

Abstract
A novel clinical study design was used to evaluate the blockade of a selective short-acting μ-opioid agonist (remifentanil) in 24 opioid-experienced subjects. Samidorphan (3-carboxamido-4-hydroxynaltrexone) is a novel opioid modulator with μ-antagonist properties. Objective (pupil diameter) and subjective (visual analog scale) responses to repeated remifentanil and saline infusion challenges were assessed after single oral administration of placebo (day 1) and samidorphan (day 2). Complete blockade persisted with samidorphan for 24 hours for pupil miosis and 48 hours for the drug liking visual analog scale. Samidorphan effects persisted beyond measurable samidorphan exposure (t½ = 7 hours). Samidorphan was associated with complete blockade of remifentanil, and the duration supports daily administration. This study used a novel approach with multiple administrations of remifentanil to successfully demonstrate a durable effect with samidorphan and a rapid and potent blockade of physiological and subjective μ-opioid effects.
Key Words: remifentanil, samidorphan, opioid blockade, pharmacodynamic, pupillometry
μu-opioid antagonists are currently used in the treatment of alcohol and opioid dependence (eg, naltrexone)1 and in opioid overdose (eg, naloxone).2 Samidorphan (3-carboxamido-4-hydroxynaltrexone) is a novel opioid receptor antagonist with high affinity and guanosine-5′-(3-O-thio) triphosphate (GTPγ S) binding activity at the μ-opioid receptor3 and exhibits mixed agonist-antagonist activity at κ and δ receptors.4 In rat studies, oral administration of samidorphan (previously described as RDC-0313 or ALKS 33) was shown to reverse morphine-induced analgesia for more than 4 hours, supporting its ability to block opioid receptors.5,6 Whereas the opioid antagonist action of samidorphan has been demonstrated in animal studies, the onset and duration of μ-receptor blockade in humans are unknown.
Previous clinical studies evaluating the time course of opioid antagonist action have used agonists such as hydromorphone or morphine as pharmacological challenges.7–9 However, such evaluations are limited by the pharmacokinetics and duration of action of the opioid agonist. As plasma concentrations (and effect) of both the agonist and antagonist fluctuate, assessing the onset, magnitude, and duration of opioid antagonism is challenging. With an elimination half-life of approximately 5 minutes, remifentanil can be administered repeatedly and safely over time and could more precisely characterize the time course of antagonist action10 to determine the onset and duration of opioid antagonism, without risk of accumulation. Remifentanil is rapidly metabolized by tissue and plasma nonspecific esterases and therefore does not accumulate significantly in tissue (clearance of 3 L/min).11 Such rapid metabolic clearance, rather than redistribution, results in a rapid offset of action after a bolus injection (5–10 minutes depending on the dose) or an infusion (context-sensitive half-time of approximately 3–4 minutes).11–13
The primary objective of this randomized, partially blind, fixed-order, placebo-controlled study was to characterize the time course and degree of reversal of the subjective (eg, drug liking, high) and physiological (pupil miosis) response to remifentanil following single oral doses of samidorphan in healthy nondependent, opioid-experienced subjects. This population was selected as subjects could apply their prior opioid experience to provide meaningful ratings of subjective opioid effects.14 The repeated administration of remifentanil after dosing with placebo or samidorphan, as well as randomized sequential and blinded administration of remifentanil or saline (ie, placebo for remifentanil), also permitted analysis of the reliability and reproducibility of the objective and subjective measures.
METHODS AND MATERIALS
This was a randomized, partially blind, fixed-order, placebo-controlled study to evaluate μ-opioid receptor antagonism by samidorphan. The study was conducted at Kendle Early Stage, Toronto, Ontario, Canada, in accordance with the International Conference on Harmonization Good Clinical Practice guidelines, Food and Drug Administration regulations governing clinical study conduct, and the Declaration of Helsinki (and its amendments) and was approved by the Institutional Review Board Services (Aurora, Ontario, Canada).
Subjects
All study participants provided written informed consent prior to any study procedures. Eligible subjects were healthy male and female recreational drug users with nontherapeutic experience with opioids, 18 to 55 years of age. Recreational opioid experience was defined as having used opioids for nontherapeutic purposes on at least 10 occasions in the past year and had used opioids at least 3 times in the 12 weeks prior to the screening visit. Subjects were excluded from participation if physically dependent on opioids, as assessed by medical history and Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, criteria, or had ever been in a substance rehabilitation program.
At screening, body mass index of 19 to 30 kg/m2, physical examination, medical history, allergy to opioids or opioid antagonists, vital signs, clinical laboratory assessments, 12-lead elec trocardiogram (ECG), and pregnancy test for females were assessed. Concomitant medications (except acetaminophen, vitamin/mineral supplements, birth control, and hormone replacement) were prohibited during the study. Following the screening visit, eligible subjects entered into a 10-day in-house dosing and assessment period. All subjects underwent a urine drug screen and alcohol breath test at screening and at admission (day −1) to the inpatient phase.
Procedures
All subjects received a single oral dose of placebo (0.01% quinine sulfate; Spectrum Chemicals & Laboratory Products, Gardena, Calif) in the morning of day 1 and were randomized in a double-blind fashion to receive either 10 or 20 mg samidorphan (Alkermes, Inc, Waltham, Mass) in the morning of day 2. Administration of study drug on days 1 and 2 was conducted in a single-blind manner; where subjects were blind to treatment. Both samidorphan and its matching placebo were administered as a solution using an amber dosing syringe. Subjects consumed 200 mL water after each dose.
Remifentanil (Ultiva; Abbott Pharmaceuticals, St-Laurent, Quebec, Canada) was reconstituted with sterile saline at a concentration of 1 mg/mL and further diluted to a concentration of 100 μg/mL. Remifentanil administration (1.0 μg/kg) was accomplished by further dilution on an individualized subject basis such that it was administered via intravenous infusion in a volume of 10 mL over 1 minute. Remifentanil challenges were administered at 0.25, 1, 2, 4, and 8 hours after placebo/samidorphan dosing, with pharmacodynamic and pharmacokinetic assessments conducted at each challenge timepoint. To reduce subject expectancy effects, saline challenges were interspersed between remifentanil challenges at 3 and 5 hours after dose. The remifentanil dose was selected based on safety and pharmacodynamic results,10 whereas the fixed volume of administration allowed for blinded administration of remifentanil or saline at each challenge session. On days 3 to 9, subjects received single daily challenges of remifentanil and saline in a randomized order, each approximately 1 hour apart; these infusions were scheduled to occur at approximately the same time as the samidorphan dosing time as recorded on day 2 (ie, 24, 48, 72, 96, 120, 144, and 168 hours following samidorphan administration).
Pharmacodynamic Assessments
Pharmacodynamic measures, including pupillometry and visual analog scale (VAS) of subjective drug effects, were administered at 2, 5, 15, and 25 minutes after each remifentanil/saline challenge; pupillometry and non–drug-specific VAS were also administered at predose and pre–remifentanil/saline challenge. Subjects rated their subjective state and the effects of each challenge infusion using 100-point “at the moment” VAS. The bipolar VAS of drug liking was used to measure balance of effects, in which 0 = “strong disliking,” 50 = neutral, and 100 = “strong liking.” Unipolar VAS was used to measure positive (high, good effects), negative (bad effects, feeling sick), sedative (sedation), and any subjective effects, with 0 = “definitely not” and 100 = “definitely so.” The VAS scores were captured using proprietary software (Scheduled Measurement System; Kendle Early Stage). Pupil diameter was measured under mesopic lighting conditions using a pupillometer (Neuroptics, Irvine, Calif).
Bioanalysis
Blood samples were collected to measure plasma concentrations of samidorphan at predose and 0.25, 1, 2, 4, 6, and 8 hours post–samidorphan dose and prior to the first daily challenge on days 3 to 9; to help maintain blinding for subjects, plasma samples were collected on day 1 (placebo). Plasma samples were processed using protein precipitation followed by analysis using liquid chromatography with mass spectrometry using a validated method (Battelle Memorial Institute, Columbus, Ohio) consisting of an isocratic mobile phase (10 mM aqueous ammonium acetate (pH 9):acetonitrile [50:50]), a Phenomenex Gemini C18 (5 μM 110 A, 2 × 100 mm) column, and a Sciex API 4000 Mass Spectrometer equipped with a TurboSpray ionization source operating in positive mode. Naltrexone hydrochloride (40 ng/mL) was utilized as an internal standard. Multiple reaction monitoring transitions for samidorphan and naltrexone were 371 > 336 amu and 342 > 324 amu, respectively. The lower and upper limits of quantification for the assay were 0.25 and 100 ng/mL, respectively, utilizing a 100-μL aliquot of plasma. The validated method met acceptance criteria for within- and between-day variability of less than 15%. Plasma concentrations of remifentanil were not analyzed as the pharmacokinetics of remifentanil are well understood, and the observed pharmacodynamic effects were consistent with those previously reported. 11
Safety
Safety was evaluated through assessment of adverse events (AEs), vital signs, and 12-lead ECG and clinical laboratory assessments. Cardiac telemetry was used to monitor heart rate and oxygen saturation continuously for up to 9 hours after dose on days 1 and 2 and up to 2 hours after the first challenge of the day on days 3 to 9.
Pharmacokinetic Analysis
Pharmacokinetic parameters for samidorphan were calculated using noncompartmental techniques. Actual elapsed time from dosing was used to estimate individual plasma pharmacokinetic parameters using Kinetica (version 4.4.1; Thermo Scientific, Waltham, MA).
Pharmacodynamic Analysis
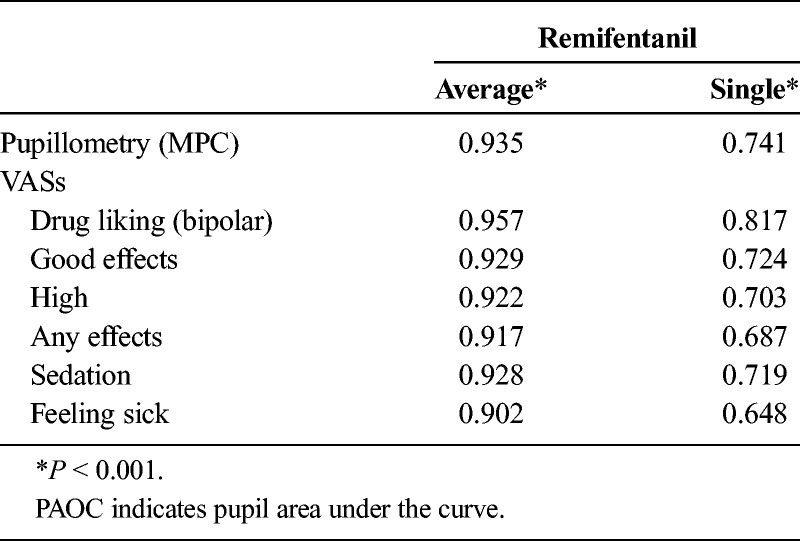
Peak effects were derived for each challenge timepoint. For pupillometry, this was maximum pupil constriction (MPC), calculated as the difference between the prechallenge pupil diameter and the smallest observed postchallenge pupil diameter. For subjective effects, the maximum effect score (Emax) was derived for each VAS. Peak effects (Emax) and MPC at each infusion on day 1 (placebo) were analyzed for test-retest reliability using intraclass correlations (ICCs) for single (individual) and average (group) measures.
To evaluate onset of samidorphan blockade, the end points for pupillometry and VAS for each corresponding challenge timepoint across day 1 (placebo) and day 2 (samidorphan) were compared using a general linear mixed-effects analysis of (co)variance model15,16 that accounts for the correlation present across the repeated measures within each subject, as well as treating the subject-specific deviations from the overall curve as random effects. PROC MIXED was used to conduct these analyses (SAS version 9.2; SAS Institute, Cary, NC). The model included samidorphan dose (10 or 20 mg), day (day 1 or day 2), challenge timepoint (0.25, 1, 2, 3, 4, 5, 8 hours after study drug administration), and their interactions as fixed effects and baseline measurements as a covariate, where applicable; subject was considered a random effect. Pairwise contrasts between day 1 and 2 were performed by matched timepoint separately for each samidorphan dose.
Duration of full blockade was assessed through comparison of pharmacodynamic parameters derived for each remifentanil and saline challenge on days 3 to 9 (ie, 24 to 168 hours after study drug administration). The dependent variable was the paired difference between the remifentanil and saline challenges (same day) on each of the measures. The general linear mixed-effects analysis of (co)variance model included samidorphan dose (10 or 20 mg) and day (days 3–9), and their interaction as fixed effects, with subject as a random effect. The first statistically significant difference indicated the loss of full blockade.
To determine the upper limit of blockade, a supportive analysis was conducted to compare pharmacodynamic parameters collected after onset of blockade (determined per above analysis) to those derived from the first remifentanil exposure following placebo; this remifentanil challenge timepoint was selected since it would be least vulnerable to any potential carryover effects. End of blockade was demonstrated by the first timepoint at which a nonsignificant difference was observed compared with the level of response following the first remifentanil challenge on day 1. From these models, least-squares means and 95% confidence intervals were estimated for the differences. No adjustment for type I error was applied in this exploratory study. Statistical significance was set at α = 0.05.
RESULTS
Eligible subjects (n = 25) were enrolled and randomized to receive placebo on day 1 and samidorphan 10 mg (n = 13) or 20 mg (n = 12) on day 2. Twenty-one subjects received samidorphan and were administered at least 1 remifentanil challenge (1.0 μg/kg, intravenously) and were included in the pharmacodynamic analyses. Mean age of the subjects was 33.2 years (range, 19–53 years), 24 subjects were male, and mean body mass index was 25.8 kg/m2 (range, 20.5–29.4 kg/m2). Three subjects were discontinued on day 1, prior to administration of samidorphan: 1 subject withdrew consent, and 2 subjects were discontinued because of AEs related to remifentanil administration. One subject experienced hyperhidrosis, dizziness, somnolence, and bradycardia, and the other vomiting. Three subjects were discontinued following administration of samidorphan: 1 subject (samidorphan 10 mg) experienced drug withdrawal and was therefore discontinued from the study, and another subject (samidorphan 20 mg) was discontinued because of AEs of decreased oxygen saturation and somnolence. One subject (samidorphan 20 mg) withdrew consent. All subjects reported recreational drug experience with at least 1 type of opioid or morphine derivative drug. The majority (60%) also had experience with various types of stimulants.
Pharmacokinetics
Mean (±SD) peak plasma concentrations were reached at approximately 2 hours postdose following samidorphan 10 mg (19.3 ± 4.9 ng/mL) and 1 hour postdose after 20 mg (47.3 ± 10.1 ng/mL) (Fig. 1). Median Tmax was 2 hours (range, 1–4 hours) with 10 mg and 1 hour (range, 1–2 hours) with 20 mg. Mean area under the time concentration curve for 0 to infinity (AUC0-inf) was 184 ± 58.8 for the 10-mg dose and 381 ± 101.0 for 20-mg dose. Samidorphan concentrations decreased linearly over time, with an observed mean (±SD) elimination half-life of 7.4 ± 1.5 hours for the 10-mg dose and 7.0 ± 1.4 hours for the 20-mg dose. By 72 hours, plasma concentrations were negligible for both the 10- and 20-mg samidorphan doses, and by 96 hours postdose, plasma concentrations were below the limit of quantification for all subjects.
FIGURE 1.
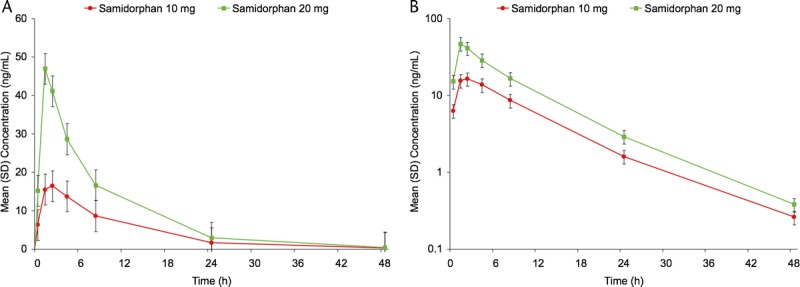
Samidorphan concentration-time profile following administration of 10 or 20 mg on day 2. Linear scale (left; mean ± SD) and log-linear (right; mean ± SD).
Pharmacodynamics
After placebo administration on day 1, each remifentanil challenge induced a rapid and short-lasting decrease in pupil diameter, consistent with its rapid pharmacokinetics, whereas saline administration produced no notable pupil constriction (Fig. 2). A statistically significant difference was observed between MPC following the first remifentanil (0.25 hour after placebo) and saline (3 hours after placebo) challenges, confirming validity of MPC as a measure of remifentanil-induced miosis (P < 0.001).
FIGURE 2.
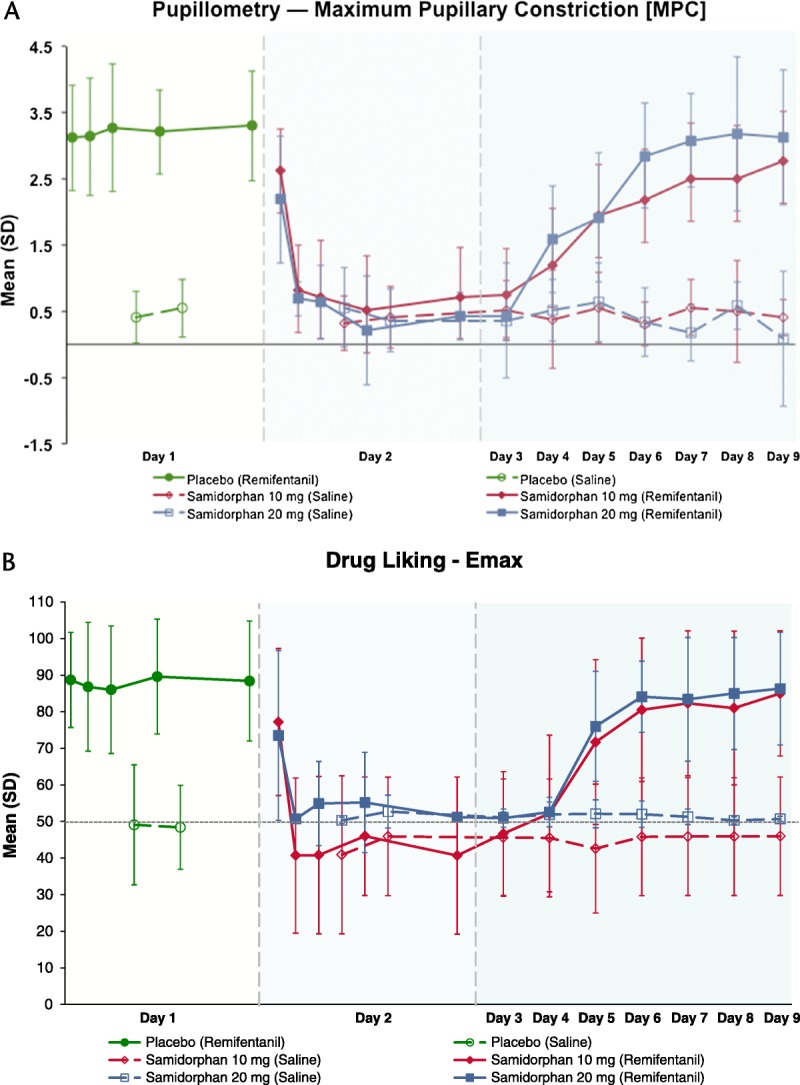
Maximum pupillary constriction and drug liking VAS scores (Emax [mean ± SD]) induced by remifentanil (closed symbols) and saline (open symbols) following administration of placebo (day 1; green) and samidorphan (day 2; 10 mg in red; 20 mg in blue). Following placebo (day 1) or samidorphan (day 2) administration, remifentanil challenges were administered at 0.25, 1, 2, 4, and 8 hours postdose, and saline challenges were administered at 3 and 5 hours postdose. On days 3 through 9, remifentanil and saline challenges were administered in a randomized sequence 1 hour apart, centered around the same time of day as samidorphan administration on day 2. Maximum pupillary constriction was derived from repeated measurements taken at 2, 5, 10, 15, and 25 minutes following each remifentanil challenge.
Analysis of variance results revealed a significant dose × day × time interaction (P < 0.001). Pairwise contrasts showed that MPC was significantly reduced by 1 hour postdose for 10 mg samidorphan and 0.25 hour postdose for 20 mg samidorphan, indicating rapid onset of blockade of remifentanil-induced miosis (Table 1). This reduction remained significant for all remaining contrasts between corresponding timepoints on day 2.
TABLE 1.
Onset of Blockade Following Samidorphan Administration: Comparison of Remifentanil-Induced MPC and Emax of Drug Liking VAS on Day 1 (Placebo) and Day 2 (Samidorphan)
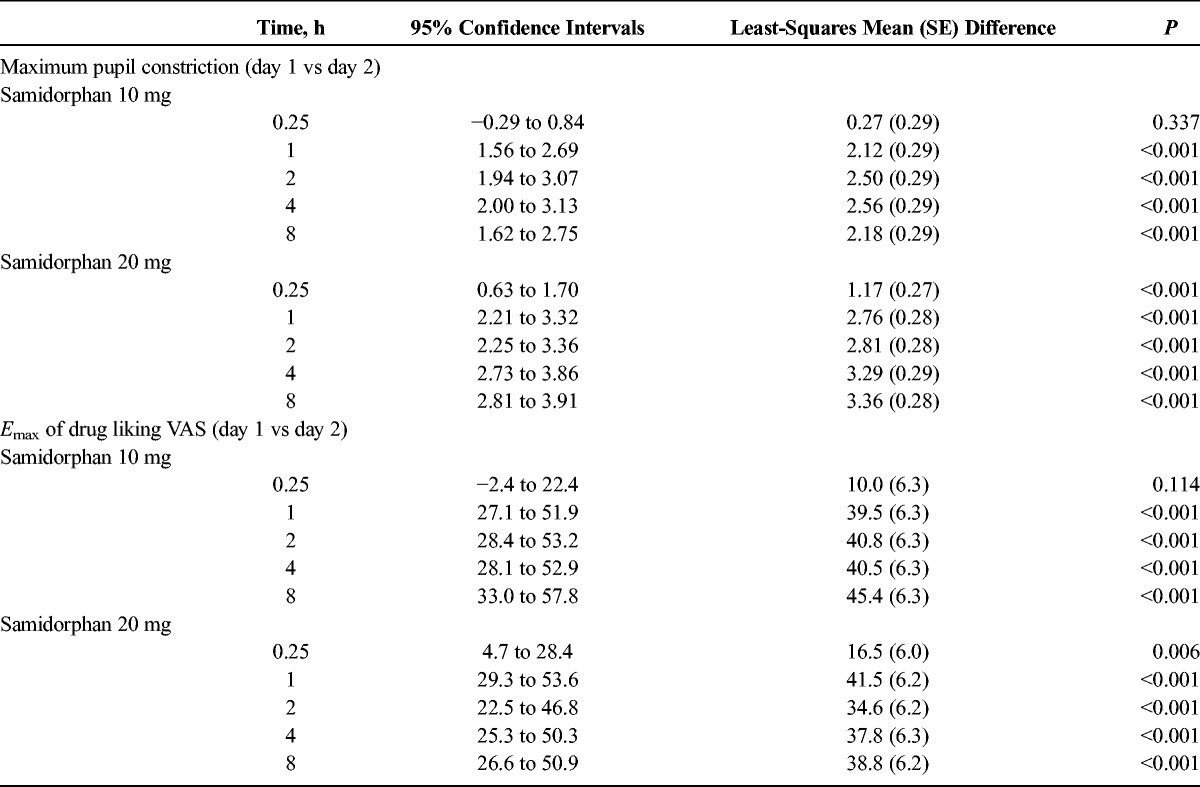
On day 1 following placebo, Emax (>80) of drug liking VAS generally occurred at 2 minutes after each remifentanil challenge, and scores declined rapidly to ∼50 (neutral) by the last VAS assessment at 25 minutes postchallenge. In comparison, the saline challenges resulted in VAS scores remaining around 50, that is, indicating no liking or disliking (data not shown). A statistically significant dose × day × time interaction was observed for drug liking (P < 0.001). Following placebo, a statistically significant difference in Emax between saline (3 hours postdose) and remifentanil (0.25 hour postdose) challenges showed that remifentanil elicited distinct positive effects compared with saline (P < 0.001) (Fig. 2). After samidorphan, the decline in Emax for drug liking was significant by 1 hour postdose for 10 mg and at 0.25 hour postdose for the 20-mg dose (Table 1). This effect persisted for all remaining contrasts between corresponding timepoints on days 1 and 2. No changes in Emax were seen following saline challenges after samidorphan dosing, indicating the samidorphan-induced blockade was specific to the subjective effects of remifentanil (Fig. 2).
The time course for any effects, good effects, high, and sedation VAS was similar to that of drug liking VAS (data not shown). Remifentanil produced significant short-lasting effects following placebo, which were completely blocked after dosing with samidorphan (Emax: dose × day × time, P < 0.001). The onset of samidorphan-induced blockade of any or positive drug effects occurred at 0.25 hour following samidorphan 20 mg, as shown by a statistically significant decrease in Emax. Following samidorphan 10 mg, onset was observed at 0.25 hour (High VAS) or 1 hour (any effects, sedation, and good effects VAS).
Peak negative effects (bad effects, feeling sick VAS) of remifentanil were low and variable following placebo (data not shown). Emax declined significantly (main effect of day, P < 0.001) and were less variable following administration of samidorphan, but no differences were observed between samidorphan and saline.
Analysis of paired differences showed that the full miosis-blocking effects of samidorphan persisted up to 24 hours after samidorphan dosing, but by 48 hours after samidorphan dosing, MPC could be clearly distinguished between remifentanil and saline challenges, independent of samidorphan dose (Table 2). Approximately 50% blockade persisted for 48 to 72 hours following samidorphan administration; remifentanil-induced miosis returned to levels seen at first exposure by 120 hours postdose, independent of dose (Fig. 2).
TABLE 2.
Duration of Blockade Following Samidorphan Administration: Comparison of MPC and Emax for Drug Liking Following Saline and Remifentanil Challenge Infusions on Days 3 to 9*
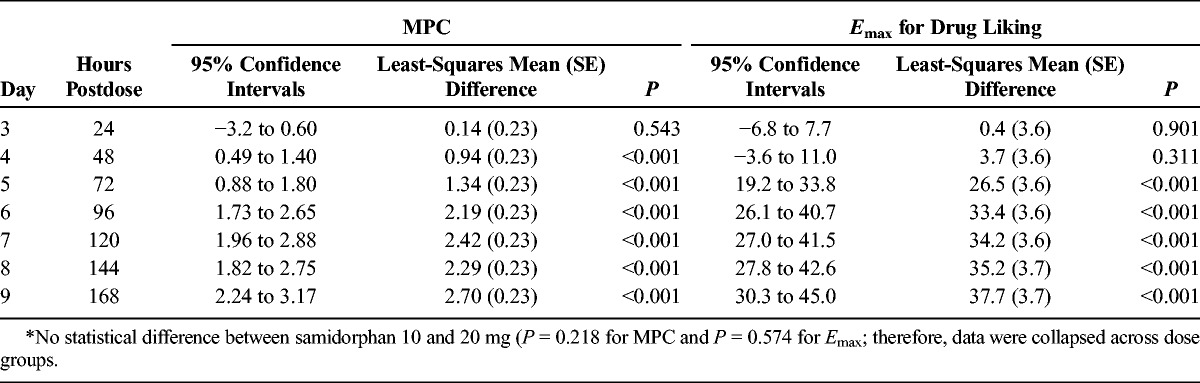
Maximal blockade of Emax drug liking VAS by samidorphan endured up to 48 hours postdose (Table 2). By 72 hours postdose, mean Emax of drug liking VAS for the remifentanil challenge increased to ∼72 and 76 for subjects in the 10- and 20-mg samidorphan groups, respectively (Fig. 2), and could be clearly discriminated from that following the saline challenge (∼43 and 52, respectively; P < 0.001). Full blockade of any, positive, or sedative effects of remifentanil persisted until 24 hours postdose, but became distinguishable from saline beginning by 48 hours postdose (data not shown). No difference between samidorphan doses was observed.
The Emax for drug liking VAS of remifentanil returned to levels seen at first exposure by 96 hours postdose (Fig. 2), whereas Emax for any effects and good effects VAS returned to initial response levels by 72 hours postdose. The feelings of high and sedation following remifentanil challenge did not return to day 1 challenge levels until at least 120 hours after samidorphan (data not shown).
Hysteresis plots of plasma samidorphan concentrations versus MPC and Emax of drug liking VAS obtained at each remifentanil challenge session after samidorphan dosing revealed counterclockwise and clockwise hysteresis loops, respectively (Fig. 3). A minimal lag phase was noted between increases in plasma concentration and observed effect, demonstrating that samidorphan reaches and acts at its effect site rapidly; however, the pharmacodynamic effects lingered beyond detectable plasma samidorphan concentrations, indicating slow receptor dissociation. Visual analysis of the hysteresis plots suggests that maximal samidorphan-induced blockade of remifentanil effects was observed at mean plasma samidorphan concentrations of ∼15 ng/mL, which is consistent with the plasma concentrations at onset of full blockade.
FIGURE 3.
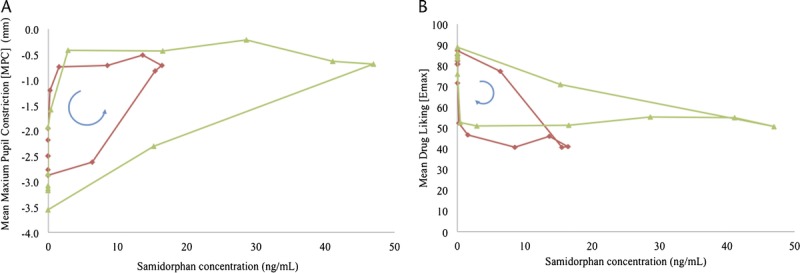
Hysteresis plot of pupillary responses (MPC) and subjective response (Emax on drug liking VAS) to remifentanil versus plasma samidorphan concentrations following administration of 10 and 20 mg samidorphan. The direction of time is presented by the arrow, beginning at a plasma samidorphan concentration of 0. All post–samidorphan remifentanil challenge timepoints on days 2 through 9 (168 hours postdose) are represented. The red line represents samidorphan 10 mg, and the green line represents samidorphan 20 mg.
For Emax of remifentanil-induced subjective effects measured using VAS, both single (individual) and average (group) ICCs were significant (P < 0.001) and ranged between R = 0.69 to 0.82 and R = 0.92 to 0.96, respectively (Table 3). For MPC (pupillometry), single and average measure ICCs were also significant (P < 0.001), with values of R = 0.74 and 0.94, respectively. For saline infusions, ICCs were generally lower on most measures (R = −0.225 to 0.393), but other measures such as drug liking VAS demonstrated significant ICCs (R = 0.702–0.825, P < 0.001).
TABLE 3.
Summary of Test-Retest Reliability: Intraclass Correlations for MPC and Emax of Subjective Effects VAS End Points Following Repeated Remifentanil Infusions
Safety
Adverse events were mostly mild in severity; those judged to be of moderate severity occurred on day 1 (ie, after placebo). The most common AEs (>10% of subjects) related to the remifentanil challenges were euphoric mood, feeling hot, somnolence, dizziness, pruritus, nausea, and nasal discomfort; 1 subject experienced reduced oxygen saturation after a remifentanil infusion. On day 2, following samidorphan, the overall incidence of AEs related to the remifentanil challenge infusions decreased. For example, the incidence of euphoric mood decreased from 100% to 40% and 46% following samidorphan 10 and 20 mg, respectively. The most common AEs (>10% but <30% of subjects) related to samidorphan were dysgeusia (related to the bitter taste of the liquid formulation), somnolence, nausea, dizziness, headache, and anorexia. There were no clinical laboratory, vital signs, or ECG findings related to samidorphan or remifentanil. One subject experienced a serious AE of opioid withdrawal following samidorphan 10 mg. This subject was an experienced opioid user that had a negative urine drug screen prior to samidorphan, but had reported extensive opioid use in the previous 12 months. Withdrawal symptoms were severe at 1 hour postdose, and the subject was admitted to hospital for observation and withdrawal-symptom management. This subject was subsequently discontinued from the study.
DISCUSSION
The objective of the current study was to characterize the magnitude and time course of blockade of μ-opioid receptor antagonism by the new chemical entity, samidorphan. A novel approach using multiple administrations of remifentanil demonstrated the ability of samidorphan to rapidly and potently block physiological and subjective μ-opioid effects and the duration of the blockade.
Remifentanil infusions resulted in significant reductions in pupil diameter and increases in subjective effects expected with opioid administration (ie, drug liking, positive drug effects, and sedation). These effects were short-lasting (<25 minutes) and consistent with the known pharmacologic profile of remifentanil.10 Samidorphan-induced blockade of remifentanil effects could be detected as early as 15 minutes (20 mg) and 1 hour (10 mg) postdose with samidorphan. Maximal blockade was independent of samidorphan dose and persisted for up to 24 or 48 hours postdose depending on the measure. Interestingly, the blockade of remifentanil-induced miosis persisted up to 24 hours postdose, whereas the subjects’ liking of remifentanil was blocked up to 48 hours postdose. This temporal dissociation between antagonism of physiological and subjective effects of opioids is consistent with previous reports using naltrexone.7,17 The return to initial remifentanil response levels was first observed at 72 or 96 hours post–samidorphan dose, well after samidorphan plasma concentrations were no longer detectable.
Considering that full blockade was maintained even at low circulating concentrations of samidorphan, for example, up to 48 hours postdose, the slower return to baseline opioid response is likely related to both plasma concentrations and the affinity of samidorphan for central μ-opioid receptors. This lag was also evident when examining the hysteresis plots, which showed that the onset of effect was more closely related to plasma concentrations than its offset. Similar findings observed that central opioid antagonist activity extended beyond measurable plasma concentrations of naltrexone.7,18 The hysteresis plots and plasma concentrations observed at the onset of blockade suggest that the minimum concentration of samidorphan that could provide maximal blockade of μ-opioid effects was ∼15 ng/mL. Therefore, a dose of samidorphan as low as 10 mg, which results in peak concentrations of ∼19 ng/mL, may be sufficient to fully block potent opioid effects up to 48 hours. In both the MPC and Emax of drug liking VAS hysteresis plots, and for both samidorphan dose levels, the hysteresis loops are “flat” for a period of time where maximal effects are observed even as samidorphan concentrations decline.
The current study had some methodological limitations. Compared with the single-blind fixed-order administration of placebo and samidorphan, a randomized double-blind crossover design would have avoided potential bias and controlled for sequence effects. However, placebo was primarily used as a control to evaluate the onset and magnitude of samidorphan blockade, and continued evaluation following placebo would have resulted in unnecessary exposure of the subjects to remifentanil. Results of pupillometry, an objective measure, argue against the possibility of observer or subject bias because these also showed no samidorphan dose-related differences in magnitude or duration of action. Importantly, samidorphan dose assignment was double-blind to ensure that any potential dose effects would not be biased.
The study results show that samidorphan has potent and persistent opioid antagonist properties in humans, supporting in vitro studies demonstrating that samidorphan has high binding affinity to μ-receptors (Ki = 0.05 nM), even greater than naltrexone (0.11 nM).4 Compared with naltrexone, samidorphan also has a longer elimination half-life following oral administration: ∼7 hours for samidorphan versus ∼4 hours for naltrexone.19 Taken together, the higher relative affinity and longer residence time of samidorphan may enable prolonged opioid receptor antagonism at lower doses; maximal blockade was evident following a 10-mg samidorphan dose.
Responding to remifentanil was highly reproducible, as demonstrated by the very large and statistically significant ICCs. These results support the high test-retest reliability of the measures 20 and also suggest that potential limitations, such as the absence of a predose baseline response to remifentanil and possible development of tolerance with repeated administration of remifentanil at short intervals, are unlikely to have confounded the results. Return to initial responding to remifentanil after samidorphan dosing occurred for all but 1 measure for the 20-mg samidorphan group, further supporting that tolerance did not develop to the effects of remifentanil under the schedule used in the current study.
Previous studies evaluating central activity of an opioid antagonist have used morphine or other opioids.7–9 However, these studies may be limited in demonstrating the time course of onset and duration of antagonist effect due to the longer residence time of these opioid agonists. As shown in the current study, the rapid onset and short half-life of remifentanil make it an attractive alternative to characterize the magnitude and time course profile of an opioid antagonist as it can be administered safely and repeatedly without accumulation in experienced opioid users.
CONCLUSIONS
This study confirmed the mechanism of samidorphan as a high-affinity μ-opioid receptor antagonist using remifentanil as an opioid agonist challenge. With repeated administration of remifentanil, the rapid onset and prolonged duration of blockade by samidorphan were successfully demonstrated using physiological and subjective measures of μ-opioid effects. Based on the current results, remifentanil provides a significant advantage in characterizing the profile of an antagonist compared with longer-acting opioids previously used in studies of this type.
ACKNOWLEDGMENTS
The authors acknowledge the contributions of Liying Zhang (Biostatistics, former affiliation: Kendle Early Stage), Ari Illeperuma (Biostatistics, former affiliation: Alkermes, Inc), Halima Thompson (Project Manager, INC Early Stage), and Keith Krenz (Drug Safety, Alkermes). The authors also acknowledge the editorial assistance of Richard S. Perry, PharmD, in the preparation of this manuscript.
AUTHOR DISCLOSURE INFORMATION
M.J.S. was an employee of INC Research at the time of study conduct and was involved in the study design, data analysis, and interpretation of the data. B.S., E.E., and R.T. are employees of Alkermes, Inc, and were involved in the study design, data analysis, and interpretation of the data. All authors were involved in critical review the manuscript for scientific content, and all authors approved submission of the final version.
Footnotes
This study was funded by Alkermes, Inc, Waltham, MA, and was conducted at INC Research, Toronto, Ontario, Canada (formerly Kendle Early Stage) under the direction of E.M.S.
REFERENCES
- 1. Ray LA, Chin PF, Miotto K. Naltrexone for the treatment of alcoholism: clinical findings, mechanisms of action, and pharmacogenetics. CNS Neurol Disord Drug Targets. 2010; 9: 13– 22. [DOI] [PubMed] [Google Scholar]
- 2. Veilleux JC, Colvin PJ, Anderson J, et al. A review of opioid dependence treatment: pharmacological and psychosocial interventions to treat opioid addiction. Clin Psychol Rev. 2010; 30: 155– 166. [DOI] [PubMed] [Google Scholar]
- 3. Wentland MP, Sun X, Bu Y, et al. Redefining the structure-activity relationships of 2,6-methano-3-benzazocines, part 3: 8-Thiocarboxamido and 8-thioforamido derivatives of cyclazocine. Bioorg Med Chem Let. 2005; 15: 2547– 2551. [DOI] [PubMed] [Google Scholar]
- 4. Wentland MP, Lou R, Lu Q, et al. Synthesis of novel high affinity ligands for opioid receptors. Bioorg Med Chem Let. 2009; 19: 2289– 2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cunningham JI, Dean RL, Todtenkopf MS, et al. Samidorphan attenuates drug-induced increases in extracellular dopamine concentrations and drug self-administration in rats. Presented at the European College of Neuropsychopharmacology Annual Meeting, Berlin, Germany; October 18–21, 2014. [Google Scholar]
- 6. Todtenkopf MS, Dean RL, Turncliff RZ, et al. In vitro and in vivo characterization of novel opioid antagonists. Alcohol Clin Exp Res. 2008; 32: 11A– 271A. [Google Scholar]
- 7. Schuh KJ, Walsh SL, Stitzer ML. Onset, magnitude and duration of opioid blockade produced by buprenorphine and naltrexone in humans. Psychopharmacology (Berl). 1999; 145: 162– 174. [DOI] [PubMed] [Google Scholar]
- 8. Strain EC, Walsh SL, Bigelow GE. Blockade of hydromorphone effects by buprenorphine/naloxone and buprenorphine. Psychopharmacology (Berl). 2002; 159: 161– 166. [DOI] [PubMed] [Google Scholar]
- 9. Yuan CS, Foss JF, O’Connor M, et al. Efficacy of orally administered methylnaltrexone in decreasing subjective effects after intravenous morphine. Drug Alcohol Depend. 1998; 52: 161– 165. [DOI] [PubMed] [Google Scholar]
- 10. Baylon GJ, Kaplan HL, Somer G, et al. Comparative abuse liability of intravenously administered remifentanil and fentanyl. J Clin Psychopharmacol. 2000; 20: 597– 606. [DOI] [PubMed] [Google Scholar]
- 11. Egan TD. Remifentanil pharmacokinetics and pharmacodynamics. A preliminary appraisal. Clin Pharmacokinet. 1995; 29: 80– 94. [DOI] [PubMed] [Google Scholar]
- 12. Minto CF, Schnider TW, Egan TD, et al. Influence of age and gender on the pharmacokinetics and pharmacodynamics of remifentanil. I. Model development. Anesthesiology. 1997; 86: 10– 23. [DOI] [PubMed] [Google Scholar]
- 13. Minto CF, Schnider TW, Shafer SL. Pharmacokinetics and pharmacodynamics of remifentanil. II. Model application. Anesthesiology. 1997; 86: 24– 33. [DOI] [PubMed] [Google Scholar]
- 14. Griffiths RR, Bigelow GE, Ator NA. Principles of initial experimental drug abuse liability assessment in humans. Drug Alcohol Depend. 2003; 70 (suppl 3): S41– S54. [DOI] [PubMed] [Google Scholar]
- 15. Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982; 38: 963– 974. [PubMed] [Google Scholar]
- 16. Verbeke G, Molenberghs G. Linear Mixed Models in Practice: A SAS-Oriented Approach. New York: Springer-Verlag, 1997. [Google Scholar]
- 17. Verebey K, Volavka J, Mulé SJ, et al. Naltrexone: disposition, metabolism, and effects after acute and chronic dosing. Clin Pharmacol Ther. 1976; 20: 315– 328. [DOI] [PubMed] [Google Scholar]
- 18. Lee MC, Wagner HN, Jr, Tanada S, et al. Duration of occupancy of opiate receptors by naltrexone. J Nucl Med. 1988; 29: 1207– 1211. [PubMed] [Google Scholar]
- 19. Dunbar JL, Turncliff RZ, Dong Q, et al. Single- and multiple-dose pharmacokinetics of long-acting injectable naltrexone. Alcohol Clin Exp Res. 2006; 30: 480– 490. [DOI] [PubMed] [Google Scholar]
- 20. Shram MJ, Schoedel KA, Turncliff R, et al. Test-retest reliability of pharmacodynamic measures used in abuse potential testing: evidence from repeated administration of remifentanil, a short-acting potent opioid agonist. Presented at the 2010 International Association for the Study of Pain, August 29 to September 2, 2010, Montreal, Quebec, Canada. [Google Scholar]