

Abstract:
Major depression and cardiovascular diseases are 2 of the most prevalent health problems in Western society, and an association between them is generally accepted. Although the specific mechanism behind this comorbidity remains to be elucidated, it is clear that it has a complex multifactorial character including a number of neuronal, humoral, immune, and circulatory pathways. Depression-associated cardiovascular abnormalities associate with cardiac dysfunctions and with changes in peripheral resistance. Although cardiac dysfunction in association with depression has been studied in detail, little attention was given to structural and functional changes in resistance arteries responsible for blood pressure control and tissue perfusion. This review discusses recent achievements in studies of depression-associated abnormalities in resistance arteries in humans and animal experimental models. The changes in arterial structure, contractile and relaxing functions associated with depression symptoms are discussed, and the role of these abnormalities for the pathology of major depression and cardiovascular diseases are suggested.
Key Words: resistance arteries, systemic peripheral resistance, major depression, hypertension, tissue perfusion, cardiac output
COMORBIDITY BETWEEN MAJOR DEPRESSION DISORDER AND CARDIOVASCULAR DISEASES
Major depression disorder (MDD) and cardiovascular disease (CVD) are 2 of the most prevalent health problems in Western society, and an association between them is generally accepted.1–3 Several prospective studies showed that the presence of MDD increases the likelihood of developing an adverse cardiovascular event in otherwise healthy patients.4–6 Moreover, patients often experience severe depression after cardiovascular events,2,7 and a significant number of them remain depressed years after the event.8 CVD is characterized by circulatory and neuroendocrine changes and recognized as a significant risk factor for MDD,9 whereas MDD, characterized by depressed mood, anhedonia (a reduced responsiveness to pleasurable stimuli), and hopelessness, is recognized as a risk factor for CVD.4,9–13 The mechanism that underlies this bidirectional association is not fully elucidated. It remains unclear whether MDD and CVD are independently induced by similar factors or these 2 disorders tightly interact with each other. Several factors are common for both disorders, for example, dysfunctions of the hypothalamic–pituitary–adrenal axis and the autonomic nervous system (ANS), activation of the renin–angiotensin–aldosterone system, and increased concentration of proinflammatory cytokines.1,2,14–16 Importantly, all these factors are capable of inducing both MDD and CVD thereby creating a malignant cycle that usually leads to chronic disease.17 The changes in these factors and their role in comorbidity of MDD and CVD are discussed in a number of excellent recent reviews1,2,14–16,18–26 and are out of scope here. It should be noted, however, that the presence of stressful environment (chronic, social, and early life stressors) is a very important variable influencing both MDD and CVD.17 Different forms of stressors can be experienced, but their common feature is that they lead to a feeling of strain and pressure, although this does not necessarily provoke pathological changes in mood state or the circulation. Depression is a complex disorder, in part, because the diagnostic criteria do not distinguish between different behavioral facets of depression, and therefore, there are different subsets of depressive patients. The late-onset depression stands out in term of its clear vascular pathology in the form of ischemic lesions but otherwise cannot be completely segregated from other forms of depression.27 The Diagnostic and Statistical Manual, Fourth Edition (DSM-IV) defines depression by the presence of at least 1 of 2 core symptoms: anhedonia—a decreased ability to experience pleasures and depressive mood lasting minimally 2 weeks.28 Because anhedonia is a cardinal phenomenon of depressive disorders, which can be evoked in rodents,29 the hedonic deficit is considered as a primary feature to be addressed in the preclinical models of depression.30,31
ANIMAL MODELS FOR STUDYING THE COMORBIDITY BETWEEN MDD AND CVD
In combination with findings from human studies, the use of well-validated, reliable, and relevant preclinical models is a useful approach in studies of the mechanisms underlying the comorbidity of MDD and CVD.30,32 Research using animal models allows for a high level of experimental control, as well as exploitation of integrative methods and analyses. The established stress dependence of both diseases can be a useful instrument in addressing the mechanisms involved in their comorbidity.
Several animal models for mood disorders have been developed and validated. These, mainly rodent models of learned helplessness,33 exposure to chronic mild unpredictable stressors (CMS),29,34 behavioral despair,35 and negative social experience,36–39 were used to study behavioral and physiological consequences of MDD. These models have been shown to successfully imitate depression-like symptoms and other changes in behavior, nervous and cardiovascular systems, immune responses, hormonal and oxidative states known to characterize patients with MDD. The construct, face, and predictive validities of these models have been comprehensively presented elsewhere.30,31,34,36,40 The CMS and negative social experience models for depression are considered currently the most realistic models with a very high validity. Importantly, the CMS model also demonstrates the individual variability in responses to chronic stress, which is very characteristic for humans. A subgroup of rats does not develop depression-like symptoms (resilient rats) during exposure to chronic unpredictable stress, whereas others develop depression-like symptoms.34 The resilient subgroup is now considered an excellent control, for more refined modeling of depression.31
Importantly, animal models for CVD are equally useful in studies of comorbidity between MDD and CVD. Thus, in accordance with the suggested comorbidity in humans, rat models for heart diseases (heart failure) show behavioral changes matching well with depression symptoms often seen in postinfarct patients.41 Similarly, rat models of cerebral ischemia42 exhibit features similar to those of depressed individuals. Thus, the bidirectional interactions between MDD and CVD should be addressed in animal models from both a psychological and a cardiovascular point of view providing an integrative understanding of mechanisms behind their comorbidity.
DEPRESSION SYMPTOMS AND SYSTEMIC VASCULAR RESISTANCE: HUMAN STUDIES
Consistent with comorbidity between MDD and CVD, an elevation in systemic arterial pressure, higher sympathetic tone and circulating levels of noradrenaline, and elevations in blood viscosity are often observed in association with increased prevalence in MDD.1,2,14–16 An increased vasoconstriction and/or blood viscosity would, in accordance with Poiseuille's law, lead to an increase in systemic vascular resistance (SVR) in patients with MDD. In fact, the elevated SVR was found in patients with MDD,43–45 although no change in resting mean arterial pressure (MAP) was reported.46 The authors suggested that depressive symptoms were associated with reduced parasympathetic and increased sympathetic tone47 resulting in a broad range of hemodynamic changes.43–45 This is supported by findings of elevated noradrenaline metabolites in plasma in response to stressor stimuli.13,48,49 Thus, although, in depressed patients, additional stress often associate with increased MAP and heart rate,22,43 under resting conditions depressive symptoms associate with reduced cardiac output (CO) and unchanged MAP suggesting elevated SVR.44 In addition to being a hemodynamic background for increased blood pressure, increased vascular resistance can be a critical factor for controlling sufficient organ perfusion even under conditions when MAP is not changed, for example, due to simultaneous reduction in CO. Accordingly, some CVDs, for example, some forms of heart failure, are not associated with elevated blood pressure but have significantly increased myocardial vascular resistance leading to insufficient blood supply to the heart.50
An additional mechanism behind elevation of SVR in MDD patients may be long-term elevation in blood viscosity and decreased plasma volume due to chronic stress exposure.16 Hemoconcentration resulting from increased capillary hydrostatic pressure and decrements in plasma volume has been postulated as an additional potential mechanism linking CVD and MDD.51 This increased filtration of fluid out of the plasma into the interstitial space has been suggested to be either a result of sympathetic nervous system activation and release of catecholamines during psychological stress16 or atrial natriuretic peptide release from the atria and from the hypothalamus.52 Accordingly, antidepressant treatment normalizes stress-induced hemoconcentration and has a positive cardiovascular impact, which correlates with relief of depressive symptoms.53 Enhanced blood viscosity is also likely to be associated with the increased blood coagulation and fibrinolysis, D-dimer, and plasminogen activator inhibitor-1 protein and platelet activation in MDD patients.54
Interestingly, although MDD patients are reported to have normal MAP43–45,49,55,56 and even reduced MAP57 under resting conditions, the treatment with some antidepressants [ie, tricyclic antidepressants (TCAs) and serotonin–noradrenaline reuptake inhibitors (SNRIs)] leads to significant elevation of blood pressure57,58 suggesting that this treatment might be associated with further elevation of SVR.46 In contrast, treatment with specific serotonin reuptake inhibitors (SSRI) does not affect MAP.55 The mechanism behind this controversy is under debate.46
It has been shown that SSRI treatment reduces vagal activity in patients with MDD.55 Because the reduction in vagal activity measured by the reduction in heart rate variability is a known risk factor for the outcome of different forms of heart disease,47 this action of SSRI antidepressants can lead to increased cardiac risk in MDD patients. Interestingly, SSRI treatment reduced the low-frequency heart rate variability,55,59 which has been suggested to reflect the modulation of sympathetic and vagal tone by baroreflex activity,60 whereas the high-frequency heart rate variability, which can be interpreted as a specific measure of parasympathetic control,61 is not affected. This is in contrast with reduced high-frequency heart rate variability seen in untreated depressive patients in comparison with healthy controls and explained by diminished parasympathetic control of the heart rate.45 A reduction of the low-frequency heart rate variability was also seen after treatment with TCAs and SNRIs, and this effect can be explained similarly to SSRIs by normalization of the increased sympathetic activity seen in depressed patients.59 However, in addition to its central effects, which normalizes sympathetic outflow, TCAs and SNRIs may have peripheral effects on noradrenaline transporter activity, which was further contributed to a reduction of sympathetic nervous activity.46,57
Surprisingly, out of the 2 principal possibilities to modulate blood pressure, that is, CO and SVR, only CO received substantial attention.62 Although depression-associated changes in vascular structure and function were studied both in vivo and in vitro,43–45 most of them are focused on large arteries which do not have any significant impact for blood pressure and SVR.63–67 Only few studies were focused on small resistance arteries in MDD patients.68,69 Currently, the main source of knowledge about depression-associated abnormalities in resistance artery structure and function is provided by animal experimental models.
DEPRESSION SYMPTOMS AND SVR: ANIMAL STUDIES
The fact that experimental models for depression are able to mirror cardiovascular abnormalities reported for MDD patients supports the validity of these models.31,32,36,40 Although the animal models for negative social experience36 and disruption of social bonds39 resemble changes in ANS regulation of heart rhythm and its variability,70 other important hemodynamic parameters (ie, MAP and SVR) were not studied in these models. It will be important to know whether CO, SVR, and MAP are altered in association with chronic social stress. Without knowing these parameters, it may be too ambitious to suggest that depression-like symptoms in these models are associated with vascular abnormalities. The available data on heart rate variability suggest only a dysbalance in sympathetic and parasympathetic activities.71 Importantly, sympathetically mediated elevation of heart rate in rats can under certain conditions be associated with a reduction in CO.72 The characteristic changes in heart rate suggest that in rodent models with depression-like symptoms induced by social stress, the hemodynamic abnormalities32,36,39,40 are similar to those seen in other model of MDD, for example, CMS.
Cardiovascular changes reported previously for the CMS model of MDD have been suggested to be associated with sympathetic/parasympathetic dysbalance.72 Interestingly, inhibition of both sympathetic and parasympathetic effects revealed a similar heart rate for CMS and stress-unchallenged groups suggesting that CO was also normalized.73,74 Higher sympathetic nervous activity was directly recorded from CMS rats in comparison with stress-unchallenged control rats.75 This sympathetic nervous system activation increased heart rate and a reduced CO which were accompanied with unchanged72,74,76 or slightly elevated MAP75 suggesting an elevation of SVR.76,77 SVR is derived from MAP and CO. SVR is increased when MAP rises with unchanged or elevated CO and when reduced CO is accompanied with unchanged of reduced MAP. The latter pattern is in accordance with experimental results from the CMS model of MDD.76,77
CMS rats demonstrated cardiovascular hyperreactivity manifested by further elevation of heart rate in response to stressful stimuli, and this was accompanied by increased MAP.73 These findings suggest a stronger reserve for increasing MAP in CMS rats in comparison with stress-unchallenged controls on activation of the sympathetic nervous system by stressors. Thus, inhibition of β-adrenoceptors with propranolol completely abolishes the effect of air jet stress on heart rate but was without any effect on the pressor response.72–74 Although the role of α-adrenergic transmission has not been tested, other neurohumoral factors, such as angiotensin, corticosterone, and vasopressin, could also elevate MAP.73 This is in accordance with clinical studies showing increased noradrenaline concentration and elevated components of the hypothalamic–pituitary–adrenal axis (eg, cortisol and adrenocorticotropic hormone) in response to stressors and orthostatic challenges, suggesting a hyperactive stress response in association with depression.78,79 Interestingly, although the behavioral changes associated with CMS exposure recovered after cessation of the stressors, the reported cardiovascular abnormalities persist over longer time.73,74 This suggests that simple recovery from depressive symptoms was not associated with alleviation of cardiovascular abnormalities. This is also supported by the finding that although SSRI treatment normalizes behavior, there was only partial normalization of CO, whereas MAP remained to be similar with stress-unchallenged controls.72 This is in accordance with clinical data showing an improvement of heart rate and heart rate variability in the treated MDD patients which never returns to normal levels observed in healthy individuals.80
We have previously analyzed the relation between the CMS-induced hemodynamic changes and stress susceptibility of rats and have found that only rats showing depressive behavior have elevated heart rate.76 This suggests, in line with previous studies,72 a reduction in CO only in rats with depression-like symptoms, whereas CO of resilient rats was unaffected.76 The experimental design did not allow for concluding whether these hemodynamic changes are inherited or a consequence of the CMS protocol, but they clearly indicate that elevation of SVR is associated with development of depression-like symptoms and is not a stress response in general.
These SVR changes can be a result of stress-induced activation of ANS, neurohumoral, and inflammatory factors, but there is also a possibility that inherited changes in vascular resistance, especially in the brain circulation, can predispose/perpetuate the depressive state (ie, vascular depression). The increased SVR are very likely associated with changes in small arteries, which provide the major resistance to blood flow through the vascular tree.81 Thus, an increase in SVR should be associated with either structural changes narrowing arterial lumen (inward remodeling) or/and with functional abnormalities leading to pronounced contraction or suppressed vasorelaxation. The possibilities for studying resistance arteries in vivo are limited, but the structure and function of these arteries can be efficiently studies in vitro.81 Surprisingly, until recently, no study addressing the structure and function of resistance arteries in association with depression symptoms was reported.
STRUCTURAL CHANGES IN RESISTANCE ARTERIES IN MDD PATIENTS
In contrast to the comprehensive literature regarding cardiac abnormalities in association with MDD, the information regarding vascular changes is very limited. Moreover, with a few exceptions, only data on large arteries suggesting endothelial dysfunction and atherosclerosis are currently available. Thus, endothelial dysfunction has been shown to occur in large arteries of untreated patients82 and those under antidepressant therapy.83 Moreover, compared with healthy individuals, a greater prevalence and incidence of increased carotid intima-media thickness was reported in MDD patients.67,84–86 Clearly, these results from large arteries cannot explain the development of microvascular pathologies and changes in SVR.
The knowledge about resistance arteries in MDD patients is mostly collected for late-life form of depression characterized by white matter damage due to ischemia.87 Ischemia can be a result of vascular abnormalities including systemic dysfunctions and localized vascular damage. Although these findings strongly suggest that the late-life depression should be called vascular depression, human cerebral vessels are difficult to access, and only postmortem studies of their structure and function are possible. Therefore, studies were performed on arteries from subcutaneous biopsies.64,68,69 These subcutaneous arteries are clearly different from cerebral vessels; however, previous studies show that changes observed in human cerebral arteries in association with several pathologies, for example, hypertension,88 are similar to those seen in subcutaneous arteries. It has been shown that patients with late-life depression have an impaired resistance artery function and abnormal wall structure compared with healthy individuals.64,68,69 Importantly, patients with late-life depression had normal blood pressure with resistance artery wall hypertrophy normally associated with hypertension.81,89 This could be particularly relevant because resistance artery hypertrophy is a prognostic indicator for subsequent incidence of cardiovascular events.88,90 A similar wall hypertrophy, without narrowing of the lumen, has previously been shown in patients with diabetes.91 The observed structural changes can lead to reduced autoregulatory capacity of resistance arteries, as it has been shown previously,92,93 and it can be highly relevant for regulation of cerebral perfusion.94
FUNCTIONAL CHANGES IN RESISTANCE ARTERIES IN MDD PATIENTS
Functional analyses of subcutaneous artery revealed unchanged responses to noradrenaline stimulation,68 although authors have not performed a detailed analysis of the contractile function. Subcutaneous arteries from patients with late-life depression demonstrated highly impaired endothelial function,64,69 which cannot be explained by a dysfunctional NO-dependent pathway only.68 This suggests that other pathways, that is, endothelium-dependent hyperpolarization and cyclooxygenase (COX) products, also play a role in this dysfunction. Further detailed analysis need to be done to fully understand the mechanisms behind this pronounced endothelial dysfunction in association with MDD. This is obviously of importance because it will have crucial significance for tissue perfusion and blood pressure control. To gain a mechanistic insight in resistance artery function, valid and realistic experimental models are needed because arterial biopsies do not allow a large number of participants to be included and several other manipulations are not feasible in clinical studies either.
STRUCTURAL CHANGES IN RESISTANCE ARTERIES IN ANIMAL MODELS OF DEPRESSION
Changes in both vascular structure and/or a balance in contraction and relaxation can be critical for alterations in SVR. Similar to human data,64,68,69 we were unable to find any changes in passive lumen diameter of resistance arteries from rats with depression-like symptoms.76 We have, however, recently found that depression-like symptoms in CMS-exposed rats associate with increased media thickness of cerebral arteries (Matchkov V.; DMScs; 02-2014; unpublished observation), but not of peripheral arteries.77 Although these changes cannot be responsible for elevation of SVR, the elevated media thickness will affect the contractile and myogenic properties of cerebral arteries and thus can play a critical role in regulation of cerebral blood flow.
FUNCTIONAL CHANGES IN RESISTANCE ARTERIES IN ANIMAL MODELS OF DEPRESSION
Responses to Contractile Agonists
Similar to previous observations, made on human subcutaneous arteries from patients with late-life depression,68 we have not previously found any differences in agonist-induced contractile responses between small arteries isolated from nonstressed rats, rats with depression-like symptoms after CMS and rats resilient to CMS.76 These results were similar for resistance arteries from several peripheral vascular beds including small mesenteric and skeletal muscle arteries. Similarly, the unchanged agonist-induced contraction was previously shown for large arteries from CMS mice with depression-like symptoms.95,96 The extent of arterial constriction is determined by the concentration of agonist at the receptors.97,98 It was found that depression-like symptoms in rats are associated with suppressed extraneuronal uptake of monoamines that compensates for an elevated neuronal reuptake76 (Fig. 1). These bimodal changes provide a reciprocal modulation of agonist concentration near their receptors, making overall contractile responses similar to those seen in arteries from nonstressed rats. Moreover, the simultaneous inhibition of neuronal and extraneuronal reuptakes clearly indicates that the intrinsic ability of smooth muscles to contract in response to noradrenaline is unaffected.76 The observed changes were associated with alterations in the expression of proteins responsible for these transporters, that is, neuronal noradrenaline transporter and organic cation transporter 2 (OCT-2), respectively.76 Interestingly, the elevation of neuronal reuptake was remarkably similar to changes seen in association with hypertension.81 Moreover, the elevated neuronal reuptake has been described not only for hypertensive rats99 and patients,100 but also in normotensive young individuals having a family history of hypertension.101 It has been suggested that these reuptake changes might be a consequence of the increased sympathetic tone associated with CMS,72,74,75 that is, a compensation for the procontractile changes associated with the increased sympathetic activity.102,103 In fact, the increased sympathetic activity has been suggested to induce proportional changes in neuronal noradrenaline transporter expression.104–107 Our previous findings demonstrated suppression of extraneuronal catecholamine uptake associated with CMS (Fig. 1).76 Importantly, we have demonstrated that this was due to suppressed expression of the OCT-2, which is known to be sensitive to corticosterone inhibition.108 Taken into account the significant stress-induced elevation of corticosterone in CMS rats (especially in those with depression-like symptoms),34,109,110 it may not be surprising that CMS leads to reduced OCT-2 protein expression.76
FIGURE 1.
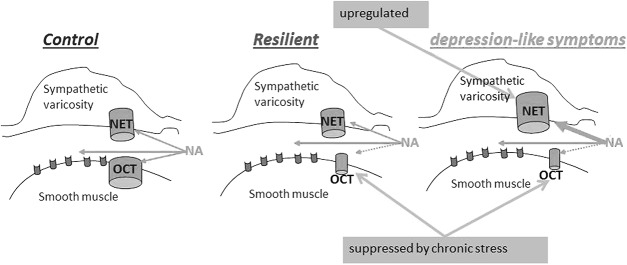
The proposed model for disturbances in catecholamines homeostasis. Based on our experimental results,68 we propose the model for disturbances in catecholamine homeostasis in the arterial wall from resilient rats and rats with depression-like symptoms (anhedonic) after CMS exposure. The chronic stress exposure suppressed corticosterone-sensitive extraneuronal monoamine uptake (OCT) in the arterial wall. Rats with depression-like symptoms upregulate neuronal noradrenaline transporter (NET) which compensates for increased sympathetic activity and reduced extraneuronal uptake. Inhibition of NET with antidepressants such as TCAs and serotonin–noradrenaline transporter inhibitors may lead to a dysbalance in released and recovered noradrenaline and vasoconstriction.
The significance of neuronal and extraneuronal monoamine uptakes varies in different vascular beds, and the neuronal monoamine reuptake is known to play a less significant role in the poorly innervated cerebral circulation. It is therefore possible to suggest that neuronal reuptake will not be able to fully compensate for increased agonist stimulation in these arteries. Additionally, an elevation of the circulating stress hormone, corticosterone, inhibits extraneuronal uptake of monoamines, further increasing the concentration of agonist at the receptors. Altogether, this could make the brain circulation particularly sensitive to circulating monoamine concentration during stress exposure. Changes in cerebral vasculature in a rodent model of MDD have not been reported yet. This indicates the importance of such future comprehensive studies of cerebral artery tone control in depression, especially keeping in mind that this is impossible to obtain from humans. Moreover, it will be important to analyze how antidepressant treatment would affect the monoamine homeostasis and whether there is a difference between antidepressants and responsiveness to antidepressant treatment. In accordance to previously reported prohypertensive effects of some antidepressants, for example, TCAs and SNRIs,57 an elevation of vasoconstrictor activity due to suppressed neuronal monoamine transport might be expected.76
Altered Endothelium-dependent Responses
Endothelial dysfunction is a complex term describing changes in several endothelium-dependent pathways. Three major endothelium-dependent pathways important for regulation of vascular diameter are mediated through NO release, synthesis of prostanoids, and endothelium-dependent hyperpolarisation (EDH).111 The contribution of these factors varies depending on vessel caliber, vascular bed, and health state. Thus, the contribution of NO signaling decreases with vessel diameter, whereas EDH exhibits an inverse pattern being more prominent in small arteries.112 Endothelial dysfunction has been shown previously to be an important factor in pathological conditions, for example, diabetes mellitus, hypertension, and atherosclerosis.111 Endothelial dysfunction is also one of the factors suggested to be involved in cardiovascular abnormalities seen in depressed patients.1,17,21,113
Although MDD patients were previously reported to have endothelial dysfunction,83,114,115 the mechanism behind this association is unknown.113 In human depression-associated endothelial dysfunction was manifested as an impaired forearm flow-mediated vasodilation.66,83,114–118 Endothelium dysfunction in the CMS model of depression has previously been studied only in large conduit arteries of rats119,120 and mice.95,121 We have previously shown the reduced endothelium-dependent relaxation to acetylcholine of resistance arteries isolated from rats with depression-like symptoms,77 but the mechanism behind this attenuation of relaxation seems to be different from those described for large arteries in human and rodents.66,83,95,114–121 In fact, resistance arteries from CMS rats with depression-like symptoms have an increased NO-dependent vasorelaxation pathway due to upregulation of endothelial NO synthase.77 This is in contrast to the results on large arteries,95,119–121 but it is in accordance with previous reports on resistance artery endothelial function in the chronic social stress model of depression where the increase in both the NO-dependent pathway and endothelial NO synthase activity were reported.122–125
It remains unclear whether the observed upregulation of NO signaling in resistance arteries is primary or compensatory to the changes in endothelium-dependent relaxation. It is tempting to speculate that suppressed EDH signaling in arteries from rats with depression-like symptoms is a major defect in endothelial dysfunction in these arteries, whereas an elevated NO response has a compensatory character.77 An attenuation of EDH-like response in resistance arteries from rats with depression-like symptoms is in contrast to the previously reported increase in EDH signaling in aorta from mice exposed to CMS.121 A significant EDH signaling in aorta is surprising by itself,112 but the absence of this signaling pathway in aorta from nonstressed mice suggest that this might be a consequence of chronic stress.121 Moreover, opposite changes in NO and EDH signaling in this study, suggest a counterbalancing interaction between these 2 pathways (Fig. 2). This counterbalancing interaction seems to be the case for both large arteries121 and small resistance arteries.77 Being one of the major pathways for endothelium-dependent relaxation in resistance arteries, EDH is an important factor for control of SVR and tissue perfusion. Accordingly, several cardiovascular pathologies have been reported in association with diminished EDH response including some forms of arterial hypertension and diabetes.126 Moreover, we have recently identified that EDH suppression is a product of reduced expression of 1 of 2 fingerprint proteins for this pathway, the intermediate conductance Ca2+-activated K+ channels, whereas the small conductance Ca2+-activated K+ channels were not affected.77 Importantly, similar pathological changes were previously reported for resistance arteries from type 2 diabetic rats.127
FIGURE 2.
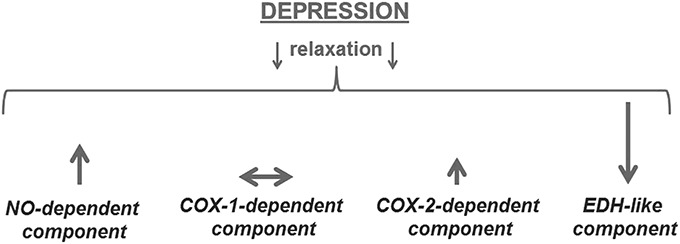
Depression-associated changes in different components of endothelium-dependent relaxation in resistance arteries.70 ↑ indicates elevation of signaling pathway impact; ↓ indicates suppression of signaling pathway impact. The size of arrow suggests the significance of the changes.
The most intriguing part of depression-associated endothelium dysfunction relates to COX-dependent signaling. COX-dependent production of eicosanoids in the vascular wall is characterized by a high degree of divergence and plasticity128,129 as well as by a broad spectrum of eicosanoid receptors with limited ligand specificity.130 It has been shown that changes in both enzymatic pathways and receptors could shift between relaxing and contractile effects of COX activation.128 Our recent study on mesenteric small arteries from rats with depression-like symptoms suggests that the activity of COX-1 and COX-2 enzymes provide 2 distinct endothelium-dependent responses.77 We did not find significant changes in the procontractile activity of the COX-1 pathway in association with depression-like symptoms (Fig. 2). However, arteries from rats with depression-like symptoms had an increased production of relaxing prostanoids through the COX-2 pathway. The mechanism for these changes and the prostanoid identity is unclear, and it cannot be explained by changes in cyclooxygenase (COX) expression.77 Other enzymes involved in prostanoid synthesis cascade could play a role in the changes in COX-2 signaling as well as changes in receptor expression and/or responsiveness.128
The limited knowledge on the detailed mechanism of depression-associated endothelial dysfunction in resistance arteries suggest that the suppression of the EDH pathway cannot be fully compensated by elevation of NO- and COX-2-dependent relaxations (Fig. 2). This could be one of the reasons for elevated SVR and perfusion abnormalities in association with MDD. Experiments with chronic antidepressant treatment could help in understanding the mechanism of association between endothelial dysfunction and psychological factors. It has previously been shown that endothelial dysfunction in aorta could by reversed by fluoxetine treatment.121
PERSPECTIVES
The clinical significance of comorbidity between MDD and CVD is well recognized, but the therapeutic potential of this knowledge is limited because of the unresolved mechanism behind this association.1–3 It is clear now that the association between these 2 diseases has a multilevel character with numerous factors common for both disorders. Cardiovascular abnormalities in association with MDD affect both cardiac function and the peripheral vasculature, and both components of the circulation are important and interdependent. However, although cardiac dysfunctions in association with MDD are intensively studied, little is known about the changes in resistance arteries. It has recently been shown in animal models that depression-like symptoms are associated with changes in catecholamines homeostasis and endothelial dysfunction in resistance arteries.76,77,123 These animal studies grant a unique opportunity to provide mechanistic understanding of cardiovascular abnormalities in association with depression symptoms. Moreover, these studies suggest a mechanism relevant for the unwanted circulatory effects of some antidepressants in depressive patients. This knowledge will be of great value for improving antidepressant treatment accounting for comorbid CVD. All these assumptions are only possible for truly translational studies where abnormalities in experimental models reproduce the pathophysiology of patients. Although the experimental support from resistance arteries of depressive patients is limited, studies of subcutaneous arteries from patients with late-life depression support similarities between resistance arteries in experimental models and in MDD patients. We suggest that the changes in SVR due to abnormalities in resistance arteries could contribute to the comorbidity of the 2 disorders. Further studies of resistance arteries are necessary to achieve a potential therapeutic benefit from this part of the circulation for the comorbidity between MDD and CVD.
(See editorial: Kuala Lumpur Emerging in Vascular Biology by Paul M. Vanhoutte. Journal of Cardiovascular Pharmacology, 2015 65:4;297–298)
Supported by the Lundbeck Foundation (R118-A11567).
The authors report no conflicts of interest.
REFERENCES
- 1.Joynt KE, Whellan DJ, O'Connor CM. Depression and cardiovascular disease: mechanisms of interaction. Biol Psychiatry. 2003;54:248–261. [DOI] [PubMed] [Google Scholar]
- 2.Hare DL, Toukhsati SR, Johansson P, et al. Depression and cardiovascular disease: a clinical review. Eur Heart J. 2013;35:1365–1372. [DOI] [PubMed] [Google Scholar]
- 3.Elderon L, Whooley MA. Depression and cardiovascular disease. Prog Cardiovasc Dis. 2013;55:511–523. [DOI] [PubMed] [Google Scholar]
- 4.Everson SA, Goldberg DE, Kaplan GA, et al. Hopelessness and risk of mortality and incidence of myocardial infarction and cancer. Psychosom Med. 1996;58:113–121. [DOI] [PubMed] [Google Scholar]
- 5.Barefoot JC, Schroll M. Symptoms of depression, acute myocardial infarction, and total mortality in a community sample. Circulation. 1996;93:1976–1980. [DOI] [PubMed] [Google Scholar]
- 6.Anda R, Williamson D, Jones D, et al. Depressed affect, hopelessness, and the risk of ischemic heart disease in a cohort of U.S. adults. Epidemiology. 1993;4:285–294. [DOI] [PubMed] [Google Scholar]
- 7.Cassem NH, Hackett TP. Psychological aspects of myocardial infarction. Med Clin North Am. 1977;61:711–721. [DOI] [PubMed] [Google Scholar]
- 8.Krishnan KR. Broken heart: depression in cardiovascular disease. Dialogues Clin Neurosci. 2003;5:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freedland KE, Rich MW, Skala JA, et al. Prevalence of depression in hospitalized patients with congestive heart failure. Psychosom Med. 2003;65:119–128. [DOI] [PubMed] [Google Scholar]
- 10.Perk J, De BG, Gohlke H, et al. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012): the Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Atherosclerosis. 2012;223:1–68. [DOI] [PubMed] [Google Scholar]
- 11.Frasure-Smith N, Lesperance F, Gravel G, et al. Social support, depression, and mortality during the first year after myocardial infarction. Circulation. 2000;101:1919–1924. [DOI] [PubMed] [Google Scholar]
- 12.Frasure-Smith N, Lesperance F, Talajic M. Depression following myocardial infarction. Impact on 6-month survival. JAMA. 1993;270:1819–1825. [PubMed] [Google Scholar]
- 13.Carney RM, Blumenthal JA, Stein PK, et al. Depression, heart rate variability, and acute myocardial infarction. Circulation. 2001;104:2024–2028. [DOI] [PubMed] [Google Scholar]
- 14.Whooley MA. Depression and cardiovascular disease: healing the broken-hearted. JAMA. 2006;295:2874–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lang UE, Borgwardt S. Molecular mechanisms of depression: perspectives on new treatment strategies. Cell Physiol Biochem. 2013;31:761–777. [DOI] [PubMed] [Google Scholar]
- 16.Lippi G, Montagnana M, Favaloro EJ, et al. Mental depression and cardiovascular disease: a multifaceted, bidirectional association. Semin Thromb Hemost. 2009;35:325–336. [DOI] [PubMed] [Google Scholar]
- 17.Johnson AK, Grippo AJ. Sadness and broken hearts: neurohumoral mechanisms and co-morbidity of ischemic heart disease and psychological depression. J Physiol Pharmacol. 2006;57(suppl 11):5–29. [PubMed] [Google Scholar]
- 18.El-Hage W, Leman S, Camus V, et al. Mechanisms of antidepressant resistance. Front Pharmacol. 2013;4:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith DF, Jakobsen S. Molecular neurobiology of depression: PET findings on the elusive correlation with symptom severity. Front Psychiatry. 2013;4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chopra K, Kumar B, Kuhad A. Pathobiological targets of depression. Expert Opin Ther Targets. 2011;15:379–400. [DOI] [PubMed] [Google Scholar]
- 21.Lett HS, Blumenthal JA, Babyak MA, et al. Depression as a risk factor for coronary artery disease: evidence, mechanisms, and treatment. Psychosom Med. 2004;66:305–315. [DOI] [PubMed] [Google Scholar]
- 22.Kibler JL, Ma M. Depressive symptoms and cardiovascular reactivity to laboratory behavioral stress. Int J Behav Med. 2004;11:81–87. [DOI] [PubMed] [Google Scholar]
- 23.Tafet GE, Bernardini R. Psychoneuroendocrinological links between chronic stress and depression. Prog Neuropsychopharmacol Biol Psychiatry 2003;27:893–903. [DOI] [PubMed] [Google Scholar]
- 24.Wulsin LR, Singal BM. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med. 2003;65:201–210. [DOI] [PubMed] [Google Scholar]
- 25.Rutledge T, Hogan BE. A quantitative review of prospective evidence linking psychological factors with hypertension development. Psychosom Med. 2002;64:758–766. [DOI] [PubMed] [Google Scholar]
- 26.Rugulies R. Depression as a predictor for coronary heart disease. a review and meta-analysis. Am J Prev Med. 2002;23:51–61. [DOI] [PubMed] [Google Scholar]
- 27.Alexopoulos GS, Meyers BS, Young RC, et al. “Vascular depression” hypothesis. Arch Gen Psychiatry. 1997;54:915–922. [DOI] [PubMed] [Google Scholar]
- 28.Hamilton M. Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol. 1967;6:278–296. [DOI] [PubMed] [Google Scholar]
- 29.Willner P. Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology. 2005;52:90–110. [DOI] [PubMed] [Google Scholar]
- 30.Willner P. The validity of animal models of depression. Psychopharmacology (Berl). 1984;83:1–16. [DOI] [PubMed] [Google Scholar]
- 31.Strekalova T, Couch Y, Kholod N, et al. Update in the methodology of the chronic stress paradigm: internal control matters. Behav Brain Funct. 2011;7:7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grippo AJ. Mechanisms underlying altered mood and cardiovascular dysfunction: the value of neurobiological and behavioral research with animal models. Neurosci Biobehav Rev. 2009;33:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maier SF, Watkins LR. Stressor controllability and learned helplessness: the roles of the dorsal raphe nucleus, serotonin, and corticotropin-releasing factor. Neurosci Biobehav Rev. 2005;29:829–841. [DOI] [PubMed] [Google Scholar]
- 34.Wiborg O. Chronic mild stress for modeling anhedonia. Cell Tissue Res. 2013;354:155–169. [DOI] [PubMed] [Google Scholar]
- 35.Cryan JF, Valentino RJ, Lucki I. Assessing substrates underlying the behavioral effects of antidepressants using the modified rat forced swimming test. Neurosci Biobehav Rev. 2005;29:547–569. [DOI] [PubMed] [Google Scholar]
- 36.Grippo AJ, Moffitt JA, Sgoifo A, et al. The integration of depressive behaviors and cardiac dysfunction during an operational measure of depression: investigating the role of negative social experiences in an animal model. Psychosom Med. 2012;74:612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartolomucci A, Palanza P, Costoli T, et al. Chronic psychosocial stress persistently alters autonomic function and physical activity in mice. Physiol Behav. 2003;80:57–67. [DOI] [PubMed] [Google Scholar]
- 38.Sgoifo A, Pozzato C, Costoli T, et al. Cardiac autonomic responses to intermittent social conflict in rats. Physiol Behav. 2001;73:343–349. [DOI] [PubMed] [Google Scholar]
- 39.Grippo AJ, Lamb DG, Carter CS, et al. Social isolation disrupts autonomic regulation of the heart and influences negative affective behaviors. Biol Psychiatry. 2007;62:1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grippo AJ. The utility of animal models in understanding links between psychosocial processes and cardiovascular health. Soc Personal Psychol Compass. 2011;5:164–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schoemaker RG, Smits JF. Behavioral changes following chronic myocardial infarction in rats. Physiol Behav. 1994;56:585–589. [DOI] [PubMed] [Google Scholar]
- 42.Bantsiele GB, Bentue-Ferrer D, Saikali S, et al. Behavioral effects of four antidepressants on an ischemic rat model of emotional disturbances. Behav Brain Res. 2009;201:265–271. [DOI] [PubMed] [Google Scholar]
- 43.Matthews SC, Nelesen RA, Dimsdale JE. Depressive symptoms are associated with increased systemic vascular resistance to stress. Psychosom Med. 2005;67:509–513. [DOI] [PubMed] [Google Scholar]
- 44.Hamer M, Tanaka G, Okamura H, et al. The effects of depressive symptoms on cardiovascular and catecholamine responses to the induction of depressive mood. Biol Psychol. 2007;74:20–25. [DOI] [PubMed] [Google Scholar]
- 45.Ehrenthal JC, Herrmann-Lingen C, Fey M, et al. Altered cardiovascular adaptability in depressed patients without heart disease. World J Biol Psychiatry. 2010;11:586–593. [DOI] [PubMed] [Google Scholar]
- 46.Dawood T, Schlaich M, Brown A, et al. Depression and blood pressure control: all antidepressants are not the same. Hypertension. 2009;54:e1. [DOI] [PubMed] [Google Scholar]
- 47.Stein PK, Kleiger RE. Insights from the study of heart rate variability. Annu Rev Med. 1999;50:249–261. [DOI] [PubMed] [Google Scholar]
- 48.Rechlin T, Weis M, Spitzer A, et al. Are affective disorders associated with alterations of heart rate variability? J Affect Disord. 1994;32:271–275. [DOI] [PubMed] [Google Scholar]
- 49.Guinjoan SM, Bernabo JL, Cardinali DP. Cardiovascular tests of autonomic function and sympathetic skin responses in patients with major depression. J Neurol Neurosurg Psychiatry. 1995;59:299–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bagi Z, Broskova Z, Feher A. Obesity and coronary microvascular disease—implications for adipose tissue-mediated remote inflammatory response. Curr Vasc Pharmacol. 2014;12:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allen MT, Patterson SM. Hemoconcentration and stress: a review of physiological mechanisms and relevance for cardiovascular disease risk. Biol Psychol. 1995;41:1–27. [DOI] [PubMed] [Google Scholar]
- 52.Patterson SM, Matthews KA, Allen MT, et al. Stress-induced hemoconcentration of blood cells and lipids in healthy women during acute psychological stress. Health Psychol. 1995;14:319–324. [DOI] [PubMed] [Google Scholar]
- 53.Wong ML, Dong C, Esposito K, et al. Elevated stress-hemoconcentration in major depression is normalized by antidepressant treatment: secondary analysis from a randomized, double-blind clinical trial and relevance to cardiovascular disease risk. PLoS One. 2008;3:e2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Geiser F, Meier C, Wegener I, et al. Association between anxiety and factors of coagulation and fibrinolysis. Psychother Psychosom. 2008;77:377–383. [DOI] [PubMed] [Google Scholar]
- 55.Dawood T, Lambert EA, Barton DA, et al. Specific serotonin reuptake inhibition in major depressive disorder adversely affects novel markers of cardiac risk. Hypertens Res. 2007;30:285–293. [DOI] [PubMed] [Google Scholar]
- 56.Salomon K, Clift A, Karlsdottir M, et al. Major depressive disorder is associated with attenuated cardiovascular reactivity and impaired recovery among those free of cardiovascular disease. Health Psychol. 2009;28:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Licht CM, de Geus EJ, Seldenrijk A, et al. Depression is associated with decreased blood pressure, but antidepressant use increases the risk for hypertension. Hypertension. 2009;53:631–638. [DOI] [PubMed] [Google Scholar]
- 58.Terhardt J, Lederbogen F, Feuerhack A, et al. Heart rate variability during antidepressant treatment with venlafaxine and mirtazapine. Clin Neuropharmacol. 2013;36:198–202. [DOI] [PubMed] [Google Scholar]
- 59.Licht CM, de Geus EJ, Zitman FG, et al. Association between major depressive disorder and heart rate variability in the Netherlands Study of Depression and Anxiety (NESDA). Arch Gen Psychiatry. 2008;65:1358–1367. [DOI] [PubMed] [Google Scholar]
- 60.Koizumi K, Terui N, Kollai M. Effect of cardiac vagal and sympathetic nerve activity on heart rate in rhythmic fluctuations. J Auton Nerv Syst. 1985;12:251–259. [DOI] [PubMed] [Google Scholar]
- 61.Katona PG, Jih F. Respiratory sinus arrhythmia: noninvasive measure of parasympathetic cardiac control. J Appl Physiol. 1975;39:801–805. [DOI] [PubMed] [Google Scholar]
- 62.Barton DA, Dawood T, Lambert EA, et al. Sympathetic activity in major depressive disorder: identifying those at increased cardiac risk? J Hypertens. 2007;25:2117–2124. [DOI] [PubMed] [Google Scholar]
- 63.Eller NH, Netterstrom B, Gyntelberg F, et al. Work-related psychosocial factors and the development of ischemic heart disease: a systematic review. Cardiol Rev. 2009;17:83–97. [DOI] [PubMed] [Google Scholar]
- 64.Paranthaman R, Greenstein A, Burns AS, et al. Relationship of endothelial function and atherosclerosis to treatment response in late-life depression. Int J Geriatr Psychiatry. 2012;27:967–973. [DOI] [PubMed] [Google Scholar]
- 65.Garcia RG, Zarruk JG, Barrera C, et al. Plasma nitrate levels and flow-mediated vasodilation in untreated major depression. Psychosom Med. 2011;73:344–349. [DOI] [PubMed] [Google Scholar]
- 66.Lavoie KL, Pelletier R, Arsenault A, et al. Association between clinical depression and endothelial function measured by forearm hyperemic reactivity. Psychosom Med. 2010;72:20–26. [DOI] [PubMed] [Google Scholar]
- 67.Eller NH, Netterstrom B, Allerup P. Progression in intima media thickness–the significance of hormonal biomarkers of chronic stress. Psychoneuroendocrinology. 2005;30:715–723. [DOI] [PubMed] [Google Scholar]
- 68.Greenstein AS, Paranthaman R, Burns A, et al. Cerebrovascular damage in late-life depression is associated with structural and functional abnormalities of subcutaneous small arteries. Hypertension. 2010;56:734–740. [DOI] [PubMed] [Google Scholar]
- 69.Paranthaman R, Greenstein AS, Burns AS, et al. Vascular function in older adults with depressive disorder. Biol Psychiatry. 2010;68:133–139. [DOI] [PubMed] [Google Scholar]
- 70.Lahmeyer HW, Bellur SN. Cardiac regulation and depression. J Psychiatr Res. 1987;21:1–6. [DOI] [PubMed] [Google Scholar]
- 71.Sheffield D, Krittayaphong R, Cascio WE, et al. Heart rate variability at rest and during mental stress in patients with coronary artery disease: differences in patients with high and low depression scores. Int J Behav Med. 1998;5:31–47. [DOI] [PubMed] [Google Scholar]
- 72.Grippo AJ, Beltz TG, Weiss RM, et al. The effects of chronic fluoxetine treatment on chronic mild stress-induced cardiovascular changes and anhedonia. Biol Psychiatry. 2006;59:309–316. [DOI] [PubMed] [Google Scholar]
- 73.Grippo AJ, Moffitt JA, Johnson AK. Cardiovascular alterations and autonomic imbalance in an experimental model of depression. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1333–R1341. [DOI] [PubMed] [Google Scholar]
- 74.Grippo AJ, Beltz TG, Johnson AK. Behavioral and cardiovascular changes in the chronic mild stress model of depression. Physiol Behav. 2003;78:703–710. [DOI] [PubMed] [Google Scholar]
- 75.Grippo AJ, Moffitt JA, Johnson AK. Evaluation of baroreceptor reflex function in the chronic mild stress rodent model of depression. Psychosom Med. 2008;70:435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bouzinova EV, Moller-Nielsen N, Boedtkjer DB, et al. Chronic mild stress-induced depression-like symptoms in rats and abnormalities in catecholamine uptake in small arteries. Psychosom Med. 2012;74:278–287. [DOI] [PubMed] [Google Scholar]
- 77.Bouzinova EV, Norregaard R, Boedtkjer DM, et al. Association between endothelial dysfunction and depression-like symptoms in chronic mild stress model of depression. Psychosom Med. 2014;76:268–276. [DOI] [PubMed] [Google Scholar]
- 78.Asnis GM, Halbreich U, Ryan ND, et al. The relationship of the dexamethasone suppression test (1 mg and 2 mg) to basal plasma cortisol levels in endogenous depression. Psychoneuroendocrinology. 1987;12:295–301. [DOI] [PubMed] [Google Scholar]
- 79.Nemeroff CB, Widerlov E, Bissette G, et al. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. 1984;226:1342–1344. [DOI] [PubMed] [Google Scholar]
- 80.Carney RM, Freedland KE, Stein PK, et al. Change in heart rate and heart rate variability during treatment for depression in patients with coronary heart disease. Psychosom Med. 2000;62:639–647. [DOI] [PubMed] [Google Scholar]
- 81.Mulvany MJ, Aalkjaer C. Structure and function of small arteries. Physiol Rev. 1990;70:921–961. [DOI] [PubMed] [Google Scholar]
- 82.Rajagopalan S, Brook R, Rubenfire M, et al. Abnormal brachial artery flow-mediated vasodilation in young adults with major depression. Am J Cardiol. 2001;88:196–198. [DOI] [PubMed] [Google Scholar]
- 83.Broadley AJ, Korszun A, Jones CJ, et al. Arterial endothelial function is impaired in treated depression. Heart. 2002;88:521–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen CS, Chen CC, Kuo YT, et al. Carotid intima-media thickness in late-onset major depressive disorder. Int J Geriatr Psychiatry. 2006;21:36–42. [DOI] [PubMed] [Google Scholar]
- 85.Smith PJ, Blumenthal JA, Babyak MA, et al. Intima-media thickness and age of first depressive episode. Biol Psychol. 2009;80:361–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paranthaman R, Burns AS, Cruickshank JK, et al. Age at onset and vascular pathology in late-life depression. Am J Geriatr Psychiatry. 2012;20:524–532. [DOI] [PubMed] [Google Scholar]
- 87.Baldwin RC. Is vascular depression a distinct sub-type of depressive disorder? A review of causal evidence. Int J Geriatr Psychiatry. 2005;20:1–11. [DOI] [PubMed] [Google Scholar]
- 88.Rizzoni D, De CC, Porteri E, et al. Altered structure of small cerebral arteries in patients with essential hypertension. J Hypertens. 2009;27:838–845. [DOI] [PubMed] [Google Scholar]
- 89.Prewitt RL, Rice DC, Dobrian AD. Adaptation of resistance arteries to increases in pressure. Microcirculation. 2002;9:295–304. [DOI] [PubMed] [Google Scholar]
- 90.Buus NH, Mathiassen ON, Fenger-Gron M, et al. Small artery structure during antihypertensive therapy is an independent predictor of cardiovascular events in essential hypertension. J Hypertens. 2013;31:791–797. [DOI] [PubMed] [Google Scholar]
- 91.Rosei EA, Rizzoni D. Small artery remodelling in diabetes. J Cell Mol Med. 2010;14:1030–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Machkov VV, Vlasova MA, Tarasova OS, et al. Responses to noradrenaline of tail arteries in hypertensive, hypotensive and normotensive rats under different regimens of perfusion: role of the myogenic response. Acta Physiol Scand. 1998;163:331–337. [DOI] [PubMed] [Google Scholar]
- 93.Machkov VV, Tarasova OS, Timin EN, et al. Effect of noradrenaline on tail arteries of SHR and WKY under perfusion at constant flow and constant pressure. Acta Physiol Scand. 1997;161:41–46. [DOI] [PubMed] [Google Scholar]
- 94.Izzard AS, Graham D, Burnham MP, et al. Myogenic and structural properties of cerebral arteries from the stroke-prone spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol. 2003;285:H1489–H1494. [DOI] [PubMed] [Google Scholar]
- 95.Isingrini E, Surget A, Belzung C, et al. Altered aortic vascular reactivity in the unpredictable chronic mild stress model of depression in mice: UCMS causes relaxation impairment to ACh. Physiol Behav. 2011;103:540–546. [DOI] [PubMed] [Google Scholar]
- 96.d'Audiffret AC, Frisbee SJ, Stapleton PA, et al. Depressive behavior and vascular dysfunction: a link between clinical depression and vascular disease? J Appl Physiol. 2010;108:1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mulvany MJ, Hansen OK, Aalkjaer C. Direct evidence that the greater contractility of resistance vessels in spontaneously hypertensive rats is associated with a narrowed lumen, a thickened media, and an increased number of smooth muscle cell layers. Circ Res. 1978;43:854–864. [DOI] [PubMed] [Google Scholar]
- 98.Eisenhofer G. The role of neuronal and extraneuronal plasma membrane transporters in the inactivation of peripheral catecholamines. Pharmacol Ther. 2001;91:35–62. [DOI] [PubMed] [Google Scholar]
- 99.Mulvany MJ, Aalkjaer C, Christensen J. Changes in noradrenaline sensitivity and morphology of arterial resistance vessels during development of high blood pressure in spontaneously hypertensive rats. Hypertension. 1980;2:664–671. [DOI] [PubMed] [Google Scholar]
- 100.Aalkjaer C, Heagerty AM, Petersen KK, et al. Evidence for increased media thickness, increased neuronal amine uptake, and depressed excitation–contraction coupling in isolated resistance vessels from essential hypertensives. Circ Res. 1987;61:181–186. [DOI] [PubMed] [Google Scholar]
- 101.Aalkjaer C, Heagerty AM, Bailey I, et al. Studies of isolated resistance vessels from offspring of essential hypertensive patients. Hypertension. 1987;9(6 pt 2):155–158. [DOI] [PubMed] [Google Scholar]
- 102.Schlaich MP, Lambert E, Kaye DM, et al. Sympathetic augmentation in hypertension: role of nerve firing, norepinephrine reuptake, and angiotensin neuromodulation. Hypertension. 2004;43:169–175. [DOI] [PubMed] [Google Scholar]
- 103.Lambert E, Dawood T, Straznicky N, et al. Association between the sympathetic firing pattern and anxiety level in patients with the metabolic syndrome and elevated blood pressure. J Hypertens. 2010;28:543–550. [DOI] [PubMed] [Google Scholar]
- 104.Brock JA, Yeoh M, McLachlan EM. Enhanced neurally evoked responses and inhibition of norepinephrine reuptake in rat mesenteric arteries after spinal transection. Am J Physiol Heart Circ Physiol. 2006;290:H398–H405. [DOI] [PubMed] [Google Scholar]
- 105.Parrish DC, Gritman K, Van Winkle DM, et al. Postinfarct sympathetic hyperactivity differentially stimulates expression of tyrosine hydroxylase and norepinephrine transporter. Am J Physiol Heart Circ Physiol. 2008;294:H99–H106. [DOI] [PubMed] [Google Scholar]
- 106.Luo M, Hess MC, Fink GD, et al. Differential alterations in sympathetic neurotransmission in mesenteric arteries and veins in DOCA-salt hypertensive rats. Auton Neurosci. 2003;104:47–57. [DOI] [PubMed] [Google Scholar]
- 107.Li W, Knowlton D, Woodward WR, et al. Regulation of noradrenergic function by inflammatory cytokines and depolarization. J Neurochem. 2003;86:774–783. [DOI] [PubMed] [Google Scholar]
- 108.Hayer-Zillgen M, Bruss M, Bonisch H. Expression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2 and hOCT3. Br J Pharmacol. 2002;136:829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Christiansen S, Bouzinova EV, Palme R, et al. Circadian activity of the hypothalamic pituitary adrenal axis is differentially affected in the rat chronic mild stress model of depression. Stress. 2012;15:547–657. [DOI] [PubMed] [Google Scholar]
- 110.Grippo AJ, Francis J, Beltz TG, et al. Neuroendocrine and cytokine profile of chronic mild stress-induced anhedonia. Physiol Behav. 2005;84:697–706. [DOI] [PubMed] [Google Scholar]
- 111.Vanhoutte PM, Shimokawa H, Tang EH, et al. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf). 2009;196:193–222. [DOI] [PubMed] [Google Scholar]
- 112.Shimokawa H, Yasutake H, Fujii K, et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol. 1996;28:703–711. [DOI] [PubMed] [Google Scholar]
- 113.Toda N, Nakanishi-Toda M. How mental stress affects endothelial function. Pflugers Arch. 2011;462:779–794. [DOI] [PubMed] [Google Scholar]
- 114.Wagner JA, Tennen H, Mansoor GA, et al. History of major depressive disorder and endothelial function in postmenopausal women. Psychosom Med. 2006;68:80–86. [DOI] [PubMed] [Google Scholar]
- 115.Sherwood A, Hinderliter AL, Watkins LL, et al. Impaired endothelial function in coronary heart disease patients with depressive symptomatology. J Am Coll Cardiol. 2005;46:656–659. [DOI] [PubMed] [Google Scholar]
- 116.Wagner J, Tennen H, Mansoor G, et al. Endothelial dysfunction and history of recurrent depression in postmenopausal women with type 2 diabetes: a case-control study. J Diabetes Complications. 2009;23:18–24. [DOI] [PubMed] [Google Scholar]
- 117.Broadley AJ, Korszun A, Abdelaal E, et al. Inhibition of cortisol production with metyrapone prevents mental stress-induced endothelial dysfunction and baroreflex impairment. J Am Coll Cardiol. 2005;46:344–350. [DOI] [PubMed] [Google Scholar]
- 118.Broadley AJ, Korszun A, Abdelaal E, et al. Metyrapone improves endothelial dysfunction in patients with treated depression. J Am Coll Cardiol. 2006;48:170–175. [DOI] [PubMed] [Google Scholar]
- 119.Neves VJ, Moura MJ, Almeida BS, et al. Chronic stress, but not hypercaloric diet, impairs vascular function in rats. Stress. 2012;15:138–148. [DOI] [PubMed] [Google Scholar]
- 120.Neves VJ, Moura MJ, Tamascia ML, et al. Proatherosclerotic effects of chronic stress in male rats: altered phenylephrine sensitivity and nitric oxide synthase activity of aorta and circulating lipids. Stress. 2009;12:320–327. [DOI] [PubMed] [Google Scholar]
- 121.Isingrini E, Belzung C, Freslon JL, et al. Fluoxetine effect on aortic nitric oxide-dependent vasorelaxation in the unpredictable chronic mild stress model of depression in mice. Psychosom Med. 2012;74:63–72. [DOI] [PubMed] [Google Scholar]
- 122.Puzserova A, Slezak P, Balis P, et al. Long-term social stress induces nitric oxide-independent endothelial dysfunction in normotensive rats. Stress. 2013;16:331–339. [DOI] [PubMed] [Google Scholar]
- 123.Puzserova A, Bernatova I. Chronic social stress increases nitric oxide-dependent vasorelaxation in normotensive rats. Interdiscip Toxicol. 2010;3:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bernatova I, Puzserova A, Navarova J, et al. Crowding-induced alterations in vascular system of Wistar-Kyoto rats: role of nitric oxide. Physiol Res. 2007;56:667–669. [DOI] [PubMed] [Google Scholar]
- 125.Puzserova A, Csizmadiova Z, Andriantsitohaina R, et al. Vascular effects of red wine polyphenols in chronic stress-exposed Wistar-Kyoto rats. Physiol Res. 2006;55:S39–S47. [DOI] [PubMed] [Google Scholar]
- 126.Grgic I, Kaistha BP, Hoyer J, et al. Endothelial Ca+-activated K+ channels in normal and impaired EDHF-dilator responses–relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol. 2009;157:509–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brondum E, Kold-Petersen H, Simonsen U, et al. NS309 restores EDHF-type relaxation in mesenteric small arteries from type 2 diabetic ZDF rats. Br J Pharmacol. 2010;159:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Feletou M, Huang Y, Vanhoutte PM. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol. 2011;164:894–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. [DOI] [PubMed] [Google Scholar]
- 130.Tsuboi K, Sugimoto Y, Ichikawa A. Prostanoid receptor subtypes. Prostaglandins Other Lipid Mediat. 2002;68-69:535–556. [DOI] [PubMed] [Google Scholar]