

Abstract
Transcription factors can either activate or repress target genes by binding onto short nucleotide sequence motifs in the promoter regions of these genes. Here, we present POBO, a promoter bootstrapping program, for gene expression data. POBO can be used to detect, compare and verify predetermined transcription factor binding site motifs in the promoters of one or two clusters of co-regulated genes. The program calculates the frequencies of the motif in the input promoter sets. A bootstrap analysis detects significantly over- or underrepresented motifs. The output of the program presents bootstrapped results in picture and text formats. The program was tested with published data from transgenic WRKY70 microarray experiments. Intriguingly, motifs recognized by the WRKY transcription factors of plant defense pathways are similarly enriched in both up- and downregulated clusters. POBO analysis suggests slightly modified hypothetical motifs that discriminate between up- and downregulated clusters. In conclusion, POBO allows easy, fast and accurate verification of putative regulatory motifs. The statistical tests implemented in POBO can be useful in eliminating false positives from the results of pattern discovery programs and increasing the reliability of true positives. POBO is freely available from http://ekhidna.biocenter.helsinki.fi:9801/pobo.
INTRODUCTION
Current high-throughput functional genomic techniques allow for the production of massive amounts of gene expression data (1,2). These include large gene expression screens using in situ synthesized oligo-arrays and two-color microarray techniques. These techniques have been used to address a variety of biological questions including expression profiling, comparative genomics and transcriptome analysis.
One of the questions arising from these genomic experiments is how gene regulation is controlled. The two major phenomena that are assumed to be responsible for gene regulation are chromatin remodeling (3,4) and transcription factors (5). Transcription factors can either activate or repress target genes by binding onto short and often specific nucleotide sequences, transcription factor elements or motifs, found in the promoter regions of these genes (5). The binding of transcription factor proteins to DNA and the consequent genome-wide effects on gene regulation are still not well understood. However, transcription factors have been identified to be specific for genes or gene families and typically couple transcription to the physiological needs of the cell (6), which leads to the assumption that co-regulated genes might share a similar control mechanism (5).
Working from the assumption that the cell possesses a common control mechanism for genes of similar function, one would expect to find common control sequences in co-expressed genes. This would allow the cell to initiate the expression of a whole range of genes that are required for a particular function, such as enzymes in a common metabolic pathway, and permit gene expression to be regulated in an efficient, concerted fashion (7). A corollary of the above assumption is that if common control sequences are found in one co-expressed cluster, then these control sequences should be underrepresented in other gene expression clusters. Furthermore, if the complete set of control elements is known, it is not possible that an exactly identical combination of control elements causes upregulation in one set of genes and, at the same time, downregulation in another if all genes are located in a region where the chromatin structure is open.
There are numerous programs available for motif discovery in the promoters of a set of co-expressed genes (8–13). Whereas some programs use probabilistic sequence models (AlignAce, MEME, MotifSampler), others use regular expression pattern matching (PROSPECT, SPEXS and oligo-analysis) (14). The aim of these motif discovery programs is to report motifs that are either overrepresented in the input set or found more often in it than in the background. A motif that exceeds the cutoff limit is reported to the user, while others are not. Problems can arise when searches of every motif use the same background model, because the frequency of motifs in the background cannot always be approximated by probabilistic models (14). This also influences the comparison of motif frequencies, which is the key event in the analysis.
In this paper, we introduce an accurate, fast and easy-to-use application program aimed at biologists. The program, POBO, can be used to validate results from motif discovery programs and to test preliminary hypotheses about motifs that might be responsible for the co-regulation of a given set of genes. In POBO, the frequency of the motif in either one or two clusters of genes is compared with the frequency of the motif in the background data. The background contains the promoter regions of all known genes from a chosen genome. The empirical background model guarantees higher accuracy than approximations derived from nucleotide composition. The search motifs can be arbitrarily complex regular expressions or matrices, and the background model is generated specifically for the search motif in every run. POBO also provides statistical confidence levels for the findings. A microarray experiment typically yields a cluster of upregulated genes and another cluster of downregulated genes, which can be used as input to a novel three-way comparison by POBO. The idea is that the control elements recognized by a repressor or an activator should be more depleted in one cluster than in the background data and, at the same time, more enriched in the other cluster than in the background data. The relative frequencies of the motif in the up- and downregulated sets compared with the background are easily checked in the graphical output of POBO. To summarize, we believe that our background model allows an accurate determination of valid and informative motifs, and second, that the ability to compare two clusters at the same time can be used to increase the explanatory power of the motifs found.
MATERIALS AND METHODS
Principles of the program
The program analyses whether a particular motif is enriched in the user's cluster(s) or not. The user can input either one or two promoter sets, which are then compared against the background data. If there are two input sets, these are also compared with each other.
Currently, there are six different background data sets: Arabidopsis thaliana, Homo sapiens, Mus musculus, Drosophila melanogaster, Caenorhabditis elegans and Saccharomyces cerevisiae. These data sets contain all promoter regions from the currently known genes of each organism. The user's input sets are compared with a background sample, referred to as the ‘background model’, which is generated on the fly from a chosen background data set. The program compares statistics on promoter sequences of the same length both in the background sample and in the user's input sets.
The statistical tests in POBO are based on comparing the total occurrences of the motif in clusters rather than comparing average occurrences of motifs in genes. In the first step, POBO counts the frequency of the motif in each of the promoters in the input sets and in the background data set. In the second step, the significance of the differences between the sets is analyzed using a bootstrap method (15). The bootstrap method generates a number of artificial promoter sets (pseudoclusters) where each pseudocluster is generated by random sampling with replacement. This step is performed for each original input and background data set. For example, if the occurrences of a motif in 10 genes are 0, 1, 1, 1, 2, 2, 2, 3, 4 and 5, then possible values for the occurrences per pseudocluster can vary from 0 to 50 if the size of a pseudocluster is 10. The most frequent value is near the average, which in this case is 21. The number of pseudoclusters and the number of promoters in each pseudocluster are set by the user. The bootstrap method enables a reliable comparison of original data sets of different sizes. The pseudoclusters have equal size and the counts of occurrences are normally distributed. Bootstrapping is followed by an analysis of variances (ANOVA) and an independent t-test, which are performed by using pseudocluster values. Also basic statistical features such as mean, mode, median and standard deviation are calculated.
ANOVA is a technique for dividing the total variation in a response into a number of components attributable to different sources, and analyzing those components (Equations 1–5) (16).
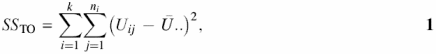


![]()
![]()
Here, the total variation is measured by the sum of squares total (SSTO), which is divided into two components. One of these is the sum of squares error (SSE) and the second is the sum of squares model (SSM). In Equations 1–5 i refers to data sets (input sets 1, 2 or background), k is total number of data sets, ni is the total number of artificial promoter clusters (pseudoclusters) of the i-th data set and j is an index of pseudoclusters. Therefore, Uij refers to the number of motif occurrences in the j-th pseudocluster in the i-th data set. Ū.. is the mean of all observations and Ūi. is the mean of all observations in the i-th data set (16). For example, if there is one pseudocluster with 23 motif occurrences, five pseudoclusters each with 33 occurrences, ten pseudoclusters each with 45 occurrences and so on in the data set, the values of Uij are 23, 33, 33, 33, 33, 33, 45, 45, 45 and so on. The F-value, which is SSM divided by SSE, indicates differences between samples. The larger the F-value, the better the search motif discriminates between the input sets and the background.
To enable direct comparison of any two data sets alone, POBO calculates the t-test value for this purpose. This can be useful when users want to compare the upregulated cluster only with the downregulated cluster to find out whether the difference in means is significant or not. t-tests can also be used when POBO is run with one input set to determine the difference between this input set and the background data set. The statistical test used here is the t-test for independent populations with unknown variances (Equations 6 and 7). In the equations, Ū is the mean of the data set, ![]() is the estimated standard error, S2 is the variance of the data set and n is the size of data set (16):
is the estimated standard error, S2 is the variance of the data set and n is the size of data set (16):
![]()

Background models
Our background data sets contain every known promoter in the genome of six representative organisms. A.thaliana promoters were downloaded from TAIR (the Arabidopsis information resource) (http://www.arabidopsis.org/); the length of the promoters was 3000 bp (17). S.cerevisiae promoters were downloaded from the rsa-tools web page (http://rsat.ulb.ac.be/rsat/); the length of the promoters was 800 bp (18). H.sapiens, M.musculus, D.melanogaster and C.elegans promoters were downloaded from Ensmart (http://www.ensembl.org/EnsMart/) (19) using the following parameters: known genes, one output per gene, 5′ upstream only and 5′ Flank 3000 bp. There were 19 599 genes for H.sapiens, 16 515 for M.musculus, 13 525 for D.melanogaster and 19 873 for C.elegans that passed these criteria.
To achieve the best accuracy, a new background model is generated on the fly depending on the chosen length of the promoters and the motif itself. This guarantees that the frequency of the motif in the input sets is always compared with an appropriate background model. For example, we examined the mean frequencies of all 6mers in the A.thaliana background data (parameters were: length either 1500 or 3000 bp, 1000 pseudoclusters and 50 promoters). In some cases, the average frequencies of 6mers varied with the length of the promoters. Motifs that had approximately the same mean in the 1500 bp run had completely different means in the 3000 bp run. For example, cluster means at 1500 and 3000 bp, respectively, for the CCCTGG motif were 4.96 and 12.32 (ratio 2.48); for CGGCCC, 5.14 and 8.71 (ratio 1.69); and for CGGACG, 5.06 and 10.28 (ratio 2.03). A probabilistic model assuming a uniform distribution of motif occurrences over the whole promoter region yields an expected ratio of 2, and would be inaccurate in some cases. Moreover, the distributions of motifs in the promoter region are non-linear. The real cluster means of the previously described motifs in 2250 bp long promoters are 8.51, 8.21 and 7.55, whereas linear interpolation between the data at 1500 and 3000 bp would have given the following means: 8.63, 6.97 and 7.72. For the above-mentioned motifs, it is impossible to create probabilistic models without severe approximations. In our view, the empirical background model yields the best possible reference for assessing the significance of results.
Inputs and outputs
There are only a few input parameters to set before running POBO. First, the user has to provide the sequences of the promoter set or sets (upstream sequences of co-regulated genes) in FASTA format. The complementary strand will be generated automatically by the program. Other parameters that have to be selected are the length of promoters to include in the analysis, the number of pseudoclusters to be generated, the number of promoters in a pseudocluster and the query motif to be searched for as a consensus string or in matrix form. When using the matrix form, a threshold score for acceptable hits must be provided also (Figure 1). Consensus strings can be selected from a list of known motifs. The web server includes a list of known motifs from TRANSFAC public version 6.0 (all matrix consensus sequences) and the plant database PLACE (20,21). It is also possible to search for the user's own consensus string or matrix-form motifs using POBO. The syntax for specifying a consensus string motif uses ‘[]’ to indicate alternative nucleotides and ‘{}’ to mark the number of repeats. For example, AG[CT]GA corresponds to AGCGA or AGTGA, AG[CT]{2}GA corresponds to AGCCGA, AGCTGA, AGTCGA or AGTTGA, and AG[ACGT]{0,2}GA corresponds to AGGA, AGNGA or AGNNGA. The search is performed using Perl regular expression matching with linear reading of the sequence. The syntax for specifying matrix presentations of motifs uses five rows, where the first row stands for the name of matrix and each of the remaining four rows for one nucleotide. Matrices can have a variable number of columns, presenting the positions of each nucleotide. Motifs input as matrices are searched for by summing the score value from the matrix for each nucleotide position in the data sets and by comparing this score value with the threshold value (22). In both cases, overlapping motifs are counted only once. For example, the motif TATA is found once in the sequence CCCTATATACCC. This makes sense biologically, since a transcription factor recognizing the TATA motif could only bind to the above segment one at a time.
Figure 1.

POBO input interface.
As output, POBO draws a PNG-formatted picture of the distributions of the motifs and writes the results of statistical analyses into the web pages. A random motif should yield overlapping distributions (Figure 2A), while a ‘good’ motif separates up- and downregulated clusters from the background (Figure 2B). POBO also writes a tab-delimited file from the results that can, for example, be uploaded into Excel. This file contains the bootstrap results (clusters sum and occurrence) and positions where each motif was found in the input sets.
Figure 2.


POBO output interface. (A) With a ‘bad’ motif, when the sample distribution is uniform. (B) With a ‘good’ motif, when the sample distribution is spread.
Availability and running the program
POBO is written in the Perl language. A MySQL (http://www.mysql.com/) database is used to store the background promoters. The output graph is drawn with the GNUPLOT program (http://www.gnuplot.info/). POBO runs successfully on the LINUX operating system. The server has been tested with the Microsoft Explorer (6.0), Netscape (7.1) and Opera (7.11) browsers without problems. For the server version, the maximum input size for clusters is limited to 62 000 characters in each set. Currently two 60 000-character promoter sets (20 promoters, length 3000 bp) can be analyzed, when using consensus string motifs, in ∼30 s (parameters: length 3000, pseudoclusters 1000 and number of promoters in a pseudocluster 20). With matrix presentations, the performance time depends on the size of the matrix. The web server, a tutorial and source codes of the web version and a standalone program are available from the group's web site (http://ekhidna.biocenter.helsinki.fi:9801/pobo).
RESULTS
We illustrate the functionality of POBO by reanalyzing data sets from the literature. WRKY is a transcription factor superfamily in A.thaliana that binds to T{0,2}TGAC[CT] or TTGACA-motifs (23,24). WRKY factors have been implicated in plant defense, where they trigger the expression of defense-related genes. WRKY factors are also involved in plant senescence and response to various environmental stresses (25). It was reported that when the WRKY70 transcription factor, a member of the WRKY superfamily, was continuously overexpressed in the plant, a set of defense-related genes was either up- or downregulated (25). We performed POBO analysis for different motifs for the genes in group 1, which contains 24 upregulated genes, and in group 4, which contains 10 downregulated genes (25). The results are summarized in Table 1. The motif TTGAC[CT] was enriched in the upregulated cluster, but this motif was also enriched in the downregulated cluster. If we assume that WRKY70 has equal affinity for the TTGAC[CT] motif in either set and acts alone, we cannot explain its frequent occurrence in the promoters of downregulated genes. The second motif, TTGACA, was depleted in the downregulated cluster but found in the background almost as often as in the upregulated cluster. Thus, this motif alone cannot be responsible for upregulation. We conclude that neither of the previously determined motifs is able to explain the expression pattern by itself. Using POBO, we discovered two hypothetical motifs that suggest a possible solution to this puzzle. These motifs fulfilled our criteria for ‘good’ motifs: their frequencies in the up- and downregulated clusters peak on opposite sides of the background, and they are present in most promoters of the target genes of either activation or repression. The TTGAC[AC]A motif is enriched in the downregulated cluster, and TTGAC[AC][CGT] is enriched in the upregulated cluster. The latter motif has the largest F-value (Table 1). It might be possible that the adenine at the last position of the TTGAC[AC]A motif is able to block the binding or working of WRKY70, or that there is competition between other unknown transcription factors and WRKY70 on this side. The ‘functional’ motif TTGAC[AC][CGT] does not contain this additional adenine. It is also possible that other, unknown repressors or activators bind into promoters elsewhere and thus produce the repression or activation of the target genes. In this case, however, these motifs should be found enriched in only one of the clusters.
Table 1. Promoter analysis of differentially expressed defense-related genes in WRKY70 transgenic plants.
| Motif | Background |
Upregulated cluster |
Downregulated cluster |
F | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Occ | Avg | Stdev | Occ | Avg | Stdev | Occ | Avg | Stdev | ||
| TTTGAC[CT] | 13 781 | 10.72 | 3.37 | 13 | 10.49 | 3.05 | 6 | 12.03 | 2.75 | 73.20 |
| TGAC | 28 073 | 131.40 | 12.49 | 24 | 151.86 | 12.12 | 10 | 146.03 | 10.50 | 806.73 |
| TTGAC[AC] | 23 170 | 27.26 | 5.43 | 23 | 31.98 | 6.12 | 9 | 22.41 | 3.12 | 897.09 |
| TTGACA | 18 860 | 16.99 | 4.20 | 16 | 20.06 | 5.66 | 6 | 10.43 | 2.42 | 1307.79 |
| TTGAC[CT] | 22 721 | 26.59 | 5.64 | 21 | 41.30 | 8.17 | 9 | 34.64 | 4.91 | 1327.08 |
| TGAC[CT] | 27 594 | 65.61 | 8.90 | 24 | 87.97 | 9.96 | 10 | 79.41 | 9.33 | 1439.90 |
| TTGAC[ACT] | 26 243 | 43.94 | 7.39 | 24 | 61.31 | 9.01 | 9 | 45.24 | 5.62 | 1678.35 |
| TTGAC[AC]A | 14 413 | 10.82 | 3.44 | 8 | 6.92 | 3.00 | 7 | 15.08 | 2.91 | 1709.38 |
| TTGAC | 26 904 | 51.08 | 7.85 | 24 | 76.70 | 9.73 | 10 | 57.15 | 5.10 | 2947.19 |
| TTGAC[AC][CGT] | 18 094 | 16.10 | 4.18 | 19 | 25.16 | 5.10 | 3 | 7.49 | 3.04 | 4443.95 |
The background contains all 28 088 genes from A.thaliana, the upregulated cluster contains 24 genes and the downregulated cluster contains 10 genes (25). All analysed promoter sequences were 1500 bp long and POBO was run with the following parameters: number of pseudoclusters 1000, number of promoters in a pseudocluster 15 and length of the background promoters 1500 bp. Occ is the number of promoters containing the given motif, Avg is the mean number of motifs in a cluster, Stdev is standard deviation of the mean and F is the F-value from ANOVA.
DISCUSSION
The principal aim was to create an easy-to-use computer program that would aid in the quantification and confirmation of predetermined motifs in co-regulated gene clusters.
POBO offers flexible pattern matching. Unlike most current programs, POBO can also find and quantify motifs with a variable number of wildcards in the middle—for example, TGAN{1,2}TGA corresponding to TGANTGA or TGANNTGA—or find motifs combined using wildcards, for example the SP1 motif followed by 0–100 wildcards followed by the TATA box.
POBO can also be used to jointly analyze two user-determined promoter sets. This comparison allows users to analyze whether a search motif discriminates up- and downregulated clusters from each other and the background model. Our idea is that control elements should not be found in two oppositely regulated promoter sets as strongly if the expression is caused by these elements. To the best of our knowledge, the ability to perform these three-way comparisons is unique to POBO. The input promoter sets are not restricted to the up- and downregulated genes from a single microarray experiment. The three-way comparison can be applied, for example, to test whether functionally related gene clusters from two species respond to the same motif.
The empirical background model is as accurate and reliable as the current gene prediction programs are. It is free from the approximation errors of probabilistic models, because it is not approximated, and it works optimally for every motif that is searched for. The current background data sets were selected for versatile use. For example, we believe that most plant researchers are able to use A.thaliana background data, and M.musculus can also be generalized for other species such as Rattus norvigus. In the future, new and more specific background data can be added to the web server. Users of the standalone version can create their own background data sets. For example, chromatin structure might be involved in tissue-specific gene expression clusters and the background model should ideally include only that subset of the genome which is open for transcription.
Currently, POBO is limited to lists of known transcription factor binding sites or user-specified motifs. We are working on combining the statistical tests used by POBO with a systematic screening of motif space in order to discover and evaluate new and putative motifs.
We believe that POBO will prove particularly valuable for biologists who wish to examine sets of co-expressed or functionally related genes easily without the need to optimize numerous parameters, and for those who wish quickly to test their preliminary hypothesis about motifs. It also provides flexible pattern matching and a ‘three-way comparison’ enabling the comparison of up- and downregulated clusters at the same time.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Günter Brader, Andreas Heger, Swapan Mallick, Christopher Wilton and other group members for helpful discussion and for providing data. This work was supported by a grant from the Ministry of Education to M.K.
REFERENCES
- 1.Schena M., Shalon,D., Davis,R.W. and Brown,P.O. (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science, 270, 467–470. [DOI] [PubMed] [Google Scholar]
- 2.Lipshutz R.J., Fodor,S.P.A., Gingeras,T.R. and Lockhart,D.J. (1999) High density synthetic oligonucleotide arrays. Nature Genet., 21, 20–24. [DOI] [PubMed] [Google Scholar]
- 3.Meyer P. (2001) Chromatin remodeling. Curr. Opin. Plant Biol., 4, 457–462. [DOI] [PubMed] [Google Scholar]
- 4.Meyer P. (2000) Transcriptional transgene silencing and chromatin components. Plant Mol. Biol., 43, 221–234. [DOI] [PubMed] [Google Scholar]
- 5.Ping Q. (2003) Recent advances in computational promoter analysis in understanding the transcriptional regulatory network. Biochem. Biophys. Res. Commu., 309, 495–501. [DOI] [PubMed] [Google Scholar]
- 6.Kornberg R.D. (1999) Eukaryotic transcriptional control. Trends Genetics, 15, 46–49. [PubMed] [Google Scholar]
- 7.Boardman P.E., Oliver,S.G. and Hubbard,S.J. (2003) SiteSeer: visualisation and analysis of transcription factor binding sites in nucleotide sequences. Nucleic Acids Res., 31, 3572–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thijs G., Lescot,M., Marchal,K., Rombauts,S., De Moor,B., Rouzé,P. and Moreau Y. (2001) A higher order background model improves the detection of regulatory elements by Gibbs Sampling. Bioinformatics, 17, 1113–1122. [DOI] [PubMed] [Google Scholar]
- 9.Hughes J.D., Estep,P.W., Tavazoie,S. and Church,G.M. (2000) Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol., 296, 1205–1214. [DOI] [PubMed] [Google Scholar]
- 10.Bailey T.L. and Elkan,C. (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. In Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology, AAAI Press, Menlo Park, CA, pp. 28–36. [PubMed] [Google Scholar]
- 11.Fujibuchi W., Anderson,J.S. and Landsman,D. (2001) PROSPECT improves cis-acting regulatory element prediction by integrating expression profile data with consensus pattern searches. Nucleic Acids Res., 29, 3988–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vilo J. (2002) Pattern discovery from biosequences. PhD Thesis, Department of Computer Science, University of Helsinki, Finland. Series of Publications A, Report A-2002-3 Helsinki.
- 13.van Helden J., André,B. and Collado-Vides,J. (1998) Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J. Mol. Biol., 281, 827–842. [DOI] [PubMed] [Google Scholar]
- 14.Kreps J., Budwoth,P., Goff,S. and Wang,R. (2003) Identification of putative plant cold responsive regulatory elements by gene expression profiling and pattern enumeration algorithm. Plant Biotechnol. J., 1, 345–352. [DOI] [PubMed] [Google Scholar]
- 15.Efron B. and Tibshirani,R.J. (1993) An Introduction to the BootStrap. Chapman & Hall/CRC, New York. [Google Scholar]
- 16.Petruccelli J.D., Nandram,B. and Chen,M. (1999) Applied Statistics for Engineers and Scientists. Prentice-Hall inc, Upper Saddle River, NJ. [Google Scholar]
- 17.Huala E., Dickerman,A.W., Garcia-Hernandez,M., Weems,D., Reiser,L., LaFond,F., Hanley,D., Kiphart,D., Zhuang,M., Huang,W. et al. (2001) The Arabidopsis Information Resource (TAIR): a comprehensive database and web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Res., 29, 102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van helden J. (2003) Regulatory sequence analysis tools. Nucleic Acids Res., 31, 3593–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbard T., Barker,D., Birney,E., Cameron,G., Chen,Y., Clark,L., Cox,T., Cuff,J., Curwen,V., Down,T. et al. (2002) The Ensembl genome database project. Nucleic Acids Res., 30, 38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wingender E., Chen,X., Fricke,E., Geffers,R., Hehl,R., Liebich,I., Krull,M., Matys,V., Michael,H., Ohnhäuser,R., Pruss,M., Schacherer,F., Thiele,S. and Urbach,S. (2001) The TRANSFAC system on gene expression regulation. Nucleic Acids Res., 29, 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higo K., Ugawa,Y., Iwamoto,M. and Korenaga,T. (1999) Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res., 27, 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hertz G.Z. and Stormo,G.D. (1999) Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics, 15, 563–577. [DOI] [PubMed] [Google Scholar]
- 23.Eulgem T., Rushton,P.J., Robatzek,S. and Somssich,I.E. (2000) The WRKY superfamily of plant transcription factors. Trends Plant Sci., 5, 199–206. [DOI] [PubMed] [Google Scholar]
- 24.Maleck K., Levine,A., Eulgem,T., Morgan,A., Schmid,J., Lawton,K.A., Dangl,J.L. and Dietrich,R.A. (2000) The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nature Genet., 26, 403–410. [DOI] [PubMed] [Google Scholar]
- 25.Li J., Brader,G. and Palva,E.T. (2004) The WRKY70 transcription factor: a node of convergence for jasmonate-mediated and salicylate-mediated signals in plant defense. Plant Cell, 16, 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.