bromides and aryl iodides. Although the G2DenP-Pd nanoparticles provided high catalytic efficiency, Fan was able to conclude that higher generational dendrons provided increased catalytic activity.
Figure 2.

Synthesis and TEM images of GnDenP-Pd catalysts. Reproduced from Ref. [98] with permission from The American Chemical Society.
Pyridyl halides and phenylboronic acid also coupled successfully with conversion rates higher than 84%. Fan postulated the higher activities of G3DenP-PdNPs could be attributed to the higher fractions of surface palladium atoms that were hampered by the phosphine dendrons. Many alkenes including styrene, phenylacetylene, and cyclohexene were also easily hydrogenated in homogeneous conditions. As for the recyclability of these materials, GnDenP-PdNPs required precipitation with methanol to collect the catalysts. Nevertheless, ICP-XPF data showed no more than 1% palladium leaching after the second and third cycles. The performance of the recycled catalyst showed no deficiency in activity.
Amine-capped Pd nanoparticle catalysts
Oleyamine is one of the most commonly used ligands for the stabilization of metal nanoparticles.[99] By using either the pure form or the combination of oleylamine and alkylammonium alkylcarbamate (AAAC) during the reduction of palladium(II) acetylacetonate (Pd(acac)2) with borane-tert-butylamine complex, amine-capped PdNPs with different shapes such as spheres, tetrahedral, and multipods could be prepared.[100] Cyclohexene hydrogenation results for different shaped PdNPs suggested that the surface structures (atomic steps, edges, etc) of the PdNPs lead to differences in catalytic activities. The multipods with many surface defects showed the highest catalytic activity and the thermodynamically stable spherical PdNPs exhibited the lowest activity during the catalytic reaction.
Zamborini and his coworkers investigated the catalytic activity and selectivity of alkylamine-stabilized Pd nanoparticles with different chain lengths (C8, C12, and C16NH2).[101] Alkylamine-capped PdNPs were synthesized using the modified Brust–Schiffrin reaction using alkylamine instead of alkanethiol. The study showed that the activity of these alkylamine-capped PdNPs for the catalytic conversion of allyl alcohol was greater than that of alkanethiol-capped PdNPs, owing to the weaker coordination to the NP. However, the selectivity of the reaction of allyl alcohol was only a 1:1 or 3:2 ratio towards the hydrogenation/isomerization products. The TOF of the reaction increased with increasing chain length of alkylamine, owing to the increased stability of PdNPs.
The reduction of [PdCl(C3H5)]2 with LiBEt3H in the presence of PAMAM dendrons with a pyridine core unit produced PdNPs stabilized by dendrons.[102] These dendron-stabilized PdNPs exhibited good activity and selectivity for mono-hydrogenation of dienes and alkynes. Increasing the dendron generation resulted in the increase in the catalytic activity for the first hydrogenation, which was likely caused by the enhanced accessibility of the substrate to the PdNP active sites. It was also found that the structure of surrounding dendron branches affected the competitive hydrogenation between 3-phenyl-2-propyn-1-ol and 1-phenyl-1-propyne. The hydrogenation of an alkyne was promoted by the hydroxyl group of the substrate, which forms hydrogen bonding with the amide groups within the dendron.
Alkyl-capped Pd nanoparticle catalysts generated from alkyl azide
Chen synthesized toluene soluble, ≈2.3 nm, palladium nano-particles protected by butylphenyl ligands.[103] Briefly, the synthesis of these materials involved the co-reduction of H2PdCl4 and butylphenyldiazonium in a toluene-THF solution by sodium borohydride. Characterization of FTIR spectra led Chen to confirm the existence of a Pd-C linkage implying butylphenyl ligands were directly imbedded to the palladium surface without the presence of any nitrogen based functional group (Figure 3). Electrochemical studies encompassed cyclic voltammograms in 0.1 m H2SO4 by PdNP-coated polished glassy carbon electrodes (5 mm). The specific electrochemical surface area (ECSA) of the PdNPs resulted in 122 m2g−1, which is 3.6 times larger than commercial palladium sources, such as “palladium black”. Based on theoretical calculations, it was determined that 50% of the PdNPs were available after the loading onto the glassy carbon electrode. Such a large ECSA augmented the electro-catalytic properties of these nanoparticles; indeed, the mass activity of these butylphenyl stabilized-PdNPs in the electro-oxidation of formic acid was discovered to be 4.5 times larger than that of “palladium black”. Moreover, the PdNP electrocatalysts outperformed Pd black by maintaining mass current densities 2.7–4.8 times higher for 600 seconds.
Figure 3.

FTIR spectra of a) the Pd-BP nanoparticles, b) n-butyl benzene, and c) 4-butylaniline. Reproduced from Ref. [103] with permission from The Royal Society of Chemistry.
Summary and Future Prospects
Unsupported palladium nanoparticles stabilized by small organic ligands provide a platform for a variety of catalytic reactions, as shown above. These studies suggest that the availability of various capping ligands enhances the overall scope of these nanoparticles for catalysis applications. The activity and selectivity of Pd nanoparticle catalysts are highly dependent upon the type and structure of surrounding organic ligands, because they control the accessibility of incoming substrates to the reactive surface atoms and the selectivity for substrates with specific interactions, much like the ligands in organometallic catalysts. Additionally, the recyclability and efficiency of these materials as shown in Table 1 potentially denotes them of high technological impact in the future. For example, the rates of hydrogenation of several Pd nanoparticle catalysts compare very well with Wilkinson’s catalyst, a common homogeneous catalyst, and Lindlar’s catalyst, a popular heterogeneous catalyst.[102,104]
Table 1.
Assessment of Pd nanoparticle catalytic systems with respect to the ligand stabilizers and the catalysis reactions.
| Catalyst | Ligand stabilizer |
Reagents for particle synthesis |
Catalysis reaction |
Highest TOF reported |
# of cycles tested |
|---|---|---|---|---|---|
| Pd nanoparticles w/2.3 nm core | dodecanethiol | PdCl2(CH3CN)2 + LiBEt3H | Suzuki–Miyaura coupling[83] | – | 6 |
| Pd nanoshells w/100 nm silica core and 20 nm Pd shell | dodecnaethiol hexadecanethiol | PdCl2 + HCl + l-ascorbic acid | Suzuki coupline[84] | – | – |
| Pd nanoparticles w/2.0 nm core | 11-mercapto-undecanoic acid | K2PdCl4 + NaBH4 + phosphoric acid | Suzuki coupline[78] | – | 5 |
| Pd nanoparticles w/3.5 nm core | thiol-β-cyclodextrin | K2PdCl4 + NaBH4 | hydrogenation of alkene[67,87] Suzuki coupling[88] |
320[a] | – |
| Pd nanoparticles w/2.0 nm core | thiol-Dendron | K2PdCl4 + NaBH4 | Heck and Suzuki coupling[89] | 2175[b] | – |
| Pd nanoparticles w/1.5–3.5 nm core | sodium S-dodecylthiosulfate sodium S-hexylthiosulfate | K2PdCl4 + NaBH4 | isomerization and hydrogenation of allyl alcohols[90–93] tandem semi-hydrogenation and isomerization of propargyl alcohols[95] |
451 [b] 380[b] |
14 - |
| Pd nanoparticles w/1.8 nm core | sodium ω-carboxyl-S-undecylthiosulfate sodium ω-carboxyl-S-hexylthiosulfate |
K2PdCl4 + NaBH4 | hydrogenation of allyl alcohol[94] | 490[b] | – |
| Pd nanoparticles w/1.7–3.5 nm core | dodecyl sulfide | PdCl2 + NaCl + NH2NH2 + NaOH | hydrogenation of alkene[96] | – | 5 |
| Pd nanoparticles w/1.2 nm core | bisphosphine-BINAP | K2PdCl4 + NaBH4 | Suzuki and Stille coupling[97] | – | – |
| Pd nanoparticles w/3–5 nm core | diphenylphosphine-dendron | Pd(acac)2 + H2 | hydrogenation of alkenes and alkynes[98] Suzuki coupling[98] |
- 677[a] |
- 9 |
| Pd spheres, tetrahedral, and multipods w/≈3, 7, and 20 nm core | oleylamine alkylammonium alkylcarbamate (AAAC) |
Pd(acac)2 + borane-tert-butylamine | hydrogenation of alkenes[100] | 160000[a] | – |
| Pd nanoparticles w/3–5 nm core | octylamine dodecylamine hexadecylamine |
K2PdCl4 + NaBH4 | isomerization and hydrogenation of allyl alcohor[101] | 605[a] | – |
| Pd nanoparticles w/5–7 nm core | pyridine-dendron | [PdCl(C3H5)]2 + LiBEt3H | semi-hydrogenation of dienes and alkynes[102] | 20[a] | – |
| Pd nanoparticles w/2.3 nm core | butylphenyldiazonium | H2PdCl4 + NaBH4 | oxidation of formic acid[103] | – | – |
mol of products per mol of Pd atoms per hour.
mol of products per mol of surface Pd atoms per hour.
Future studies should seek to enhance further understanding on critical structure-function relationships for unsupported organic ligand-capped metal nanoparticle catalysts. Compared to the plenty of attention placed on the metallic cores that determine the catalytic properties of nanoparticles, the influence of surrounding organic ligands (or capping agents) is still relatively less understood. Therefore, more systematic studies on the effect of the surface ligands with controlled structure, functionality, and conformation will help us to establish a greater understanding of the ligands’ role in the observed activity and selectivity of nanocatalysts. In addition, the potential influence of organic ligands on electronic states of metal nanoparticle surfaces or charge transfer phenomena between them would require more in-depth studies.[30] The findings can be further applied to the development of optimized metal nanoparticle catalysts with high regioselectivity, chemoselectivity, and/or stereoselectivity in various organic reactions.
Considering their size, spherical shape, and versatile ligand characteristics, the synthesis of well-designed organic ligand-capped metal nanoparticles will greatly benefit the advancement in enzyme-mimic catalysis by serving as an excellent model system. For example, by introducing different hydrophobic functional groups in the “tail” of hydrocarbon chains of organic ligands, one can adjust non-covalent interactions in the near-surface environment in a manner analogous to changing functional groups in an enzyme binding pocket.[105] Therefore, this approach might allow the investigation of the ability of surface immobilized ligands on precisely tuning catalytic selectivity through non-covalent interactions.[80] The studies would be an important step towards the expansion of the simple model system for non-covalent interaction towards more complex ones that mimics the binding pockets of enzymes. Besides their important roles in biology, catalysis also plays critical roles in environmental applications such as waste decomposition, green catalysis, and micellar catalysis, the continued investigations of small organic ligand-capped metal nanoparticle catalysts will likely have a large impact on the broad field of research in science and technology.
Acknowledgements
This research was funded by grants from the National Institute of General Medical Science (#SC3M089562) and CSULB.
Biographies

Diego J. Gavia, born in Guadalajara, Mexico, obtained his B.S. in both Chemistry and Physics from the University of California, Santa Barbara. He then received his M.S. in organic chemistry from California State University, Long Beach (2013) under the direction of Dr. Young-Seok Shon, where he researched Pd nanoparticle catalysed reactions and Au nanoscale drug delivery.

Young-Seok Shon carried out his doctoral research with T. R. Lee at the University of Houston in Texas, United States, where he received his PhD in 1999. Between 1999 and 2001 he held his postdoctoral position at the University of North Carolina at Chapel Hill (with Royce W. Murray). He began his academic career at Western Kentucky University (2001–2006), before continuing at California State University, Long Beach, where he is currently a professor of Chemistry and Biochemistry. His current main interests are the study and application of unsupported metal nanoparticle catalysts.
References
- 1.Kane J, Ong J, Saraf RF. J. Mater. Chem. 2011;21:16846–16858. [Google Scholar]
- 2.Karmakar S, Kumar S, Rinaldi R, Maruccio G. J. Phys. Conf. Ser. 2011;292:012002. [Google Scholar]
- 3.Slaughter L, Chang W-S, Link S. J. Phys. Chem. Lett. 2011;2:2015–2023. [Google Scholar]
- 4.Bolduc OR, Masson J-F. Anal. Chem. 2011;83:8057–8062. doi: 10.1021/ac2012976. [DOI] [PubMed] [Google Scholar]
- 5.Bunker CE, Smith MJ. J. Mater. Chem. 2011;21:12173–12180. [Google Scholar]
- 6.Pundt A. Adv. Eng. Mater. 2004;6:11–21. [Google Scholar]
- 7.Yamauchi M, Kobayashi H, Kitagawa H. ChemPhysChem. 2009;10:2566–2576. doi: 10.1002/cphc.200900289. [DOI] [PubMed] [Google Scholar]
- 8.Vons VA, Leegwater H, Legerstee WJ, Eijt SWH, Scmidt-Ott A. Int. J. Hydrogen Energy. 2010;35:5479–5489. [Google Scholar]
- 9.Namiki Y, Fuchigami T, Tada N, Kawamura R, Matsunuma S, Kitamoto Y, Nakagawa M. Acc. Chem. Res. 2011;44:1080–1093. doi: 10.1021/ar200011r. [DOI] [PubMed] [Google Scholar]
- 10.Rosarin FS, Mirunalini S. J. Bioanal. Biomed. 2011;3:085–091. [Google Scholar]
- 11.Thakor AS, Jokerst J, Zavaleta C, Massoud TF, Gambhir SS. Nano Lett. 2011;11:4029–4036. doi: 10.1021/nl202559p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy CJ, Gole AM, Stone JW, Sisco PN, Alkilany AM, Goldsmith EC, Baxter SC. Acc. Chem. Res. 2008;41:1721–1730. doi: 10.1021/ar800035u. [DOI] [PubMed] [Google Scholar]
- 13.Mahmoudi M, Lynch I, Ejtehadi MR, Monopoli MP, Bombelli FB, Laurent S. Chem. Rev. 2011;111:5610–5637. doi: 10.1021/cr100440g. [DOI] [PubMed] [Google Scholar]
- 14.Shemetov AA, Nabiev I, Sukhanova A. ACS Nano. 2012;6:4585–4602. doi: 10.1021/nn300415x. [DOI] [PubMed] [Google Scholar]
- 15.Toshima N, Yonezawa T. New J. Chem. 1998;22:1179–1201. [Google Scholar]
- 16.Crooks RM, Zhao M, Sun L, Chechik V, Yeung LK. Acc. Chem. Res. 2001;34:181–190. doi: 10.1021/ar000110a. [DOI] [PubMed] [Google Scholar]
- 17.Moreno-Mañas M, Pleixats R. Acc. Chem. Res. 2003;36:638–643. doi: 10.1021/ar020267y. [DOI] [PubMed] [Google Scholar]
- 18.Dhakshinamoorthy A, Garcia H. Chem. Soc. Rev. 2012;41:5262–5284. doi: 10.1039/c2cs35047e. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Cui X, Shi F, Deng Y. Chem. Rev. 2012;112:2467–2505. doi: 10.1021/cr200260m. [DOI] [PubMed] [Google Scholar]
- 20.Stratakis M, Garcia H. Chem. Rev. 2012;112:4469–4506. doi: 10.1021/cr3000785. [DOI] [PubMed] [Google Scholar]
- 21.Scholten JD, Leal BC, Dupont J. ACS Catal. 2012;2:184–200. [Google Scholar]
- 22.Mikami Y, Dhakshinamoorthy A, Alvaro M, Garcia H. Catal. Sci. Technol. 2013;3:58–69. [Google Scholar]
- 23.Klabunde KJ, Richards RM. Nanoscale Materials in Chemistry. 2nd ed. New Jersey: Wiley, Hoboken; 2009. [Google Scholar]
- 24.Chee SW, Sharma R. Micron. 2012;43:1181–1187. doi: 10.1016/j.micron.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 25.Wu Z, Chen J, Di Q, Zhang M. Catal. Commun. 2012;18:55–59. [Google Scholar]
- 26.Rueping M, Koenigs RM, Borrmann R, Zoller J, Weirich TE, Mayer J. Chem. Mater. 2011;23:2008–2010. [Google Scholar]
- 27.Ghosh Chaudhuri R, Paria S. Chem. Rev. 2012;112:2373–2433. doi: 10.1021/cr100449n. [DOI] [PubMed] [Google Scholar]
- 28.Wegner K, Vinati S, Piseri P, Antonini A, Zelioli A, Barborini E, Ducati C, Milani P. Nanotechnology. 2012;23:185603. doi: 10.1088/0957-4484/23/18/185603. [DOI] [PubMed] [Google Scholar]
- 29.Tsunoyama H, Sakurai H, Negishi Y, Tsukuda T. J. Am. Chem. Soc. 2005;127:9374–9375. doi: 10.1021/ja052161e. [DOI] [PubMed] [Google Scholar]
- 30.Tsunoyama H, Ichikuni N, Sakurai H, Tsukuda T. J. Am. Chem. Soc. 2009;131:7086–7093. doi: 10.1021/ja810045y. [DOI] [PubMed] [Google Scholar]
- 31.Hou W, Dehm NA, Scott RWJ. J. Catal. 2008;253:22–27. [Google Scholar]
- 32.Zhang F, Zhao X, Feng C, Li B, Chen T, Lu W, Lei X, Xu S. ACS Catal. 2011;1:232–237. [Google Scholar]
- 33.Chowdhury B, Bando KK, Bravo-Suárez JJ, Tsubota S, Haruta M. J. Mol. Catal. A. 2012;359:21–27. [Google Scholar]
- 34.Huang J, Lima E, Akita T, Guzman A, Qi C, Takei T, Haruta M. J. Catal. 2011;278:8–15. [Google Scholar]
- 35.Alves L, Ballesteros B, Boronat M, Cabrero-Antonio JR, Concepcion P, Corma A, Correa-Duarte MA, Mendoza E. J. Am. Chem. Soc. 2011;133:10251–10261. doi: 10.1021/ja202862k. [DOI] [PubMed] [Google Scholar]
- 36.Hayakawa K, Yoshimura T, Esumi K. Langmuir. 2003;19:5517–5521. [Google Scholar]
- 37.Wunder S, Polzer F, Lu Y, Mei Y, Ballauff M. J. Phys. Chem. C. 2010;114:8814–8820. [Google Scholar]
- 38.Lizuka Y, Tode T, Takao T, Yatsu K-I, Takeuchi T, Tsubota S, Haruta M. J. Catal. 1999;187:50–58. [Google Scholar]
- 39.Sanchez-Castillo MA, Couto C, Kim WB, Dumesic JA. Angew. Chem. Int. Ed. 2004;43:1140–1142. doi: 10.1002/anie.200353238. Angew. Chem.2004, 116, 1160–1162. [DOI] [PubMed] [Google Scholar]
- 40.Feng J-J, Chen X-F, Shi M, Duan W-L. J. Am. Chem. Soc. 2010;132:5562–5563. doi: 10.1021/ja100606v. [DOI] [PubMed] [Google Scholar]
- 41.Ogura T, Yoshida K, Yanagisawa A, Imamoto T. Org. Lett. 2009;11:2245–2248. doi: 10.1021/ol900533h. [DOI] [PubMed] [Google Scholar]
- 42.Savarin C, Srogl J, Liebeskind LS. Org. Lett. 2000;2:3229–3231. doi: 10.1021/ol000231a. [DOI] [PubMed] [Google Scholar]
- 43.Alphonse F-A, Suzenet F, Keromnes A, Lebret B, Guillaument G. Org. Lett. 2003;5:803–805. doi: 10.1021/ol027453o. [DOI] [PubMed] [Google Scholar]
- 44.Denmark SE, Smith RC. J. Am. Chem. Soc. 2010;132:1243–1245. doi: 10.1021/ja907049y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoo KS, Yoon CH, Jung KW. J. Am. Chem. Soc. 2006;128:16384–16393. doi: 10.1021/ja063710z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J-H, Kim J-W, Shokouhimehr M, Lee Y-S. J. Org. Chem. 2005;70:6714–6720. doi: 10.1021/jo050721m. [DOI] [PubMed] [Google Scholar]
- 47.Mino T, Sato Y, Saito A, Tanaka Y, Saotome H, Sakamoto M, Fujita T. J. Org. Chem. 2005;70:7979–7984. doi: 10.1021/jo051035q. [DOI] [PubMed] [Google Scholar]
- 48.Zhao Y, Zhou Y, Liang L, Yang X, Du F, Zhang H. Org. Lett. 2009;11:555–558. doi: 10.1021/ol802608r. [DOI] [PubMed] [Google Scholar]
- 49.In 2010, the cost of platinum was $87,741 per kg and rhodium was $88,415 per kg, while palladium was $13,632 per kg
- 50.Dotzauer DM, Bhattacharjee S, Wen Y, Bruening ML. Langmuir. 2009;25:1865–1871. doi: 10.1021/la803220z. [DOI] [PubMed] [Google Scholar]
- 51.Dash P, Bond T, Fowler C, Hou W, Coombs N, Scott RWJ. J. Phys. Chem. C. 2009;113:12719–12730. [Google Scholar]
- 52.Lin Y, Watson KA, Fallbach MJ, Ghose S, Smith JG, Delozier DM, Cao W, Crooks RE, Connell JW. ACS Nano. 2009;3:871–884. doi: 10.1021/nn8009097. [DOI] [PubMed] [Google Scholar]
- 53.Wagener P, Schwenke A, Barcikowski S. Langmuir. 2012;28:6132–6140. doi: 10.1021/la204839m. [DOI] [PubMed] [Google Scholar]
- 54.Yu A, Liang Z, Cho J, Caruso F. Nano Lett. 2003;3:1203–1207. [Google Scholar]
- 55.Mudunkotuwa IA, Grassian VH. J. Am. Chem. Soc. 2010;132:14986–14994. doi: 10.1021/ja106091q. [DOI] [PubMed] [Google Scholar]
- 56.Wang J, Qin Y-L, Liu X, Zhang X-B. J. Mater. Chem. 2012;22:12468–12470. [Google Scholar]
- 57.Panigrahi S, Basu S, Praharaj S, Pande S, Jana S, Pal A, Ghosh SK, Pal T. J. Phys. Chem. C. 2007;111:4596–4605. doi: 10.1021/jp065740u. [DOI] [PubMed] [Google Scholar]
- 58.Aldeman JR, Boyd DA, Goodwin DG, Psaltis D. Nano Lett. 2009;9:4417–4423. doi: 10.1021/nl902711n. [DOI] [PubMed] [Google Scholar]
- 59.Mai Y, Eisenberg A. Acc. Chem. Res. 2012;45:1657–1666. doi: 10.1021/ar2003144. [DOI] [PubMed] [Google Scholar]
- 60.Myers VS, Weir MG, Carino EV, Yancey DF, Pande S, Crooks RM. Chem. Sci. 2011;2:1632–1646. [Google Scholar]
- 61.Deraedt C, Astruc D. Acc. Chem. Res. 2014;47:494–503. doi: 10.1021/ar400168s. [DOI] [PubMed] [Google Scholar]
- 62.Zhang H, Cui H. Langmuir. 2009;25:2604–2612. doi: 10.1021/la803347h. [DOI] [PubMed] [Google Scholar]
- 63.Roucoux A. Top. Organomet. Chem. 2005;16:261–279. [Google Scholar]
- 64.Hornstein BJ, Finke RG. Chem. Mater. 2004;16:139–150. [Google Scholar]
- 65.Castro EG, Salvatierra RV, Schreiner WH, Oliveira MM, Zarbin AJG. Chem. Mater. 2010;22:360–370. [Google Scholar]
- 66.Eklund SE, Cliffel DE. Langmuir. 2004;20:6012–6018. doi: 10.1021/la049787n. [DOI] [PubMed] [Google Scholar]
- 67.Liu J, Alvarez J, Ong W, Román E, Kaifer AE. Langmuir. 2001;17:6762–6764. [Google Scholar]
- 68.Sarathy KV, Raina G, Yadav RT, Kulkarni GU, Rao CNR. J. Phys. Chem. B. 1997;101:9876–9880. [Google Scholar]
- 69.Comeau KD, Meli MV. Langmuir. 2012;28:377–381. doi: 10.1021/la202895n. [DOI] [PubMed] [Google Scholar]
- 70.Chen C-F, Tzeng S-D, Chen H-Y, Lin K-J, Gwo S. J. Am. Chem. Soc. 2008;130:824–826. doi: 10.1021/ja0773610. [DOI] [PubMed] [Google Scholar]
- 71.Esumi K, Sarashina S, Yoshimura T. Langmuir. 2004;20:5189–5191. doi: 10.1021/la049415e. [DOI] [PubMed] [Google Scholar]
- 72.Aslam M, Schultz EA, Meade TST, Dravid VP. Cryst. Growth Des. 2007;7:471–475. doi: 10.1021/cg060656p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Isaacs SR, Cutler EC, Park J-S, Lee TR, Shon Y-S. Langmuir. 2005;21:5689–5692. doi: 10.1021/la050656b. [DOI] [PubMed] [Google Scholar]
- 74.Negishi Y, Kurashige W, Kamimura U. Langmuir. 2011;27:12289–12292. doi: 10.1021/la203301p. [DOI] [PubMed] [Google Scholar]
- 75.Horinouchi S, Yamanoi Y, Yonezawa T, Mouri T, Nishihara H. Langmuir. 2006;22:1880–1884. doi: 10.1021/la052657+. [DOI] [PubMed] [Google Scholar]
- 76.Shon Y-S. Metallic Nanoparticles Protected with Monolayers: Synthetic Methods. In: Lyshevski SE, editor. Dekker Encyclopedia of Nanoscience and Nanotechnology. New York: Taylor & Francis; 2014. pp. 2396–2407. [Google Scholar]
- 77.Simard J, Briggs C, Boal AK, Rotello VM. Chem. Commun. 2000:1943–1944. [Google Scholar]
- 78.Cargnello M, Wieder NL, Canton P, Montini T, Giambastini G, Benedetti A, Gorte RJ, Fornasiero P. Chem. Mater. 2011;23:3961–3969. [Google Scholar]
- 79.Pang SH, Schoenbaum CA, Schwartz DK, Medlin JW. ACS Catal. 2014;4:3123–3131. [Google Scholar]
- 80.Kahsar KR, Schwartz DK, Medlin JW. J. Am. Chem. Soc. 2014;136:520–526. doi: 10.1021/ja411973p. [DOI] [PubMed] [Google Scholar]
- 81.Schoenbaum CA, Schwartz DK, Medlin JW. J. Catal. 2013;303:92–99. [Google Scholar]
- 82.Gan LY, Zhang Y-X, Zhao J. J. Phys. Chem. C. 2010;114:996–1003. [Google Scholar]
- 83.Lu F, Ruiz J, Astruc D. Tetrahedron Lett. 2004;45:9443–9445. [Google Scholar]
- 84.Kim J-H, Park J-S, Chung H-W, Boote BW, Lee TR. RSC Adv. 2012;2:3968–3977. [Google Scholar]
- 85.Roth PJ, Theato P. Chem. Mater. 2008;20:1614–1621. [Google Scholar]
- 86.Simard J, Briggs C, Boal AK, Rotello VM. Chem. Commun. 2000:1943–1944. [Google Scholar]
- 87.Alvarez J, Liu J, Roman E, Kaifer AE. Chem. Commun. 2000:1151–1152. [Google Scholar]
- 88.Strimbu L, Liu J, Kaifer AE. Langmuir. 2003;19:483–485. [Google Scholar]
- 89.Gopidas KR, Whitesell JK, Fox MA. Nano Lett. 2003;3:1757–1760. [Google Scholar]
- 90.Sadeghmoghaddam E, Lam C, Choi D, Shon Y-S. J. Mater. Chem. 2011;21:307–312. [Google Scholar]
- 91.Sadeghmoghaddam E, Gaieb K, Shon Y-S. Appl. Catal. A. 2011;405:137–141. [Google Scholar]
- 92.Gavia DJ, Shon Y-S. Langmuir. 2012;28:14502–14508. doi: 10.1021/la302653u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sadeghmoghaddam E, Gu H, Shon Y-S. ACS Catal. 2012;2:1838–1845. doi: 10.1021/cs300270d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gavia DJ, Maung MS, Shon Y-S. ACS Appl. Mater. Interfaces. 2013;5:12432–12440. doi: 10.1021/am4035043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gavia DJ, Koeppen J, Sadeghmoghaddam E, Shon Y-S. RSC Adv. 2013;3:13642–13645. doi: 10.1039/c3ra42119h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ganesan M, Freemantle RG, Obare SO. Chem. Mater. 2007;19:3464–3471. [Google Scholar]
- 97.Tatumi R, Akita T, Fujihara H. Chem. Commun. 2006:3349–3351. doi: 10.1039/b605390b. [DOI] [PubMed] [Google Scholar]
- 98.Wu L, Li B-L, Huang Y-Y, Zhou H-F, He Y-M, Fan Q-H. Org. Lett. 2006;8:3605–3608. doi: 10.1021/ol0614424. [DOI] [PubMed] [Google Scholar]
- 99.Mourdikoudis S, Liz-Marzán LM. Chem. Mater. 2013;25:1465–1476. [Google Scholar]
- 100.Hu B, Ding K, Wu T, Zhou X, Fan H, Jiang T, Wang Q, Han B. Chem. Commun. 2010;46:8552–8554. doi: 10.1039/c0cc03485a. [DOI] [PubMed] [Google Scholar]
- 101.Moreno M, Kissell LN, Jasinski JB, Zamborini FP. ACS Catal. 2012;2:2602–2613. [Google Scholar]
- 102.Mizugaki T, Murata M, Fukubayashi S, Mitsudome T, Jitsukawa K, Kaneda K. Chem. Commun. 2008:241–243. doi: 10.1039/b710860e. [DOI] [PubMed] [Google Scholar]
- 103.Zhou Z-Y, Kang X, Song Y, Chen S. Chem. Commun. 2011;47:6075–6077. doi: 10.1039/c1cc11235j. [DOI] [PubMed] [Google Scholar]
- 104.Bhattacharjee S, Bruening ML. Langmuir. 2008;24:2916–2920. doi: 10.1021/la703055d. [DOI] [PubMed] [Google Scholar]
- 105.Pang SH, Schoenbaum CA, Schwartz DK, Medlin JW. Nat. Commun. 2013;4:92–99. doi: 10.1038/ncomms3448. [DOI] [PubMed] [Google Scholar]