

Abstract
CE-MC server (http://cemc.sdsc.edu) provides a web-based facility for the alignment of multiple protein structures based on C-α coordinate distances, using combinatorial extension (CE) and Monte Carlo (MC) optimization methods. Alignments are possible for user-selected PDB (Protein Data Bank) chains as well as for user-uploaded structures or the combination of the two. The whole process of generating multiple structure alignments involves three distinct steps, i.e. all-to-all pairwise alignment using the CE algorithm, iterative global optimization of a multiple alignment using the MC algorithm and formatting MC results using the JOY program. The server can be used to get multiple alignments for up to 25 protein structural chains with the flexibility of uploading multiple coordinate files and performing multiple structure alignment for user-selected PDB chains. For large-scale jobs and local installation of the CE-MC program, users can download the source code and precompiled binaries from the web server.
INTRODUCTION
Recent advances in the use of synchrotron beamlines, lab automations and robotics have resulted in high-throughput determination of protein structures using X-ray crystallography. The number of structures in the Protein Data Bank (PDB) has doubled over the past four years, creating a need to develop sophisticated bioinformatics tools and web servers to analyze protein structure data. Alignment of multiple protein structures is the first and vital step for many other structure comparison methods such as homology modeling, knowledge-based structure prediction and structure-based drug design. Since protein structures are more conserved than sequences under evolutionary pressure, alignments based on three-dimensional (3D) protein structures can break through the present limitations of sequence alignment methods (1), and the potential power of structure comparison is fully realized in the recent explosion in the size of the PDB (2).
Currently, only a handful of public web servers are available to obtain multiple protein structure alignments, such as CE (http://cl.sdsc.edu/ce.html), DALI (http://www.ebi.ac.uk/dali/), HOMSTRAD (http://www-cryst.bioc.cam.ac.uk/data/align/) CAMPASS (http://www-cryst.bioc.cam.ac.uk/~campass/) and COMPARER (http://www-cryst.bioc.cam.ac.uk/COMPARER/). The CE (3) and DALI (4) servers provide multiple alignments based on ‘pileup’ alignment of structural neighbors from ‘master–slave’ pairwise alignments; however, multiple alignments are not optimized by all-to-all comparison of protein chains. HOMSTRAD (5) and CAMPASS (6) provide multiple structural alignments only for a predefined set of protein families, not for user-selected chains.
The CE-MC web server works based on a combination of pairwise and multiple structure alignment algorithms with complete user control over the selection of the structures to be aligned. Previously, we have reported a method for multiple protein structure alignment using Monte Carlo optimization (7,8). Based on this method, here we present a web server (CE-MC) that generates multiple protein structure alignment for user-selected PDB chains as well as for user-uploaded structures, or for a combination of the two.
DESIGN AND IMPLEMENTATION
The CE-MC web server interface (Figure 1) has been designed using C/C++ and CGI code with appropriate fields hyperlinked to access help information. The user can input PDB chain identification numbers (chain ids) or upload local coordinate files. Multiple structure alignments can be performed for user-selected protein chains present in the PDB or user-uploaded protein structures that may or may not be present in the PDB, or a combination of these two cases. Initial seed alignments are assembled from pairwise alignment data based on the combinatorial extension (CE) algorithm, and alignments are iteratively optimized using Monte Carlo (MC) simulation. Since multiple structure alignments have a high computational overhead and are time consuming, jobs are scheduled in a queue on the host machine and processed in order. Hyperlinks to access the results are emailed to the user upon completion of the job, and results are stored on the host machine for 72 h.
Figure 1.

A screen shot of the CE-MC server main page.
Algorithms
The CE-MC program is based on two independent algorithms, i.e. combinatorial extension and Monte Carlo optimization. The CE algorithm (3) is used to perform an all-against-all pairwise alignments for all chains in the set. The resulting Z-scores from these alignments are used to generate a guide tree using the UPGMA (9) method. The guide tree is then used to build a progressive alignment, by sequentially aligning structures according to the tree. To combine aligned clusters, an alignment of the highest scoring structure pair between the two clusters is used to guide the alignment of the clusters. This method provides a relatively high quality initial alignment, which is suitable for subsequent refinement using the MC algorithm. A distance-based score is calculated for each eligible column in the alignment. An eligible column is one that contains residues (not gaps) in at least one-third of its rows. Geometric distances are calculated from the 3D coordinates of C-α atoms for each pair of residues in a column for R(R−1)/2 combinations, where R is the number of residues in a column. Column distances are defined as average geometric distances calculated for each column. The alignment score S is calculated from the column distances in aligned blocks, using the following scoring function:
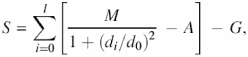
where l is the total number of eligible aligned columns, M = 20 is the maximum score of a match, di is the average distance for column i, d0 is the maximum distance which is not penalized and
![]()
G is linear gap penalty term with gap initiation and gap extension penalties of 15 and 7, respectively.
Random trial moves are performed on the alignment one residue at a time or one column at a time and a new score is calculated for each trial move. If the score improves upon the trial move, the move is always accepted and the change in the alignment becomes permanent. If the score deteriorates, the move may still be accepted or rejected based on a factor P that depends on the extent of score deterioration and the trial move count. In a nutshell, global optimization of the multiple alignment is accomplished by random and iterative exploration of the search space, with occasional excursions into the non-optimal territory, until the optimization converges. [For more details, read references (7,8)]. The final alignment after MC optimization is reformatted using the JOY program (10), which uses 3D coordinate information to display the secondary structural and local environmental features in a sequence alignment.
Input data
PDB structures with chain ids should be entered in space-delimited format. The minimum number of chains is 3 and the maximum is 25. If no coordinates files are uploaded, by default the program retrieves structure data from the PDB. Optionally, for analyzing local structures (not present in the PDB), up to eight coordinate files can be uploaded in standard PDB file format with the file extension ‘.ent’. The user can also select optional Z-score cutoff and distance cutoff parameters. The Z-score cutoff is used to filter out the chains that have average Z-score below the cutoff, where the average Z-score for each chain is calculated from all pairwise Z-score values. This option is very useful for eliminating some very distant structures in the set that may reduce the overall alignment quality. The distance cutoff is used to define the quality of multiple alignments, i.e. at smaller distance cutoff, local conserved regions are better aligned but the ratio of the aligned length to the total is lower and vice versa.
Output data
The CE-MC program outputs multiple alignments in four different formats, i.e. JOY/html, JOY/PostScript, text and FASTA formats. The JOY/html format is ideal for viewing 3D structure-based alignments in multiple sequence alignment format (Figure 2). The JOY/PostScript file is useful for editing and publication of CE-MC alignments, while the text and FASTA formats can be conveniently reformatted to further analyze CE-MC results using other programs. Results in the text format show the values for three measures of quality in the final alignment, i.e. alignment distance, alignment score and alignment length. Alignment distance is the average of all eligible column distances, alignment score is the global alignment score (S) calculated as described above, and alignment length is the number of eligible columns in the alignment compared with the total length of the alignment. In addition, transformed XYZ coordinates for all chains after superposition are also provided in standard PDB format. The results also include a log file containing a commentary on all tasks performed to aid in detecting the problem in the case of failure. The order of chain ids in the CE-MC output may not be the same as the original order of the input chain ids because the program determines the master structure on the fly and may also filter out some chains depending on the Z-score cutoff chosen. However, a separate script has been built to reorder the chain ids into the same order as the user input ids.
Figure 2.

A screen shot of CE-MC results in JOY format, for a subset of structures belonging to the aspartic proteinase family. Uppercase, solvent inaccessible; lowercase, solvent accessible; red, α-helix; blue, β-strand, maroon, 3–10 helix; bold, hydrogen bond to main chain amide; underline, hydrogen bond to main chain carbonyl; cedilla, disulphide bond; italic, positive phi.
DISCUSSION
The whole process of generating viewable multiple structure alignments involves three distinct steps, i.e. all-to-all pairwise alignment using the CE algorithm, global optimization of a multiple alignment using the MC algorithm and reformatting the MC results using the JOY program. Since CE is a local alignment algorithm, it generates quality alignments in the conserved secondary structure regions. The MC algorithm takes advantage of the CE alignments to assemble the seed alignment and performs global optimization. A four-CPU SUN–SPARC machine with 4 GB of main memory currently hosts the CE-MC server. The time taken for computing a multiple structure alignment depends on several factors such as the number and length of structure chains, the structural diversity of the family being aligned and the distance cutoff chosen. For example, it takes ∼7 min to align 10 structures of average length 250 amino acids, assuming no other jobs are ahead in the queue. Since the computational complexity of the CE-MC algorithm is quadratic, we limit the maximum number to 25 chains. However, to analyze larger datasets, we encourage users to download our UNIX standalone version of the CE-MC software, accessible from the current web server. The standalone version also provides the flexibility of manipulating more alignment parameters to suit the characteristics of the specific structural families being aligned.
Acknowledgments
ACKNOWLEDGEMENTS
The authors are grateful to Dr Kenji Mizuguchi for providing the JOY binaries and to Ms Jo-Lan Chung for her help with the user-friendliness of the server. This work was supported by a PACI-REU (Partnership for Advanced Computational Infrastructure-Research Experience for Undergraduate Students) grant (2003) and NSF grant DBI 9808706.
REFERENCES
- 1.Jia J., Huang,W., Schorken,U., Sahm,H., Sprenger,G.A., Lindqvist,Y. and Schneider,G. (1996) Crystal structure of transaldolase B from Escherichia coli suggests a circular permutation of the alpha/beta barrel within the class I aldolase family. Structure, 4, 715–724. [DOI] [PubMed] [Google Scholar]
- 2.Westbrook J., Feng,Z., Chen,L., Yang,H. and Berman,H.M. (2003) The Protein Data Bank and structural genomics. Nucleic Acids Res., 31, 481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shindyalov I.N. and Bourne,P.E. (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Prot. Eng., 11, 739–747. [DOI] [PubMed] [Google Scholar]
- 4.Holm L. and Sander,C. (1996) Mapping the protein universe. Science, 273, 595–603. [DOI] [PubMed] [Google Scholar]
- 5.Mizuguchi K., Deane,C.M., Blundell,T.L. and Overington,J.P. (1998) HOMSTRAD: a database of protein structure alignments for homologous families. Prot. Sci., 7, 2469–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sowdhamini R., Burke,D.F., Huang,J-F., Mizuguchi,K., Nagarajaram,H.A., Srinivasan,N., Steward,R.E. and Blundell,T.L. (1998) CAMPASS: a database of structurally aligned protein superfamilies. Structure, 6, 1087–1094. [DOI] [PubMed] [Google Scholar]
- 7.Guda C., Scheeff,E.D., Bourne,P.E. and Shindyalov,I.N. (2001) A new algorithm for the alignment of multiple protein structures using Monte Carlo optimization. Pac. Symp. Biocomput., 6, 275–286. [DOI] [PubMed] [Google Scholar]
- 8.Guda C., Scheeff,E.D, Bourne,P.E. and Shindyalov,I.N. (2002) Comparative analysis of protein structure: new concepts and approaches for multiple structure alignment. In Tsigelny, I.F. (ed.), Protein Structure Prediction: Bioinformatics Approach. International University Line, La Jolla, CA, pp. 451–459. [Google Scholar]
- 9.Sneath P.H.A. and Sokal,R.R. (1973) Numerical Taxonomy. W.H. Freeman and Company, San Francisco, CA, pp. 230–234. [Google Scholar]
- 10.Mizuguchi K., Deane,C.M., Blundell,T.L., Johnson,M.S. and Overington,J.P. (1998) JOY: protein sequence–structure representation and analysis. Bioinformatics, 14, 617–623. [DOI] [PubMed] [Google Scholar]