

Abstract
Ageing is a complex and multifactorial process characterized by the accumulation of many forms of damage at the molecular, cellular, and tissue level with advancing age. Ageing increases the risk of the onset of chronic inflammation-associated diseases such as cancer, diabetes, stroke, and neurodegenerative disease. In particular, ageing and cancer share some common origins and hallmarks such as genomic instability, epigenetic alteration, aberrant telomeres, inflammation and immune injury, reprogrammed metabolism, and degradation system impairment (including within the ubiquitin-proteasome system and the autophagic machinery). Recent advances indicate that damage-associated molecular pattern molecules (DAMPs) such as high mobility group box 1, histones, S100, and heat shock proteins play location-dependent roles inside and outside the cell. These provide interaction platforms at molecular levels linked to common hallmarks of ageing and cancer. They can act as inducers, sensors, and mediators of stress through individual plasma membrane receptors, intracellular recognition receptors (e.g., advanced glycosylation end product-specific receptors, AIM2-like receptors, RIG-I-like receptors, and NOD1-like receptors, and toll-like receptors), or following endocytic uptake. Thus, the DAMP Hypothesis is novel and complements other theories that explain the features of ageing. DAMPs represent ideal biomarkers of ageing and provide an attractive target for interventions in ageing and age-associated diseases.
Keywords: ageing, cancer, longevity, damage-associated molecular pattern (DAMP) molecules, receptor, biomarker
1. Introduction
The ultimate goal of biomedical research is to translate laboratory discoveries or clinical observations into new therapies to ameliorate disease and extend life expectancy and quality, namely the average total number of years of remaining meaningful life at a given age [1]. Embedded in varying environmental and social factors, ageing and disease affect life expectancy and longevity in humans. Ageing is a complex and multifactorial process characterized by the many forms of damage accumulation at the molecular, cellular, and tissue level that progress with advancing age, decreasing the body’s normal response and function. Ageing itself is not a disease, but it significantly increases the risk of the onset of chronic inflammatory-associated diseases including many forms of cancer, stroke, diabetes, neurodegenerative diseases, cardiovascular disease, osteoporosis, and others [2]. An increased understanding of the molecular nature of the ageing process is therefore critical to improving health and quality of life in an ageing population. In the past decade, a number of theories have been proposed to explain how and why we age and develop diseases. For example, ageing has been widely proposed to be caused by oxidative injury [3], wear and tear on the body [4], cellular senescence [5], or changes in the neural [6], endocrine [7], or immune systems [8]. Other theories consider ageing a predetermined process controlled by genes [9] or from the declining roll of natural selection [10]. Of course, no single theory currently exists that can explain all of the hallmarks of ageing, suggesting that ageing is a multi-step and multi-event process.
Cancer cells exhibit several characteristics distinct from normal cells [11]. At first glance, cancer and ageing have an inverse relationship because cancer cells are capable of uncontrolled growth and division, whereas ageing cells have a diminished proliferative capacity. It has been well-established that older adults have a higher risk for cancer. Ageing is involved in a number of events responsible for carcinogenesis and cancer development at the molecular, cellular, and tissue levels [12]. Indeed, ageing and cancer have common origins due to internal and environmental stress and share some common hallmarks such as genomic instability, epigenetic alteration, aberrant telomeres, inflammation and immune injury, reprogrammed metabolism, and impaired degradation of intracellular biomolecules and organelles (Figure 1) [11, 13]. Although the molecular link between ageing and cancer remains largely unknown, damage-associated molecular pattern molecules (DAMPs) play a potential role in the pathogenesis of ageing and cancer. DAMPs, sometimes termed alarmins or danger signals, are endogenous molecules released from cells in response to exogenous and endogenous stressors, especially following injury or cellular death [14, 15]. They can act as inducers, sensors, and mediators of stress and the immune response through individual plasma membrane receptors, cytosolic/intracellular recognition receptors, or following endocytic uptake. DAMPs and their receptors (e.g., advanced glycosylation end product-specific receptor [AGER/RAGE], toll-like receptors [TLRs], NOD1-like receptors [NLRs], RIG-I-like receptors [RLRs], and AIM2-like receptors [ALRs]) play multiple roles in human health and disease, especially inflammatory and immune-associated disease [16, 17]. Here, we outline six common biological hallmarks of ageing and cancer; discuss the behavior and function of DAMPs in ageing and age-associated disease; and highlight the multifaceted role of two nuclear families (e.g., high mobility group box 1 [HMGB1] and histone) and two cytosolic families (e.g., S100 proteins and heat shock proteins [HSPs]) acting collectively as DAMPs in the pathogenesis of ageing and cancer.
Figure 1.
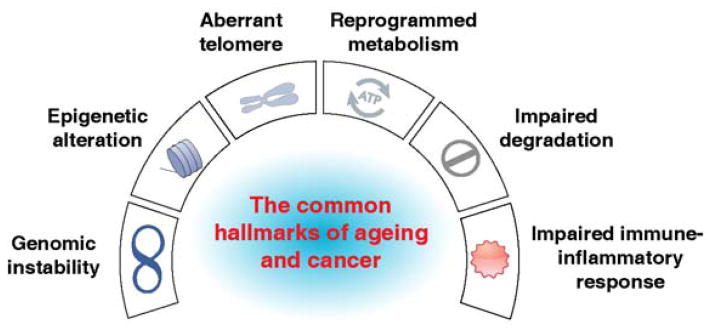
The six common hallmarks of ageing and cancer
2. The Common Hallmarks of Ageing and Cancer
2.1 Genomic Instability
Genomic instability refers to a range of genetic alterations from the nucleotide, chromosomal, to nuclear architectural levels when the genetic lesion is increased and the surveillance mechanisms become significantly impaired [18]. Frequent causes of genomic instability include intrinsic damage arising from reactive oxygen species (ROS) produced during mitochondrial dysfunction or activation of nicotinamide adenine dinucleotide phosphate-oxidase, and extrinsic environmental stressors including ultraviolet radiation from the sun. ROS play a role in ageing and carcinogenesis by inducing DNA damage, lipid peroxidation, and protein structural and functional modifications. At the nucleotide level, disrupted DNA damage response pathways, including DNA repair, cell cycle checkpoints, and programmed apoptosis, can cause nucleotide substitution, insertion, and deletion, as a panoply of genetic alterations/mutations [19]. The nucleotide excision pathway, base excision repair, and mismatched base repair are essential for single-stranded DNA break repair, whereas non-homologous end joining and homologous recombination are responsible for double-stranded DNA break repair [20]. Single-stranded breaks are more frequent and can lead to ageing and neurodegenerative brain disease, while double-stranded breaks are more serious and can result in cancer [21]. At the chromosomal level, variations in chromosome number (e.g., aneuploidy, monoploidy, and euploidy) as well as deletions, duplications, inversions, and translocations of chromosome segments, contribute to genomic instability. At the nuclear architectural level, defects in the nuclear lamina, a structural component of the nuclear envelope, can also produce genomic instability [22]. Recently, an analysis of mice with targeted mutation or defective genes involved in maintaining genomic stability have provided a direct link between cancer and ageing [23]. As examples, deficiencies of DNA damage response genes (e.g., excision repair cross-complementation group 4 [ERCC4] [24], Werner syndrome, RecQ helicase-like (WRN) [25], ataxia telangiectasia mutated [ATM] [26, 27], breast cancer 1, early onset [BRCA1] [28], tumor protein p53 [TP53] [29], and sirtuin 2 [30]) and nuclear architecture genes (e.g., lamin A [LMNA] [31]) increase the risk of cancer as well as premature ageing. Nuclear DAMPs, including HMGB1, histones, and DNA, are the major components of chromosomes. On one hand, loss of nuclear DAMPs increases genomic instability in response to stress. On the other hand, the release of nuclear DAMPs is significantly increased in cells following genomic instability, which facilitates the systemic inflammatory response [15, 32]. Chronic inflammation conditions can increase the risk of cancer. Age-related accumulation of ROS from mitochondria has also been demonstrated as a major source of not only somatic mutation [33], but also DAMP release [34]. Collectively, these findings suggest that DNA oxidative damage-mediated genomic instability from both the nucleus and mitochondria may be a fundamental cause of ageing and a variety of human diseases, including cancer.
2.2 Aberrant Telomeres
Telomeres are highly-specialized nucleoprotein structures at the ends of linear chromosomes, which were first observed by Hermann Muller and Barbara McClintock in the early 1930s [35]. Telomeres are essential for maintaining genome integrity [36]. A highly-conserved telomeric repeat DNA sequence, namely (TTAGGG)n, prevents fusion between chromosome ends and ensures proper replication [37]. Telomerase, a ribonucleoprotein with enzymatic activity, adds new DNA sequence repeats to both strands of the telomere overhang [38]. In addition to the telomerase-mediated telomere lengthening pathway, some cells utilize a specific pathway called alternative lengthening of telomeres (ALT) to elongate telomeres through transferring telomere tandem repeats between sister-chromatids [39]. Furthermore, the chromosome end is protected against DNA damage by multiple telomere-binding proteins, especially a protein complex called shelterin (consisting of the six protein telomeric repeat binding factor (NIMA-interacting) 1 [TERF1/TRF1], telomeric repeat binding factor 2 [TRF2], TERF1 (TRF1)-interacting nuclear factor 2 [TINF2/TIN2], protection of telomeres 1 [POT1], telomeric repeat binding factor 2, interacting protein [TERF2IP/RAP1], and adrenocortical dysplasia homolog (mouse) [ACD/TPP1] in mammalian cells) [40]. Although the mechanism remains to be fully characterized, shelterin directly interacts with multiple classical DNA repair pathways, therefore protecting telomeres from injury [41]. Additional factors such as chromatin remodeling complex (e.g., SWItch/Sucrose NonFermentable [SWI/SNF] and INO80) contribute to telomere structure and function adaptation in response to DNA damage [42, 43]. The structural basis for this process is not clear. Telomeric dysfunction is a major mechanism for the generation of genomic instability, while telomere length is a key factor affecting cellular lifespan [44–46]. Loss of HMGB1 in cells reduces telomerase activity and decreases telomere length [47]. Epigenetic modification by histone and DNA methylation also regulates telomere length, structure, and function [48, 49]. These links between nuclear DAMP and telomere-length regulation provide important new avenues for understanding processes of cancer development and ageing.
At the cellular level, ageing refers to senescence, a process by which a cell becomes old and dies [5]. Senescence is induced from the shortening of telomeres to the point that the chromosome reaches a critical length. In normal somatic cells, including adult stem cells, shorter telomeres have become associated with the ageing process due to telomerase repression. In germ cells, high levels of telomerase activity prevent telomeres from shortening. In cancer cells, telomere shortening can be observed at early stages due to more frequent division; however, increased expression and activity of telomerase guard their immortality by maintaining telomere length [50]. These dynamic changes make telomerase inhibitors a new therapeutic approach for patients with cancer. Genetically-modified animal models have confirmed a positive relationship between telomere length and lifespan, and ageing can be reverted by restoring telomerase activation [51]. However, studies using transgenic mice suggest that telomeres and telomerase act as both tumor promoters and suppressors, depending on the cell type and genomic context [52–56]. Similarly, cellular senescence also plays both tumor suppression and promotion roles in the regulation of tumorigenesis [57]. It remains unclear how the pathological telomere attrition checkpoint is activated by oncogenetic signals at different stages.
2.3 Epigenetic Alteration
Epigenetics is defined as changes in gene activity and expression that occur without alteration in DNA sequence, namely the genetic code, itself [58]. The markers and mechanisms of epigenetic alteration include DNA methylation variation, histone posttranslational modifications (PTMs), and noncoding RNA changes. Epigenetic alterations often enhance or inhibit gene activity in a combined manner [59]. Epigenetic changes may directly lead to ageing and cancer initiation, or render cells more sensitive to subsequent genetic or epigenetic alterations [60]. DNA methylation is the process of DNA methyltransferase enzymes-mediated conversion of cytosine to 5-methylcytosine [61]. In mammals, DNA methylation typically occurs at CpG sites and most CpG sequences in the human genome are methylated [62]. There is more evidence of individual variation in age- and environmental exposure-related methylation across the life span than was previously expected [63]. The importance of DNA methylation and its mode of action may vary between sexes and change during ageing [64, 65]. As a silencing epigenetic mark, DNA methylation is required for proper embryogenesis and development, genome stability, telomere length maintenance, and gene expression [66]. DNA methylation is the first recognized epigenetic modification in ageing and ageing-related diseases and is generally characterized by decreased global DNA methylation and an increased specific gene promoter-hypermethylation [67]. These genes include tumor suppressor genes (cyclin-dependent kinase inhibitor 2A [CDKN2A], lysyl oxidase [LOX], runt-related transcription factor 3 [RUNX3], and retinoic acid receptor responder (tazarotene induced) 1 [RARRES1/TIG1]) as well as cancer-related genes such as those encoding the estrogen receptor and insulin-like growth factor 2 [IGF2] [68]. Histones are composed of core histones (H2A, H2B, H3, and H4) and linker histones (H1 and H5). As basic components of nucleosomes (the structural unit of chromatin), histone can be modified multiple times by individual PTMs or by the same PTM at different residues. These modifications are important for chromatin homeostasis and gene expression [69, 70], and are linked to ageing and cancer. For example, increased histone acetylation at H4K16, trimethylation at H4K20 or H3K4, as well as decreased methylation at H3K9 or trimethylation at H3K27, are age-associated epigenetic markers [71]. In contrast to ageing, histone hypoacetylation at H3K9 and H4K16 following aberrant expression or activity of histone deacetylases and/or histone hypermethylation at H3K9 and H3K27 by histone methyltransferases can induce cellular transformation [72]. Phosphorylation of H2A histone family member X (H2AFX/H2AX) at Ser139 is an early molecular event of the DNA damage response, which facilitates the recruitment of DNA repair proteins to DNA damage sites to act as histone guardians of the genome [73]. These results are consistent with an animal study showing that H2AFX-deficient mice have retarded growth and exhibit signs of genomic instability [74].
RNA change is as an emerging epigenetic marker involved in chromatin modification. Notably, the differential expression profile of microRNAs (miRNAs), a class of small, noncoding RNA molecules that usually lead to post-transcriptional gene silencing, are functionally related to ageing and cancer, although specificity is an issue [75–77]. miRNA action itself is not considered a mechanism of epigenetics. miRNAs can regulate the expression of proteins involved in DNA methylation and histone PTMs. Additionally, epigenetic mechanisms are also important regulators of cell type-specific miRNAs. These findings suggest interplay between miRNA and epigenetics. Similar to HMGB1 and histones, miRNAs can be released into the circulation as DAMPs in many diseases, making these molecules unusually attractive biomarkers [78, 79]. An exosome-dependent pathway has recently been shown to contribute to the release of miRNAs in cancer cells [80]. Once released, extracellular miRNAs can directly interact with TLRs in surrounding immune cells, leading to NF-κB pathway activation and increased secretion of proinflammatory cytokines that ultimately promotes cancer cell proliferation and metastatic potential [80]. Collectively, epigenetic modifications are progressive processes that could be regulated by multiple mechanisms in response to environmental stimuli. It will be interesting to define and map the epigenetic modification patterns not only inside of cells, but also outside of cells in ageing and age-associated diseases.
2.4 Inflammation and Immune-mediated Injury
Chronic inflammation and immune injury are common hallmarks of ageing and cancer [81]. The immune system protects the body from infection and injury by recognizing and responding to substances derived from foreign microbes (e.g., pathogen-associated molecular pattern molecules [PAMPs]) and endogenous molecules (e.g., DAMPs) [16]. Excessive PAMPs and DAMPs can impair immunity and the associated inflammatory response, contributing to the pathophysiology of ageing and many chronic diseases including cancer, neurodegenerative diseases, and diabetes [14]. Excessive inflammation is implicated in oxidative injury, remodeling of the extracellular matrix, activated angiogenesis, enhanced fibrosis, cell death, and altered microenvironment in diverse target tissues [82]. Also, the normal immune system has the ability to eliminate dying, dead, senescent, and hyperploid cells through various mechanisms such as monocyte/macrophage-mediated phagocytosis, natural killer cell-mediated cytotoxicity, and antigen-specific T-cell responses following dendritic cell selection and expansion [83, 84]. Moreover, decreased antioxidant capacity and an associated increased ROS affect the redox status and function of the immune cell in the setting of tissue inflammation and immune surveillance [85]. Several well-studied inflammatory pathways such as nuclear factor-κB (NF-κB) signaling [86, 87], activation of the inflammasome [88, 89], and hyperactivation of the signal transducer and activator of transcription 3 (acute-phase response factor) (STAT3) [90, 91] factors can drive production of the classical pro-inflammatory cytokines (e.g., tumor necrosis factor [TNF], interleukin[IL]-1, and IL-6), which may directly link systemic chronic inflammation to immune functional decline in ageing and cancer. Genetic and pharmacological inhibition of these inflammatory pathways generally prevents ageing and tumorigenesis.
Age-related changes in our innate and adaptive immune systems are known collectively as immunosenescence. The important contributor to decline in immune function in the elderly adult is the changes observed in adaptive immunity, including reduced production of B and T cells in the bone marrow and thymus and diminished function of mature lymphocytes in secondary lymphoid tissues [92]. Not only is the adaptive immune response altered, but it has also recently become evident that most innate immune functions, including those of neutrophils, dendritic cells, and natural killer (NK) cells, are affected at least to some extent by the ageing process. As a result, the ageing of the immune system is probably the major determinant for susceptibility to diseases including cancer. Cancer immunosurveillance involving many players (such as CD8 cytotoxic T cells, Th1 responses, NK cells, macrophages, B cells, and γδ T cells) is the immune system’s ability to recognize and destroy newly arising malignant cells. However, these newly arising malignant cells are gradually able to gain several mechanisms of immune evasion during ageing that may favor tumor development and progression [93].
The sirtuin family, a class of proteins that possess either mono-ADP-ribosyltransferase or deacylase activity, is a potential negative regulator of the inflammatory response acting partly through deacetylation of NF-κB [94]. Besides downregulating the inflammatory response, sirtuin-1, -2 and -6 can sustain genomic stability and metabolic balance, extending the lifespan of mice, and generally acting as tumor suppressors [95–98]. Thus, the decreased expression of sirtuins may be involved in the activation of these processes during ageing. In addition, interplay between the neuroendocrine and immune systems can fine-tune the inflammatory response through release of hormones and neurotransmitters [99]. Further studies are essential to define the complex regulatory networks of the inflammatory response in ageing and age-associated disease and explore how targeting these pathways could have therapeutic potential.
2.5 Reprogrammed Metabolism
Metabolism is generally divided into two counter-regulatory categories: catabolism and anabolism, or the processes of breakdown or synthesis of compounds needed by the cells, respectively. Bioenergetics, also called energy metabolism, is the general process by which living cells produce and use the energy needed to stay alive, grow, and proliferate. Many metabolic processes can result in production and utilization of energy in forms such as adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide molecules. Glycolysis and mitochondrial oxidative phosphorylation are the sole mechanisms of ATP synthesis in human cells. Normal metabolism is reprogrammed to meet the challenges of macromolecular synthesis under ageing and some diseases [100].
A well-established reprogrammed metabolism observed in cancer cells is the “Warburg effect,” namely that tumor cells can strategically generate ATP through aerobic glycolysis (in addition to oxidative phosphorylation), even when oxygen is abundant [101], enabling exaggerated tumor cell proliferation. Similarly, ageing drives large systemic reductions in oxidative phosphorylation, shifting the entire body metabolically toward aerobic glycolysis [102]. The “Warburg effect” is one of the key hallmarks of cancer and is regulated by a number of transcription factors and kinases, especially hypoxia-inducible factor 1α (HIF1α) and pyruvate kinase M2 (PKM2) [103–105]. Changes in HIF1α with age have been reported in rats and could be related to age-related pathologies [106]. Loss of HIF1α accelerates epidermal ageing in the skin [107]. Our recent study shows that the HIF1α and PKM2-mediated Warburg effect is required for HMGB1 release in activated macrophages and systemic inflammatory syndromes, providing a direct link between reprogrammed metabolism and inflammation by inducing DAMP release [108]. In addition to macrophages, metabolic reprogramming also facilitates the maturation of dendritic cells [109] and influences the differentiation of both anti-inflammatory Treg cells and pro-inflammatory Th17 cells [110, 111]. Additional studies are necessary to determine the crosstalk between metabolism reprogramming and the immune system in ageing.
Caloric restriction (CR) without malnutrition has been show to slow the ageing process, extend lifespan, and decrease onset of disease in various species, although the underlying mechanism remains unclear [112]. A possible mechanism of action of CR-induced longevity is reprogrammed metabolism with increased protein synthesis and reduced energy metabolism by transcriptional regulation [113, 114]. CR may result in protection from cancer risks, partly through limiting the “Warburg effect” [115]. In contrast to CR, high-fat and high-cholesterol diets can accelerate tumor growth and development [116]. As a protein hormone, adiponectin is not only important for enhancing glucose and fatty acid oxidation, but also responsible for the action of CR [117, 118]. Additionally, CR can improve insulin sensitivity through regulating the insulin-and IGF-1-signaling and autophagy pathways [119, 120]. CR or fasting improves systemic inflammation in sepsis and ischemic injury partly through reduction of the release of DAMPs such as HMGB1 [121, 122]. In addition, the reduction in serum HMGB1 appears to be mediated by the NAD-dependent protein deacetylase sirtuin-1-associated autophagic response [122]. Several energy sensors such as AMP-activated protein kinase (AMPK), v-akt murine thymoma viral oncogene homolog (AKT), and sirtuin 1 can balance survival and death in response to metabolic stress through regulating their downstream effectors such as the mammalian/mechanistic target of rapamycin (MTOR), TP53, foxhead box O (FOXO), and HIF1α [12]. In summary, these observations indicate that abnormal metabolism may trigger ageing-associated diseases. The translational potential of these findings remains to be further explored in humans.
2.6 Impaired Degradation
Failure to remove and dispose of defective proteins or cellular components remarkably increases susceptibility to disease. Eukaryotic cells include two major categories of degradation pathways for waste management and recycling. Whereas the ubiquitin-proteasome system (UPS) is the major nonlysosomal proteolytic pathway of intracellular proteins, autophagic pathways can selectively eradicate damaged cell organelles, protein aggregates, invasive microorganisms, or effete molecules including proteins, DNA, and RNA through delivery to digestive lysosomes [123]. Autophagy, the UPS, and molecular chaperones contribute to cellular quality control (Figure 2). The 26S proteasome, a large multi-catalytic, multi-subunit protease complex located in the cytosol and the nucleus of eukaryotic cells, constitutes the central proteolytic machinery of the UPS. Autophagy can be divided into three broad categories: macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy (hereafter referred to as autophagy), the most common type, is a highly-regulated dynamic process that includes the formation and maturation of several membrane structures such as the phagophore, autophagosome, and autolysosme. The degraded components produced from autophagy can be reused for biosynthesis or energy production and sustain metabolism during stress [124, 125]. This property makes increased autophagy generally a programmed survival pathway, although excessive autophagy may rarely cause cell death [125, 126]. Increased autophagy reduces accumulation of proteotoxic protein aggregates, improves innate immune responses, and limits apoptosis and cellular senescence. Deficient UPS and autophagy pathways are associated with most aspects of normal physiology and development, and are also involved in a broad array of pathological conditions and diseases, including ageing, cancer, and many neurodegenerative diseases including Parkinson’s and Alzheimer’s [127, 128]. Decreased proteasome activity has been observed in ageing. Of note, proteasome deficiency is both a cause and a consequence of ageing, and contributes to the general decrease in protein integrity and functionality with age. Autophagy-deficient mice exhibit decreased longevity and increases in certain types of tumorigenesis due to genome instability, inflammation, and organelle injury, whereas specific induction of autophagy (e.g., delivery of the MTOR inhibitor rapamycin and CR) can increase longevity in multiple animal species [129–135]. However, some paradoxical observations suggest that autophagy has a context-, type- and stage-dependent role in tumor development and cancer therapy [136]. For example, ATG5-mediated autophagy suppresses oncogenetic KRasG12D-driven early lung oncogenesis, but accelerates cancer progression at later stages [137]. Indeed, both autophagy inducers (e.g., rapamycin) and inhibitors (e.g., chloroquine) exhibit significant anticancer activity [138, 139]. A complex relationship exists between DAMPs and autophagy in cellular adaption to injury and unscheduled cell death [15]. On one hand, DAMP such as HMGB1 and S100 can induce autophagy, contributing to drug resistance [140]. On the other hand, autophagy can regulate the release of DAMPs in immune and cancer cells [17]. Thus, an increased understanding of the properties of autophagy and DAMPs in cancer is therefore critical for its optimal exploitation for therapeutic advantage. In contrast, proteasome activity is increased in cancer cells and inhibition of the UPS could be used as a novel approach for cancer therapy.
Figure 2. Predominant intracellular quality control strategies to sustain homeostasis in response to pathological and physiological stress.

Whereas the ubiquitin-proteasome system is the major nonlysosomal proteolytic pathway of intracellular proteins, autophagy pathways can selectively eradicate damaged cell organelles or unused molecules including protein, DNA, and RNA through lysosomes. In addition, molecular chaperones including HSPs contribute to the correction of misfolded or unfolded proteins.
3. The DAMP Hypothesis and the Danger Theory
In 1994, the “danger theory” was proposed by Polly Matzinger to explain how the immune system works to distinguish between self and non-self danger signals in response to sterile inflammation in the absence of pathogens [141]. In contrast to infection, sterile inflammation occurs in acute conditions, such as ischemia-reperfusion injury, crystal-induced arthritis, and trauma, as well as with chronic diseases such as ageing, cancer, atherosclerosis, and autoimmune diseases. In response to injury, death, and other stressors, certain endogenous molecules can be released or secreted from dead, dying, or injured cells or those exposed on the cell surface. These molecules, later named DAMPs, often act as an alarm to warn our immune system to respond to tissue damage or injury, promoting a wound healing response (Figure 3) [142, 143]. In contrast to cell-derived DAMPs, PAMPs are generated from pathogens [14]. In addition to passive release, certain DAMPs can be secreted in immune cells following PAMP stimulation. Furthermore, it has been suggested that most PAMPs are hydrophobic molecules derived from damaged or injured microbes [144]. Both PAMPs and DAMPs can be recognized by common pattern recognition receptors including TLRs, NLRs, RLRs, ALRs, and AGER to initiate an immune and inflammation response [145]. Of note, the mechanisms used by our immune system to manage dead and dying cells are complex and context-dependent. Depending on cell death type and the microenvironment in which it occurs, DAMPs can mediate either immunogenic or tolerogenic cell death, which is important for immune therapy for cancer [17, 146, 147].
Figure 3. Release and activity of DAMPs in response to injury, death, and stress.

DAMPs can be derived from any compartment of the cell, including the nucleus and the cytoplasm (e.g., mitochondria), although they vary greatly depending on the types and severity of the injury. They can act as inducers, sensors, and mediators of stress through plasma membrane receptors, intracellular recognition receptors (e.g., AGER and TLRs/NLRs/RLRs/ALRs), or endocytic uptake. CIRP: cold-inducible RNA-binding protein. TFAM: mitochondrial transcription factor A.
DAMPs can be derived from any compartment of the cell including the nucleus and cytoplasm (e.g., mitochondria, endoplasmic reticulum, and lysosome). They vary greatly depending on the type and severity of the injury. Among them, protein DAMPs include endogenous proteins, such as HMGB1, HSPs, the S100 family of calcium binding proteins, serum amyloid A, and histones, whereas other non-protein sources of DAMPs include ATP, uric acid, heparin sulfate, DNA (genomic and mitochondrial DNA), and RNA [14]. DAMPs act in a single or combined manner in other cell types besides immune cells such as fibroblasts and endothelial, cancer, and stem cells, supporting their roles in the regulation of multiple cellular processes such as growth, differentiation, survival, and death. Additionally, PTMs, especially proteolysis and oxidation, can affect extracellular activity of DAMPs, which is often associated with cell death [148, 149]. Importantly, circulating DAMP levels are increased in ageing and many diseases. They may be a clinically useful biomarker for diagnosis, risk prediction, and therapy monitoring in certain diseases. It is thus important to explore the mechanisms of DAMP release and assess their biologic roles in the settings of stress and disease [15].
4. DAMPs at the Crossroads of Ageing and Cancer
Many lines of evidence indicate that intracellular and extracellular DAMPs play a role in the regulation of ageing and cancer. These DAMPs have well-defined functions with several characteristics involved in the biology and common hallmarks of ageing and cancer. Proteins (e.g., HMGB1 and histone) and non-proteins (e.g., DNA and ATP) that can function as DAMPs have a normal function inside the cells of origin. We use the terms “intracellular DAMPs” and “extracellular DAMPs” to distinguish between the localization of DAMPs during stress. Increased stressors, especially oxidative stress, lead to DAMPs translocating and releasing into the extracellular space. Loss of intracellular DAMPs increases genomic instability, epigenetic alteration, telomere attrition, reprogrammed metabolism, and impaired degradation system, whereas increased extracellular DAMPs cause excessive inflammation and immune injury. These DAMP-mediated pathogenic changes are an important force driving the initiation and development of ageing as well as age-related diseases, including cancer (Figure 4). In addition to DAMPs regulating the hallmarks of ageing and cancer, the expression, release and activity of DAMPs can be regulated by various common hallmarks of cancer/ageing. For example, DNA damage caused by genomic instability, epigenetic alteration, and aberrant telomeres can induce release of nuclear DAMPs such as HMGB1, histones, and DNA. Lactic acid from anaerobic glycolysis can promote HMGB1 release. Cytokine and chemokine from an impaired immune-inflammatory response can induce expression and release of DAMPs. In addition, autophagy not only regulates DAMP release, but also degradation. Below, we highlight examples of DAMPs and their multiple location-dependent functions in ageing and cancer.
Figure 4. DAMPs regulate common hallmarks of ageing and cancer.
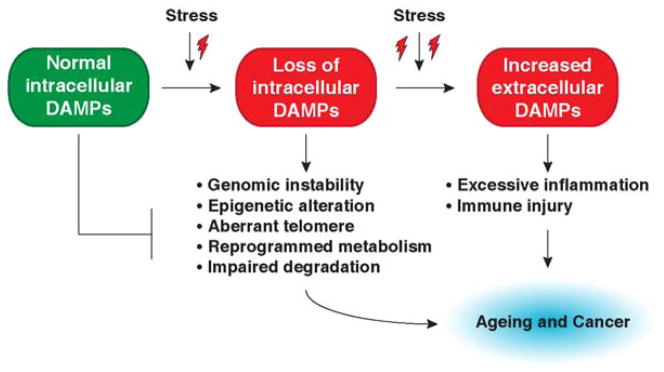
Most DAMPs are nuclear and cytosolic proteins. Increased stressors, especially oxidative stress, lead to DAMPs translocating and releasing into the extracellular space. Loss of intracellular DAMPs increase genomic instability, epigenetic alteration, telomere attrition, reprogrammed metabolism, and impaired degradation system, whereas increased extracellular DAMPs cause excessive inflammation and immune injury. These DAMP-mediated pathogenic changes are an important force driving the initiation and development of ageing as well as age-related diseases, including cancer.
4.1 HMGB1
HMGB1, one of the best-studied DAMPs, is a member of a group of non-histone nuclear proteins with high electrophoretic mobility. As a highly evolutionarily-conserved protein, HMGB1 is essential for life - global knockout HMGB1 mice dying shortly after birth [150]. About 95% of HMGB1 is normally found within the nucleus, functioning as an architectural chromatin-binding factor and DNA chaperone with DNA binding and bending activity [151]. Nuclear HMGB1 regulates a number of DNA-associated events including recombination, replication, transcription, and repair. Loss of HMGB1 in cells and tissues increases DNA damage, cell death, nuclear DAMP release, and genomic instability, which subsequently affects many cellular processes including increased inflammatory and organelle injury [32, 152, 153]. HMGB1 not only resides in the nucleus, but can also translocate to the cytoplasm (mitochondria, endosome, and lysosome) and cell membrane and from there be released into the extracellular space.
Outside the cell, HMGB1 exhibits multiple functions in the regulation of inflammation, immunity, migration, metabolism, and autophagy. In particular, HMGB1 is actively secreted by immune cells and passively released by damaged or dead cells, which mediates the inflammatory response and contributes to poor outcomes in inflammatory-associated diseases [154, 155]. Oxidative stress or oxidative injury, a leading cause of ageing and cancer [34], is a remarkably basic mechanism responsible for HMGB1 secretion and release [156]. Several antioxidants (e.g., quercetin [157], green tea [158], and N-acetylcysteine [159]) prevent or reduce HMGB1 release and are protective in the setting of experimental infection and sterile inflammation. Remarkably, the HMGB1-mediated immune response depends on many factors including its receptor, redox status, and binding partner (e.g., DNA, histone, and lipopolysaccharide). In addition to receptor-mediated activity, HMGB1 can be activated thorough direct endocytic uptake in immune [160] and cancer cells [161], which mediates pyroptosis, enhanced bioenergetics, and alters metabolism, respectively.
Whereas HMGB1 protein expression decreases, acetylated HMGB1 increases with advancing age [162–164]. DNA double-strand breaks accumulate in the mouse brain and are specifically related to downregulated HMGB1 expression in ageing [165]. These findings indicate that HMGB1 levels and modifications may reflect the chromatin state and cellular function. Apart from absolute amount of HMGB1, the distribution of HMGB1 appears to be altered in the aged brain. HMGB1 is downregulated in neurons in the aged brain, whereas it is upregulated in astrocytes, suggesting that HMGB1 may play different roles in different types of brain cells and structures [165]. One characteristic of ageing is loss of the number of nucleosomes with increased DNA damage and epigenetic alteration [166–168]. This change can lead to age-dependent reprogramming and may drive cancer initiation.
Endogenous HMGB1 is an important regulator of nucleosome biology including organization, biogenesis, and release. It is becoming increasingly clear that loss of HMGB1 in cells or tissues reduces nucleosome sliding and stability [169], decreases intracellular nucleosome number [170], increases global gene expression with a specific transcriptomic profile [170], and promotes nucleosome release in local and systemic inflammation [32]. HMGB1 is also released from dying neuronal cells or senescent cells. Extracellular HMGB1 accelerates neuro-inflammation and senescence-associated inflammation in a receptor-dependent manner. These receptors include AGER, TLR2, and TLR4 [171, 172]. Following interaction, they activate mitogen-activated protein kinases (MAPKs), NF-κB, and phosphoinositide 3-kinases (PI3Ks)/AKT signaling pathways [34]. Of note, AGER is a multiple ligand receptor and its expression is increased in ageing and cancer [173, 174]. AGER is important for the synergistic effect between HMGB1 and DNA in the innate immune response [175, 176]. In addition to HMGB1, AGER ligands include advanced glycation endproducts (AGE), certain S100 protein, matrix proteins, and β-amyloid fibrils [177]. These ligands are a common thread in the pathogenesis of ageing, cancer, diabetes, and neurodegenerative disease [178, 179]. Interestingly, extracellular HMGB1 has the ability to promote proliferation and self-renew stem and progenitor cells partly through AGER, suggesting a positive role for HMGB1 in tissue repair and regeneration, which may limit ageing [180].
The expression level of intracellular HMGB1 is related to cellular senescence [172]. Both overexpression and knockdown of HMGB1 expression through genetic engineering technology induce a TP53-dependent senescent growth arrest, proving a link between HMGB1 and TP53 in the regulation of cellular senescence [172]. In cancer cells, binding of HMGB1 to TP53 regulates their cytosolic localization and the switch between apoptosis and autophagy [181]. These findings provide insights into their reciprocal roles in ageing and carcinogenesis.
Different from ageing [209,212], the total expression of HMGB1 is often upregulated in cancer [182]. However, the nuclear HMGB1 level is decreased in several cancer cells (e.g., pancreatic cancer) compared to normal cells [161] with reciprocal increases in cytosolic HMGB1. Intracellular HMGB1 may act as a tumor suppressor. Nuclear HMGB1 may enhance tumor suppressor (e.g., retinoblastoma protein [RB]) activity through protein-protein interaction. For instance, binding of HMGB1 to RB prevents tumorigenicity in breast cancer cells through induction of RB-dependent G1 arrest and apoptosis [183]. In addition, HMGB1 deficiency increases genome instability with telomere shortening, which contributes to tumorigenesis [170, 184, 185]. Our studies demonstrate that HMGB1 is generally a positive regulator of autophagy [186–192], although an HMGB1-independent autophagy pathway exists [193, 194]. Loss of HMGB1 in cells results in autophagy dysfunction, which may cause genome instability, inflammation, and subsequent tumorigenesis.
Apart from tumor suppressor function, extracellular HMGB1 is generally a tumor promoter in the tumor microenvironment. Extracellular HMGB1 and its receptors (e.g., AGER and TLR4) facilitate tumor development by multiple mechanisms involved in many tumor biomarkers including the inflammatory microenvironment [195–197], metabolic requirements [186, 198], invasion, and metastasis [199–202], antitumor immunity [203–205], and angiogenesis [206, 207]. Thus, HMGB1 inhibition or receptor blockade can limit tumor development. Collectively, these findings demonstrate that HMGB1 performs both oncogenic and tumor-suppressive roles and this behavior may influence clinical decisions [208].
4.2 Histone
Histone has been suggested as another important nuclear DAMP. As the basic components of nucleosomes, nuclear histones and their PTMs regulate chromosome structure, function, and gene transcription [209]. Similar to loss of HMGB1 [170], loss of nucleosome in yeast results in global transcriptional upregulation and genomic instability with elevated levels of DNA damage, retrotransposition, large-scale chromosome rearrangement, and translocation during ageing [168]. These findings suggest a common biology for intracellular nuclear DAMP in the regulation of genomic stability as well as genome chromatinization. Apart from their nuclear function, emerging studies indicate that histones as well as nucleosomes can be released following infection (e.g., sepsis) [210], sterile inflammation (e.g., trauma, ischemia-reperfusion injury, and pancreatitis) [32, 211, 212], and various types of cell death (e.g., apoptosis, necrosis, and NETosis) [213]. Some TLRs (e.g., TLR2, TLR4, and TLR9) and the NLR family, pyrin domain containing 3 (NALP3) inflammasome are essential for extracellular histone activity [211, 212, 214, 215]. After binding to their receptors, extracellular histone can activate MAPKs, NF-κB, AKT, and myeloid differentiation primary response gene 88 (MyD88)-signaling pathways [216]. Dynamic changes in circulating levels of histones as well as nucleosomes, like HMGB1, serve as potential biomarkers and novel therapeutic targets in ageing and human diseases, including cancer [79, 217, 218].
The direct link between histones and ageing and cancer has been found through investigating the PTMs of histones, which establish a so-called “histone code” as epigenetic regulators [219]. Besides methylation and acetylation, histones can be modified by ubiquitination, phosphorylation, citrullination, sumoylation, biotinylation, or ADP-ribosylation at multiple sites. As critical epigenetics regulators, histone modifications are more reversible than DNA methylation, although the underlying mechanism remains unknown. With respect to regulation of chromatin status and DNA transcription, histone-associated chromatin modifications seems to be one of the driving forces of senescence, ageing, and cancer [220–222]. The changes of histone modification have been implicated in many biological processes such as stem cell differentiation [223], inflammation [224], autophagy [225], and metabolism [226] [227] which positively or negatively affect the development of ageing and cancer. In addition to specific sites of histone PTMs contributing to ageing and cancer, histone methylation at H3K4 and H3K79 and histone acetylation at H3K9, H3K56, H4K5, H4K12 and H4K16 are associated with gene activation. In contrast, histone methylation at H3K9, H3K27, and H4K20 facilitates gene silencing. Further studies are required to clarify the relationship between histone modification profile, gene activity, and molecular properties in ageing and cancer [228].
4.3 S100
The S100 protein family consists of 24 members, characterized by low molecular weights (9–13 KDa), that take homodimer, heterodimer, and oligomers forms and undergo tissue-specific expression [229, 230]. Their name is derived from the chemical property of being soluble in 100% ammonium sulfate solution [231]. S100 proteins are structurally similar to calmodulin and have two calcium-binding motifs with helix-loop-helix (“EF-hand type”) conformation. One canonical EF-hand at the C terminus is common for all EF hand proteins, and one variant at the N terminus is unique for S100 proteins [232]. The functions of intracellular S100 proteins have been extensively studied, and most members participate in the regulation of various cellular processes such as calcium homeostasis, enzyme activities, cell growth, proliferation, differentiation and migration, protein degradation, cytoskeletal interactions, protein phosphorylation, and transcriptional factor activity [229, 230, 233]. Like several other calcium-binding proteins, some S100 proteins can be released and secreted by different cells. Among them, phagocyte-specific S100A8, S100A9, S100A12, and S100B are well-documented DAMPs with proinflammatory activity in innate immunity [234, 235]. Like other DAMPs, the secretion of S100 does not depend on the classical endoplasmic reticulum-Golgi route [236]. S100A8 and S100A9 often form the heterodimer S100A8/A9 in the extracellular space. Binding of S100A8/A9 to TLR4 mediates sepsis [237], whereas S100A12 and S100B promote AGER-dependent inflammation and migration [238, 239]. Besides S100A12 and S100B, AGER is a common receptor for many S100 proteins (S100A1, S100A4, S100A6, S100A8/A9, S100A7/A15, S100A11, S100A13, and S100P), which are involved in cancer, diabetes, neurodegeneration, and other inflammatory-associated disease [177, 178, 240–242].
The expression of S100 proteins (e.g., S100B and S100A6) is increased in ageing brains and then decreased during old age. In addition, they have different distributions in the ageing brain regions [243, 244]. Knockdown of S100A6 causes cell-cycle arrest in the G2/M phase and subsequent cellular senescence and loss of numbers of endothelial cells [245] and fibroblasts [246]. Mice overexpressing the human S100B show pathological changes in their brains [247]. In addition, serum S100 protein (e.g., S100B) concentrations are increased and related to human ageing progression [248, 249]. These findings provide direct evidence linking S100 family members to ageing. S100 genes are clustered on human chromosome 1q21. This region is also frequently rearranged in various tumors, especially in human breast carcinomas [250]. Some major forms of cancer exhibit dramatic changes in the expression of S100 proteins (e.g., S100B, S100A2, S100A4, S100A6, S100A8/A9, and S100P). Serum S100 proteins such as S100B are biomarkers of certain cancers, including malignant melanoma [251]. The S100-AGER signaling pathway in the tumor microenvironment appears to be important for many tumor biology processes and especially links inflammation and cancer progression through activation of MAPK and the NF-κB pathway. Besides tumorigenesis, our study indicated that AGER is required for S100A8-mediated autophagy in leukemia cells, which contributes to chemotherapy resistance [252]. However, some S100 proteins (e.g., S100A4, S100A6, S100A8/A9, and S100A14) can trigger AGER-dependent apoptosis in several cancer cells, suggesting that AGER plays a dual role in the regulation of survival and death depending on the context [253]. It is not clear whether AGER is required for S100 (e.g., S100A4)-mediated cancer-stromal interplay [254] as well as cancer stem cell self-renewal [255]. Some of the role of S100A4 appears to involve promotion of mesenchymal stem cell proliferation and survival [256]. In some cases, TLR4 participates in S100 protein-mediated tumor growth and metastasis [257, 258]. Future studies will be required to elucidate the relationship between AGER and TLR4 in the regulation of S100-mediated promotion of tumor growth.
4.4 HSPs
HSPs are a family of conserved, ubiquitously-expressed proteins among different species. Under normal conditions, HSPs are constitutively present in various cells, but are overexpressed when cells suffer from hyperthermia, pH shift, hypoxia, toxins, or other stress. This specific cytoprotective response, generally termed heat shock response, is a homeostatic maintenance mechanism in response to environmental stress [259, 260]. Heat shock transcription factor 1 (HSF1) is the master transcription factor for regulating HSP gene expression and the heat shock response [261]. HSPs are termed and classified according to their molecular mass, and human HSPs are composed of HSP110, HSP90, HSP70, HSP40, and small HSP as well as the human chaperonin families HSP60/HSP10 and MARVEL domain containing 2 (MARVELD2/TRIC) [262]. A key function of intracellular HSPs is molecular chaperones, which contribute to the proper folding and activation of many signaling proteins [261]. Once loss or dysfunction of HSPs occurs, misfolded proteins form aggregates which may impair and eventually kill the cell, which will cause ageing, neurological degradation disorders, and cancer [259]. Apart from acting as molecular chaperones, HSPs are also involved in protein assembly, export, turn-over, and regulation. For instance, chaperone-mediated autophagy refers to heat shock 70kDa protein 8 (HSPA8/HSC70) associated with its co-chaperones (e.g., HSP40, BCL2-associated athanogene, Hip, and Hop) delivers the substrate protein (containing a KFERQ-related motif in its amino acid sequence) directly into lysosomes for degradation [263]. In addition, some HSPs can be released during cell stress and injury and act as immunodominant molecules, triggering inflammatory and immune responses [264, 265].
Ageing has long been seen as a result of cell function errors, often of several different types, which lead to cellular dysfunction and accumulation of denatured, unfolded, and damaged proteins. As molecular chaperones and autophagic regulators, HSPs are key components in regulating ageing-related cellular phenotypes as well as lifespan through increasing protein turnover and suppression of proteotoxicity [266, 267]. Several HSP genes and proteins are upregulated during normal ageing [268], whereas pathological ageing attenuates the HSF1-HSP pathway and leads to production of toxic accumulation [269, 270]. Overexpression of HSPs (e.g., HSP70, HSP90, and small HSPs) by genetic technology extends lifespan in fruit flies [271], Caenorhabditis elegans [272, 273], and yeast [274], and protects against neurodegeneration in mice [275]. HSP accumulation in the nucleus after damage to the cell protects chromosome structure and function through limiting genomic instability [276] and preventing nuclear DAMP (e.g., HMGB1) release in response to oxidative injury and inflammatory stimuli [277, 278]. HSPs such as HSP70 can also inhibit TP53-dependnent and independent senescence as well as apoptosis. Interestingly, HSF1 regulates gene expression, contributing to longevity not only by itself, but also through interplay with other transcription factors such as FOXO3 [279–282]. Thus, a complex transcriptional control network regulates the stress response.
HSPs and HSF1 are also involved in tumor formation, development, and therapy [283–286]. Numerous studies indicate that the expression of HSPs and HSF1 is increased in many human tumors (especially of epithelial origin or gliomas) and positively correlates with tumor growth, metastases, drug resistance, and poor prognosis. Thus, suppression of HSP (e.g., HSP90, HSP70, HSP60, and HSP27) expression increases chemotherapy-induced death and inhibits tumor growth and development in vivo and in vitro [287]. In contrast, overproduction of HSPs by gene transfection increases therapy resistance and promotes tumor growth. In addition to their intracellular distribution, several HSPs (e.g., HSP90 and HSP70) present on the plasma membrane components of tumor cells [288–290] and in the extracellular space during cell death [291]. Extracellular HSP90 and HSP70 can activate the adaptive immune system [292] and elicit specific anti-tumor T-cell immunity through the induction of antigen presentation [264, 265]. TLR4 is the major receptor that recognizes HSPs exposed by tumor cells, which facilitates intracellular antigen processing and presentation [293]. Other potential cell surface receptors such as TLR2, CD40, CD91, CCR5, and members of the scavenger receptor family such as LOX-1, SREC-1, and FEEL-1 also mediate binding and uptake of HSPs into antigen-presenting cells [294, 295]. Moreover, HSF1 may be an oncogene; HSF1-deficient mice are less susceptible to chemical-induced skin tumors [285]. HSF1 regulates tumor progression through multiple mechanisms involved in the regulation of the stress response, metabolism, inflammation, and oncogene activity and senescence [285, 296, 297]. Thus, targeting HSF1 may provide a new approach in cancer therapy.
5. Conclusions and Perspectives
Ageing remains a key public health concern; slowing ageing and associated morbidity and extending our lifespan is our long-term goal. Ageing is a common high risk factor for many diseases, especially human malignancies. There are various possibilities as to how loss of cell growth (ageing) and gain of cell growth (cancer) pathways can meet. A large and growing number of hallmarks are associated with ageing and cancer. In this review, we discussed six common hallmarks contributing to the initiation, maintenance, and progression of ageing and cancer. There are still substantial gaps in our knowledge of the biological changes that lead to ageing and disease. Recent studies show that aberrant expression, translocation, and release of DAMPs might act as an intriguing upstream signal, mediating crosstalk between these common hallmarks and resulting in ageing and tumorigenesis. In healthy cells, endogenous DAMPs (e.g., HMGB1, histone, and HSPs) usually contribute to sustaining cellular homeostasis in response to mild stress. Following severe or prolonged damage, loss of intracellular DAMPs increases genomic instability, epigenetic alteration, autophagic dysfunction, effete organelles and protein aggregates. Conversely, increased DAMPs are often harmful when released, mediating an excessive inflammatory response and immune dysfunction. Among them, nuclear DAMP such as HMGB1 may be an attractive target for clinical trials in cancer/other diseases associated with ageing. A number of emergent strategies such as antibodies, peptide inhibitors, endogenous hormones, and various chemical compounds have been used to inhibit HMGB1 release and activity [298]. Several antioxidative agents such as ethyl pyruvate, quercetin, green tea, and N-acetylcysteine, curcumin have not only anti-ageing properties, but also the ability to inhibit HMGB1 release [34]. In addition, natural and synthetic SIRT1 activators show promise for the treatment of ageing and age-related diseases. They are also protective in the setting of experimental inflammation, partly through attenuating systemic HMGB1 accumulation as well as activity [122, 299].
Although the ‘DAMP Hypothesis’ provides novel insights into the pathogenic mechanism linking ageing and ageing-associate disease, many specific questions remain to be answered: Do all types of DAMPs affect ageing and cancer progression in a similar manner? What is the feedback loop regulating DAMP release and activity? Regardless of the source, how do different DAMPs communicate to fine-tune the common biology of ageing and cancer? How is the structural basis of the DAMP signaling pathway involved in disease states and ageing? How do DAMPs qualitatively and quantitatively sense different forms of stressors in different cells? An improved understanding of the role of DAMPs in ageing and ageing-associated diseases is creating a new opportunity for diagnosis and therapeutic intervention.
Acknowledgments
We thank Christine Heiner (Department of Surgery/Department of Anesthesiology, University of Pittsburgh) for her critical reading of the manuscript. Work done in support of findings reviewed in this manuscript was aided by the Core Support of the UPCI (P30CA047904 to Davidson). This work was also supported by the National Institutes of Health (R01 CA160417 to DT; R01 CA181450 to HJZ/MTL), and a 2013 Pancreatic Cancer Action Network-AACR Career Development Award (Grant Number 13-20-25-TANG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tosato M, et al. The aging process and potential interventions to extend life expectancy. Clin Interv Aging. 2007;2(3):401–12. [PMC free article] [PubMed] [Google Scholar]
- 2.Harman D. The aging process: major risk factor for disease and death. Proc Natl Acad Sci U S A. 1991;88(12):5360–3. doi: 10.1073/pnas.88.12.5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 4.Jin K. Modern Biological Theories of Aging. Aging Dis. 2010;1(2):72–74. [PMC free article] [PubMed] [Google Scholar]
- 5.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nat Rev Neurosci. 2006;7(1):30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- 7.van Heemst D. Insulin, IGF-1 and longevity. Aging Dis. 2010;1(2):147–57. [PMC free article] [PubMed] [Google Scholar]
- 8.Dorshkind K, Montecino-Rodriguez E, Signer RA. The ageing immune system: is it ever too old to become young again? Nat Rev Immunol. 2009;9(1):57–62. doi: 10.1038/nri2471. [DOI] [PubMed] [Google Scholar]
- 9.Burhans WC, Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007;35(22):7545–56. doi: 10.1093/nar/gkm1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ackermann M, et al. On the evolutionary origin of aging. Aging Cell. 2007;6(2):235–44. doi: 10.1111/j.1474-9726.2007.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448(7155):767–74. doi: 10.1038/nature05985. [DOI] [PubMed] [Google Scholar]
- 13.Lopez-Otin C, et al. The hallmarks of aging. Cell. 2013;153(6):1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Q, et al. DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy. 2013;9(4):451–8. doi: 10.4161/auto.23691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang D, et al. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249(1):158–75. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou W, et al. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell death & disease. 2013;4:e966. doi: 10.1038/cddis.2013.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aguilera A, Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9(3):204–17. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 19.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25(5):409–33. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sancar A, et al. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 21.Lombard DB, et al. DNA repair, genome stability, and aging. Cell. 2005;120(4):497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 22.Burke B, Stewart CL. The nuclear lamins: flexibility in function. Nat Rev Mol Cell Biol. 2013;14(1):13–24. doi: 10.1038/nrm3488. [DOI] [PubMed] [Google Scholar]
- 23.Vijg J, Suh Y. Genome instability and aging. Annu Rev Physiol. 2013;75:645–68. doi: 10.1146/annurev-physiol-030212-183715. [DOI] [PubMed] [Google Scholar]
- 24.Niedernhofer LJ, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444(7122):1038–43. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 25.Agrelo R, et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc Natl Acad Sci U S A. 2006;103(23):8822–7. doi: 10.1073/pnas.0600645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong KK, et al. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 2003;421(6923):643–8. doi: 10.1038/nature01385. [DOI] [PubMed] [Google Scholar]
- 27.Cremona CA, Behrens A. ATM signalling and cancer. Oncogene. 2013 doi: 10.1038/onc.2013.275. [DOI] [PubMed] [Google Scholar]
- 28.Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34(5):1416–26. doi: 10.1093/nar/gkl010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodier F, Campisi J, Bhaumik D. Two faces of p53: aging and tumor suppression. Nucleic Acids Res. 2007;35(22):7475–84. doi: 10.1093/nar/gkm744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000;14(9):1021–6. [PubMed] [Google Scholar]
- 31.Broers JL, et al. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006;86(3):967–1008. doi: 10.1152/physrev.00047.2005. [DOI] [PubMed] [Google Scholar]
- 32.Kang R, et al. Intracellular hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology. 2014;146(4):1097–107. doi: 10.1053/j.gastro.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harman D. Free radical theory of aging. Mutat Res. 1992;275(3–6):257–66. doi: 10.1016/0921-8734(92)90030-s. [DOI] [PubMed] [Google Scholar]
- 34.Tang D, et al. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal. 2011;14(7):1315–35. doi: 10.1089/ars.2010.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilson E, Geli V. How telomeres are replicated. Nat Rev Mol Cell Biol. 2007;8(10):825–38. doi: 10.1038/nrm2259. [DOI] [PubMed] [Google Scholar]
- 36.O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010;11(3):171–81. doi: 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moyzis RK, et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A. 1988;85(18):6622–6. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43(2 Pt 1):405–13. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 39.Lundblad V, Blackburn EH. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell. 1993;73(2):347–60. doi: 10.1016/0092-8674(93)90234-h. [DOI] [PubMed] [Google Scholar]
- 40.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19(18):2100–10. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 41.Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336(6081):593–7. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu EY, et al. Regulation of telomere structure and functions by subunits of the INO80 chromatin remodeling complex. Mol Cell Biol. 2007;27(16):5639–49. doi: 10.1128/MCB.00418-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dror V, Winston F. The Swi/Snf chromatin remodeling complex is required for ribosomal DNA and telomeric silencing in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24(18):8227–35. doi: 10.1128/MCB.24.18.8227-8235.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boukamp P, Popp S, Krunic D. Telomere-dependent chromosomal instability. J Investig Dermatol Symp Proc. 2005;10(2):89–94. doi: 10.1111/j.1087-0024.2005.200401.x. [DOI] [PubMed] [Google Scholar]
- 45.O’Hagan RC, et al. Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell. 2002;2(2):149–55. doi: 10.1016/s1535-6108(02)00094-6. [DOI] [PubMed] [Google Scholar]
- 46.Artandi SE, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406(6796):641–5. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 47.Polanska E, et al. HMGB1 gene knockout in mouse embryonic fibroblasts results in reduced telomerase activity and telomere dysfunction. Chromosoma. 2012 doi: 10.1007/s00412-012-0373-x. [DOI] [PubMed] [Google Scholar]
- 48.Vera E, et al. Epigenetic regulation of telomeres in human cancer. Oncogene. 2008;27(54):6817–33. doi: 10.1038/onc.2008.289. [DOI] [PubMed] [Google Scholar]
- 49.Blasco MA. The epigenetic regulation of mammalian telomeres. Nat Rev Genet. 2007;8(4):299–309. doi: 10.1038/nrg2047. [DOI] [PubMed] [Google Scholar]
- 50.Xu L, Li S, Stohr BA. The role of telomere biology in cancer. Annu Rev Pathol. 2013;8:49–78. doi: 10.1146/annurev-pathol-020712-164030. [DOI] [PubMed] [Google Scholar]
- 51.Jaskelioff M, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469(7328):102–6. doi: 10.1038/nature09603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang S. Modeling aging and cancer in the telomerase knockout mouse. Mutat Res. 2005;576(1–2):39–53. doi: 10.1016/j.mrfmmm.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 53.Donate LE, Blasco MA. Telomeres in cancer and ageing. Philos Trans R Soc Lond B Biol Sci. 2011;366(1561):76–84. doi: 10.1098/rstb.2010.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet. 2005;6(8):611–22. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- 55.Blanco R, et al. Telomerase abrogation dramatically accelerates TRF2-induced epithelial carcinogenesis. Genes Dev. 2007;21(2):206–20. doi: 10.1101/gad.406207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer. 2011;11(3):161–76. doi: 10.1038/nrc3025. [DOI] [PubMed] [Google Scholar]
- 57.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192(4):547–56. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–68. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 59.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 60.Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339(6127):1567–70. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9(16):2395–402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 62.Egger G, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–63. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 63.Christensen BC, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5(8):e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fraga MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102(30):10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bollati V, et al. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. 2009;130(4):234–9. doi: 10.1016/j.mad.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11(3):204–20. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mays-Hoopes LL. DNA methylation in aging and cancer. J Gerontol. 1989;44 (6):35–6. doi: 10.1093/geronj/44.6.35. [DOI] [PubMed] [Google Scholar]
- 68.Munoz-Najar U, Sedivy JM. Epigenetic control of aging. Antioxid Redox Signal. 2011;14(2):241–59. doi: 10.1089/ars.2010.3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 70.Rando OJ, Chang HY. Genome-wide views of chromatin structure. Annu Rev Biochem. 2009;78:245–71. doi: 10.1146/annurev.biochem.78.071107.134639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23(8):413–8. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 72.Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther. 2009;8(6):1409–20. doi: 10.1158/1535-7163.MCT-08-0860. [DOI] [PubMed] [Google Scholar]
- 73.Fernandez-Capetillo O, et al. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3(8–9):959–67. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 74.Celeste A, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296(5569):922–7. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kasinski AL, Slack FJ. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11(12):849–64. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kunej T, et al. Epigenetic regulation of microRNAs in cancer: an integrated review of literature. Mutat Res. 2011;717(1–2):77–84. doi: 10.1016/j.mrfmmm.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 77.Smith-Vikos T, Slack FJ. MicroRNAs and their roles in aging. J Cell Sci. 2012;125(Pt 1):7–17. doi: 10.1242/jcs.099200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Turchinovich A, et al. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011;39(16):7223–33. doi: 10.1093/nar/gkr254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Allam R, et al. Extracellular histones in tissue injury and inflammation. Journal of molecular medicine. 2014;92(5):465–72. doi: 10.1007/s00109-014-1148-z. [DOI] [PubMed] [Google Scholar]
- 80.Wang W, Lotze MT. Good things come in small packages: exosomes, immunity and cancer. Cancer Gene Ther. 2014;21(4):139–41. doi: 10.1038/cgt.2014.14. [DOI] [PubMed] [Google Scholar]
- 81.Vasto S, et al. Inflammation, ageing and cancer. Mech Ageing Dev. 2009;130(1–2):40–5. doi: 10.1016/j.mad.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 82.Libby P. Inflammatory mechanisms: the molecular basis of inflammation and disease. Nutr Rev. 2007;65(12 Pt 2):S140–6. doi: 10.1111/j.1753-4887.2007.tb00352.x. [DOI] [PubMed] [Google Scholar]
- 83.Senovilla L, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science. 2012;337(6102):1678–84. doi: 10.1126/science.1224922. [DOI] [PubMed] [Google Scholar]
- 84.Flannagan RS, Jaumouille V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol. 2012;7:61–98. doi: 10.1146/annurev-pathol-011811-132445. [DOI] [PubMed] [Google Scholar]
- 85.Gostner JM, et al. Redox regulation of the immune response. Redox Rep. 2013;18(3):88–94. doi: 10.1179/1351000213Y.0000000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Balistreri CR, et al. NF-kappaB pathway activators as potential ageing biomarkers: targets for new therapeutic strategies. Immun Ageing. 2013;10 (1):24. doi: 10.1186/1742-4933-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 88.Youm YH, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18(4):519–32. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zitvogel L, et al. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol. 2012;13(4):343–51. doi: 10.1038/ni.2224. [DOI] [PubMed] [Google Scholar]
- 90.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ershler WB, Keller ET. Age-associated increased interleukin-6 gene expression, late-life diseases, and frailty. Annu Rev Med. 2000;51:245–70. doi: 10.1146/annurev.med.51.1.245. [DOI] [PubMed] [Google Scholar]
- 92.Montecino-Rodriguez E, Berent-Maoz B, Dorshkind K. Causes, consequences, and reversal of immune system aging. J Clin Invest. 2013;123(3):958–65. doi: 10.1172/JCI64096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fulop T, et al. Aging, immunity, and cancer. Discov Med. 2011;11(61):537–50. [PubMed] [Google Scholar]
- 94.Kawahara TL, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009;136(1):62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kanfi Y, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483(7388):218–21. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 96.Mostoslavsky R, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124(2):315–29. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 97.Satoh A, et al. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013;18(3):416–30. doi: 10.1016/j.cmet.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sauve AA, et al. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–65. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 99.Rehman HU, Masson EA. Neuroendocrinology of ageing. Age Ageing. 2001;30(4):279–87. doi: 10.1093/ageing/30.4.279. [DOI] [PubMed] [Google Scholar]
- 100.DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148(6):1132–44. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ertel A, et al. Is cancer a metabolic rebellion against host aging? In the quest for immortality, tumor cells try to save themselves by boosting mitochondrial metabolism. Cell Cycle. 2012;11(2):253–63. doi: 10.4161/cc.11.2.19006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134(5):703–7. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 104.Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol. 2011;76:347–53. doi: 10.1101/sqb.2011.76.010678. [DOI] [PubMed] [Google Scholar]
- 105.Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. 2012;23(11):560–6. doi: 10.1016/j.tem.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bianchi G, et al. p53 and p66 proteins compete for hypoxia-inducible factor 1 alpha stabilization in young and old rat hearts exposed to intermittent hypoxia. Gerontology. 2006;52(1):17–23. doi: 10.1159/000089821. [DOI] [PubMed] [Google Scholar]
- 107.Rezvani HR, et al. Loss of epidermal hypoxia-inducible factor-1alpha accelerates epidermal aging and affects re-epithelialization in human and mouse. J Cell Sci. 2011;124(Pt 24):4172–83. doi: 10.1242/jcs.082370. [DOI] [PubMed] [Google Scholar]
- 108.Yang L, et al. PKM2 Regulates the Warburg Effect and Promotes HMGB1 Release in Sepsis. Nat Commun. 2014 doi: 10.1038/ncomms5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Krawczyk CM, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115(23):4742–9. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shi LZ, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367–76. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dang EV, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146(5):772–84. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Colman RJ, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–4. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mouchiroud L, et al. Pyruvate imbalance mediates metabolic reprogramming and mimics lifespan extension by dietary restriction in Caenorhabditis elegans. Aging Cell. 2011;10(1):39–54. doi: 10.1111/j.1474-9726.2010.00640.x. [DOI] [PubMed] [Google Scholar]
- 114.Anderson RM, Weindruch R. Metabolic reprogramming in dietary restriction. Interdiscip Top Gerontol. 2007;35:18–38. doi: 10.1159/000096554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26(9):877–90. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Basen-Engquist K, Chang M. Obesity and cancer risk: recent review and evidence. Curr Oncol Rep. 2011;13(1):71–6. doi: 10.1007/s11912-010-0139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ding Q, et al. Caloric restriction increases adiponectin expression by adipose tissue and prevents the inhibitory effect of insulin on circulating adiponectin in rats. J Nutr Biochem. 2012;23(8):867–74. doi: 10.1016/j.jnutbio.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 118.Shinmura K, et al. Cardioprotective effects of short-term caloric restriction are mediated by adiponectin via activation of AMP-activated protein kinase. Circulation. 2007;116(24):2809–17. doi: 10.1161/CIRCULATIONAHA.107.725697. [DOI] [PubMed] [Google Scholar]
- 119.Canto C, Auwerx J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol Metab. 2009;20(7):325–31. doi: 10.1016/j.tem.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Minina EA, et al. Autophagy mediates caloric restriction-induced lifespan extension in Arabidopsis. Aging Cell. 2013;12(2):327–9. doi: 10.1111/acel.12048. [DOI] [PubMed] [Google Scholar]
- 121.Hasegawa A, et al. Alternate day calorie restriction improves systemic inflammation in a mouse model of sepsis induced by cecal ligation and puncture. J Surg Res. 2012;174(1):136–41. doi: 10.1016/j.jss.2010.11.883. [DOI] [PubMed] [Google Scholar]
- 122.Rickenbacher A, et al. Fasting protects liver from ischemic injury through Sirt1-mediated downregulation of circulating HMGB1 in mice. J Hepatol. 2014;61(2):301–8. doi: 10.1016/j.jhep.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 123.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584(7):1393–8. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 124.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344–8. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105–37. doi: 10.1146/annurev-pathol-020712-163918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Maiuri MC, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 127.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–62. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 128.Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol. 2006;17(7):1807–19. doi: 10.1681/ASN.2006010083. [DOI] [PubMed] [Google Scholar]
- 129.Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol. 12(9):842–6. doi: 10.1038/ncb0910-842. [DOI] [PubMed] [Google Scholar]
- 130.Harrison DE, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460 (7253):392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 132.Zhao Z, et al. A Dual Role for UVRAG in Maintaining Chromosomal Stability Independent of Autophagy. Dev Cell. 2012 doi: 10.1016/j.devcel.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Degenhardt K, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mathew R, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21(11):1367–81. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Takahashi Y, et al. Bif-1 haploinsufficiency promotes chromosomal instability and accelerates Myc-driven lymphomagenesis via suppression of mitophagy. Blood. 2013 doi: 10.1182/blood-2012-10-459826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–10. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rao S, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi: 10.1038/ncomms4056. [DOI] [PubMed] [Google Scholar]
- 138.Law BK. Rapamycin: an anti-cancer immunosuppressant? Crit Rev Oncol Hematol. 2005;56(1):47–60. doi: 10.1016/j.critrevonc.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 139.Kimura T, et al. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73(1):3–7. doi: 10.1158/0008-5472.CAN-12-2464. [DOI] [PubMed] [Google Scholar]
- 140.Liu L, et al. DAMP-mediated autophagy contributes to drug resistance. Autophagy. 2011;7(1):112–4. doi: 10.4161/auto.7.1.14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 142.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 143.Matzinger P. Friendly and dangerous signals: is the tissue in control? Nat Immunol. 2007;8(1):11–3. doi: 10.1038/ni0107-11. [DOI] [PubMed] [Google Scholar]
- 144.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4(6):469–78. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 145.Tang D, et al. PAMPs and DAMPs: Signal 0’s that Spur Autophagy and Immunity. Immunol Rev. 2012 doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Krysko O, et al. Many faces of DAMPs in cancer therapy. Cell death & disease. 2013;4:e631. doi: 10.1038/cddis.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Krysko DV, et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860–75. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 148.Carta S, et al. DAMPs and inflammatory processes: the role of redox in the different outcomes. J Leukoc Biol. 2009;86(3):549–55. doi: 10.1189/jlb.1008598. [DOI] [PubMed] [Google Scholar]
- 149.Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007;28(10):429–36. doi: 10.1016/j.it.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 150.Calogero S, et al. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat Genet. 1999;22(3):276–80. doi: 10.1038/10338. [DOI] [PubMed] [Google Scholar]
- 151.Muller S, et al. New EMBO members’ review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. Embo J. 2001;20 (16):4337–40. doi: 10.1093/emboj/20.16.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yanai H, et al. Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci U S A. 2013;110(51):20699–704. doi: 10.1073/pnas.1320808110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Huang H, et al. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection. Hepatology. 2014;59(5):1984–97. doi: 10.1002/hep.26976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 155.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 156.Tang D, et al. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. 2007;81(3):741–7. doi: 10.1189/jlb.0806540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tang D, et al. Quercetin Prevents Lipopolysaccharide-induced HMGB1 Release and Proinflammatory Function. Am J Respir Cell Mol Biol. 2009;41 (6):651–660. doi: 10.1165/rcmb.2008-0119OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li W, et al. A Major Ingredient of Green Tea Rescues Mice from Lethal Sepsis Partly by Inhibiting HMGB1. PLoS ONE. 2007;2(11):e1153. doi: 10.1371/journal.pone.0001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tsung A, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204(12):2913–23. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Xu J, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014 doi: 10.1038/cdd.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kang R, et al. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene. 2014;33(5):567–77. doi: 10.1038/onc.2012.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Prasad S, Thakur MK. In vitro acetylation of the liver HMG non-histone proteins and its modulation by spermine and dexamethasone during aging of rats. Mol Biol Rep. 1988;13(4):221–4. doi: 10.1007/BF00788174. [DOI] [PubMed] [Google Scholar]
- 163.Thakur MK, Prasad S. Analysis of age-associated alteration in the synthesis of HMG nonhistone proteins of the rat liver. Mol Biol Rep. 1991;15(1):19–24. doi: 10.1007/BF00369896. [DOI] [PubMed] [Google Scholar]
- 164.Prasad S, Thakur MK. Distribution of high mobility group proteins in different tissues of rats during aging. Biochem Int. 1990;20(4):687–95. [PubMed] [Google Scholar]
- 165.Enokido Y, et al. Age-dependent change of HMGB1 and DNA double-strand break accumulation in mouse brain. Biochem Biophys Res Commun. 2008;376(1):128–33. doi: 10.1016/j.bbrc.2008.08.108. [DOI] [PubMed] [Google Scholar]
- 166.O’Sullivan RJ, et al. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol. 2010;17(10):1218–25. doi: 10.1038/nsmb.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Feser J, et al. Elevated histone expression promotes life span extension. Mol Cell. 2010;39(5):724–35. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hu Z, et al. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014;28(4):396–408. doi: 10.1101/gad.233221.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bonaldi T, et al. The DNA chaperone HMGB1 facilitates ACF/CHRAC-dependent nucleosome sliding. Embo J. 2002;21(24):6865–73. doi: 10.1093/emboj/cdf692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Celona B, et al. Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol. 2011;9(6):e1001086. doi: 10.1371/journal.pbio.1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gao HM, et al. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci. 2011;31(3):1081–92. doi: 10.1523/JNEUROSCI.3732-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Davalos AR, et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201(4):613–29. doi: 10.1083/jcb.201206006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Simm A, et al. Age associated changes of AGE-receptor expression: RAGE upregulation is associated with human heart dysfunction. Exp Gerontol. 2004;39 (3):407–13. doi: 10.1016/j.exger.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 174.Kang R, et al. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc Natl Acad Sci U S A. 2012;109(18):7031–6. doi: 10.1073/pnas.1113865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu L, et al. HMGB1-DNA Complex-induced Autophagy Limits AIM2 Inflammasome Activation through RAGE. Biochem Biophys Res Commun. 2014 doi: 10.1016/j.bbrc.2014.06.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tian J, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–96. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 177.Sparvero LJ, et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med. 2009;7:17. doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ramasamy R, et al. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. 2005;15(7):16R–28R. doi: 10.1093/glycob/cwi053. [DOI] [PubMed] [Google Scholar]
- 179.Toth C, Martinez J, Zochodne DW. RAGE, diabetes, and the nervous system. Curr Mol Med. 2007;7(8):766–76. doi: 10.2174/156652407783220705. [DOI] [PubMed] [Google Scholar]
- 180.Sorci G, et al. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta. 2013;1833(1):101–9. doi: 10.1016/j.bbamcr.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 181.Livesey KM, et al. p53/HMGB1 Complexes Regulate Autophagy and Apoptosis. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-11-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tang D, et al. High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799(1–2):131–40. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jiao Y, Wang HC, Fan SJ. Growth suppression and radiosensitivity increase by HMGB1 in breast cancer. Acta Pharmacol Sin. 2007;28(12):1957–67. doi: 10.1111/j.1745-7254.2007.00669.x. [DOI] [PubMed] [Google Scholar]
- 184.Giavara S, et al. Yeast Nhp6A/B and mammalian Hmgb1 facilitate the maintenance of genome stability. Curr Biol. 2005;15(1):68–72. doi: 10.1016/j.cub.2004.12.065. [DOI] [PubMed] [Google Scholar]
- 185.Polanska E, et al. HMGB1 gene knockout in mouse embryonic fibroblasts results in reduced telomerase activity and telomere dysfunction. Chromosoma. 2012;121(4):419–31. doi: 10.1007/s00412-012-0373-x. [DOI] [PubMed] [Google Scholar]
- 186.Tang D, et al. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metabolism. 2011;13(6):701–11. doi: 10.1016/j.cmet.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tang D, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881–92. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Tang D, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29(38):5299–310. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Livesey KM, et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012;72(8):1996–2005. doi: 10.1158/0008-5472.CAN-11-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Huang J, et al. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res. 2012;72(1):230–8. doi: 10.1158/0008-5472.CAN-11-2001. [DOI] [PubMed] [Google Scholar]
- 191.Liu L, et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia. 2011;25(1):23–31. doi: 10.1038/leu.2010.225. [DOI] [PubMed] [Google Scholar]
- 192.Tang D, et al. High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid Redox Signal. 2011;15(8):2185–95. doi: 10.1089/ars.2010.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tang D, et al. HMGB1 Phenotypic Role Revealed with Stress. Mol Med. 2014 doi: 10.2119/molmed.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Huebener P, et al. High-mobility group box 1 is dispensable for autophagy, mitochondrial quality control, and organ function in vivo. Cell Metab. 2014;19 (3):539–47. doi: 10.1016/j.cmet.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gebhardt C, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008;205(2):275–85. doi: 10.1084/jem.20070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mittal D, et al. TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. Embo J. 2010;29(13):2242–52. doi: 10.1038/emboj.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bald T, et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature. 2014 doi: 10.1038/nature13111. [DOI] [PubMed] [Google Scholar]
- 198.Kang R, et al. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene. 2013 doi: 10.1038/onc.2012.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kuniyasu H, et al. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J Pathol. 2002;196(2):163–70. doi: 10.1002/path.1031. [DOI] [PubMed] [Google Scholar]
- 200.Sasahira T, et al. Expression of receptor for advanced glycation end products and HMGB1/amphoterin in colorectal adenomas. Virchows Arch. 2005;446(4):411–5. doi: 10.1007/s00428-005-1210-x. [DOI] [PubMed] [Google Scholar]
- 201.Taguchi A, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405(6784):354–60. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 202.Huttunen HJ, et al. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002;62(16):4805–11. [PubMed] [Google Scholar]
- 203.Kusume A, et al. Suppression of dendritic cells by HMGB1 is associated with lymph node metastasis of human colon cancer. Pathobiology. 2009;76(4):155–62. doi: 10.1159/000218331. [DOI] [PubMed] [Google Scholar]
- 204.Liu Z, Falo LD, Jr, You Z. Knockdown of HMGB1 in tumor cells attenuates their ability to induce regulatory T cells and uncovers naturally acquired CD8 T cell-dependent antitumor immunity. J Immunol. 2011;187(1):118–25. doi: 10.4049/jimmunol.1003378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.He Y, et al. Tissue damage-associated ‘danger signals’ influence T cell responses that promote the progression of pre-neoplasia to cancer. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-12-2704. [DOI] [PubMed] [Google Scholar]
- 206.van Beijnum JR, et al. Tumor angiogenesis is enforced by autocrine regulation of high-mobility group box 1. Oncogene. 2012 doi: 10.1038/onc.2012.49. [DOI] [PubMed] [Google Scholar]
- 207.Sasahira T, et al. The expression of receptor for advanced glycation end products is associated with angiogenesis in human oral squamous cell carcinoma. Virchows Arch. 2007;450(3):287–95. doi: 10.1007/s00428-006-0359-2. [DOI] [PubMed] [Google Scholar]
- 208.Kang R, et al. HMGB1 in Cancer: Good, Bad, or Both? Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-13-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–99. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 210.Xu J, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15(11):1318–21. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Huang H, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll-like receptor 9 in mice. Hepatology. 2011;54(3):999–1008. doi: 10.1002/hep.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Xu J, et al. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187(5):2626–31. doi: 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wu D, et al. Apoptotic release of histones from nucleosomes. J Biol Chem. 2002;277(14):12001–8. doi: 10.1074/jbc.M109219200. [DOI] [PubMed] [Google Scholar]
- 214.Huang H, et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J Immunol. 2013;191(5):2665–79. doi: 10.4049/jimmunol.1202733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Allam R, et al. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur J Immunol. 2013;43(12):3336–42. doi: 10.1002/eji.201243224. [DOI] [PubMed] [Google Scholar]
- 216.Chen R, et al. Release and activity of histone in diseases. Cell Death Dis. 2014 doi: 10.1038/cddis.2014.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Holdenrieder S, et al. Clinical relevance of circulating nucleosomes in cancer. Ann N Y Acad Sci. 2008;1137:180–9. doi: 10.1196/annals.1448.012. [DOI] [PubMed] [Google Scholar]
- 218.Chen R, et al. Release and activity of histone in diseases. Cell death & disease. 2014;5:e1370. doi: 10.1038/cddis.2014.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 220.Dimauro T, David G. Chromatin modifications: the driving force of senescence and aging? Aging (Albany NY) 2009;1(2):182–90. doi: 10.18632/aging.100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Waldmann T, Schneider R. Targeting histone modifications--epigenetics in cancer. Curr Opin Cell Biol. 2013;25(2):184–9. doi: 10.1016/j.ceb.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 222.Han S, Brunet A. Histone methylation makes its mark on longevity. Trends Cell Biol. 2012;22(1):42–9. doi: 10.1016/j.tcb.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lunyak VV, Rosenfeld MG. Epigenetic regulation of stem cell fate. Hum Mol Genet. 2008;17(R1):R28–36. doi: 10.1093/hmg/ddn149. [DOI] [PubMed] [Google Scholar]
- 224.Nicodeme E, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–23. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fullgrabe J, et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500(7463):468–71. doi: 10.1038/nature12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Shyh-Chang N, et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339(6116):222–6. doi: 10.1126/science.1226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Sebastian C, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151(6):1185–99. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Karlic R, et al. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 2010;107(7):2926–31. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Donato R, et al. Functions of S100 proteins. Curr Mol Med. 2013;13(1):24–57. [PMC free article] [PubMed] [Google Scholar]
- 230.Heizmann CW, Fritz G, Schafer BW. S100 proteins: structure, functions and pathology. Front Biosci. 2002;7:d1356–68. doi: 10.2741/A846. [DOI] [PubMed] [Google Scholar]
- 231.Moore BW. A soluble protein characteristic of the nervous system. Biochem Biophys Res Commun. 1965;19(6):739–44. doi: 10.1016/0006-291x(65)90320-7. [DOI] [PubMed] [Google Scholar]
- 232.Schafer BW, Heizmann CW. The S100 family of EF-hand calcium-binding proteins: functions and pathology. Trends Biochem Sci. 1996;21(4):134–40. doi: 10.1016/s0968-0004(96)80167-8. [DOI] [PubMed] [Google Scholar]
- 233.Kligman D, Hilt DC. The S100 protein family. Trends Biochem Sci. 1988;13(11):437–43. doi: 10.1016/0968-0004(88)90218-6. [DOI] [PubMed] [Google Scholar]
- 234.Foell D, et al. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81(1):28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- 235.Roth J, Goebeler M, Sorg C. S100A8 and S100A9 in inflammatory diseases. Lancet. 2001;357(9261):1041. doi: 10.1016/S0140-6736(05)71610-X. [DOI] [PubMed] [Google Scholar]
- 236.Rammes A, et al. Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J Biol Chem. 1997;272(14):9496–502. doi: 10.1074/jbc.272.14.9496. [DOI] [PubMed] [Google Scholar]
- 237.Vogl T, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13(9):1042–9. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 238.Hofmann MA, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97 (7):889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 239.Bianchi R, et al. S100B protein stimulates microglia migration via RAGE-dependent up-regulation of chemokine expression and release. J Biol Chem. 2011;286(9):7214–26. doi: 10.1074/jbc.M110.169342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Leclerc E, et al. Binding of S100 proteins to RAGE: an update. Biochim Biophys Acta. 2009;1793(6):993–1007. doi: 10.1016/j.bbamcr.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 241.Donato R. RAGE: a single receptor for several ligands and different cellular responses: the case of certain S100 proteins. Curr Mol Med. 2007;7(8):711–24. doi: 10.2174/156652407783220688. [DOI] [PubMed] [Google Scholar]
- 242.Heizmann CW, Ackermann GE, Galichet A. Pathologies involving the S100 proteins and RAGE. Subcell Biochem. 2007;45:93–138. doi: 10.1007/978-1-4020-6191-2_5. [DOI] [PubMed] [Google Scholar]
- 243.Linnemann D, Skarsfelt T. Regional changes in expression of NCAM, GFAP, and S100 in aging rat brain. Neurobiol Aging. 1994;15(5):651–5. doi: 10.1016/0197-4580(94)00060-3. [DOI] [PubMed] [Google Scholar]
- 244.Tiu SC, et al. Differential expression of S100B and S100A6(1) in the human fetal and aged cerebral cortex. Brain Res Dev Brain Res. 2000;119(2):159–68. doi: 10.1016/s0165-3806(99)00151-0. [DOI] [PubMed] [Google Scholar]
- 245.Bao L, et al. The S100A6 calcium-binding protein regulates endothelial cell-cycle progression and senescence. Febs J. 2012;279(24):4576–88. doi: 10.1111/febs.12044. [DOI] [PubMed] [Google Scholar]
- 246.Slomnicki LP, Lesniak W. S100A6 (calcyclin) deficiency induces senescence-like changes in cell cycle, morphology and functional characteristics of mouse NIH 3T3 fibroblasts. J Cell Biochem. 2010;109(3):576–84. doi: 10.1002/jcb.22434. [DOI] [PubMed] [Google Scholar]
- 247.Shapiro LA, Marks A, Whitaker-Azmitia PM. Increased clusterin expression in old but not young adult S100B transgenic mice: evidence of neuropathological aging in a model of Down Syndrome. Brain Res. 2004;1010(1–2):17–21. doi: 10.1016/j.brainres.2003.12.057. [DOI] [PubMed] [Google Scholar]
- 248.Gazzolo D, et al. Pediatric concentrations of S100B protein in blood: age- and sex-related changes. Clin Chem. 2003;49(6 Pt 1):967–70. doi: 10.1373/49.6.967. [DOI] [PubMed] [Google Scholar]
- 249.Portela LV, et al. The serum S100B concentration is age dependent. Clin Chem. 2002;48(6 Pt 1):950–2. [PubMed] [Google Scholar]
- 250.Engelkamp D, et al. Six S100 genes are clustered on human chromosome 1q21: identification of two genes coding for the two previously unreported calcium-binding proteins S100D and S100E. Proc Natl Acad Sci U S A. 1993;90 (14):6547–51. doi: 10.1073/pnas.90.14.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Harpio R, Einarsson R. S100 proteins as cancer biomarkers with focus on S100B in malignant melanoma. Clin Biochem. 2004;37(7):512–8. doi: 10.1016/j.clinbiochem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 252.Yang M, et al. S100A8 Contributes to Drug Resistance by Promoting Autophagy in Leukemia Cells. PLoS ONE. 2014;9(5):e97242. doi: 10.1371/journal.pone.0097242. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 253.Chen H, et al. S100 protein family in human cancer. American journal of cancer research. 2014;4(2):89–115. [PMC free article] [PubMed] [Google Scholar]
- 254.Schmidt-Hansen B, et al. Functional significance of metastasis-inducing S100A4(Mts1) in tumor-stroma interplay. J Biol Chem. 2004;279(23):24498–504. doi: 10.1074/jbc.M400441200. [DOI] [PubMed] [Google Scholar]
- 255.Lo JF, et al. The epithelial-mesenchymal transition mediator S100A4 maintains cancer-initiating cells in head and neck cancers. Cancer Res. 2011;71 (5):1912–23. doi: 10.1158/0008-5472.CAN-10-2350. [DOI] [PubMed] [Google Scholar]
- 256.Eisenbacher JL, et al. S100A4 and Uric Acid Promote Mesenchymal Stromal Cell Induction of IL-10+/IDO+ Lymphocytes. J Immunol. 2014;192(12):6102–10. doi: 10.4049/jimmunol.1303144. [DOI] [PubMed] [Google Scholar]
- 257.Kallberg E, et al. S100A9 interaction with TLR4 promotes tumor growth. PLoS ONE. 2012;7(3):e34207. doi: 10.1371/journal.pone.0034207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Hiratsuka S, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10(11):1349–55. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 259.Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb Symp Quant Biol. 2011;76:91–9. doi: 10.1101/sqb.2012.76.010637. [DOI] [PubMed] [Google Scholar]
- 260.Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. 2010;40(2):253–66. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 261.Feder ME, Hofmann GE. Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu Rev Physiol. 1999;61:243–82. doi: 10.1146/annurev.physiol.61.1.243. [DOI] [PubMed] [Google Scholar]
- 262.Kampinga HH, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14(1):105–11. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Chiang HL, et al. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246(4928):382–5. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- 264.Tsan MF, Gao B. Heat shock proteins and immune system. J Leukoc Biol. 2009;85(6):905–10. doi: 10.1189/jlb.0109005. [DOI] [PubMed] [Google Scholar]
- 265.Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol. 2002;2(3):185–94. doi: 10.1038/nri749. [DOI] [PubMed] [Google Scholar]
- 266.Tower J. Hsps and aging. Trends Endocrinol Metab. 2009;20(5):216–22. doi: 10.1016/j.tem.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Balch WE, et al. Adapting proteostasis for disease intervention. Science. 2008;319(5865):916–9. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 268.Wang HD, Kazemi-Esfarjani P, Benzer S. Multiple-stress analysis for isolation of Drosophila longevity genes. Proc Natl Acad Sci U S A. 2004;101(34):12610–5. doi: 10.1073/pnas.0404648101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Perez FP, et al. Longevity pathways: HSF1 and FoxO pathways, a new therapeutic target to prevent age-related diseases. Curr Aging Sci. 2012;5(2):87–95. doi: 10.2174/1874609811205020087. [DOI] [PubMed] [Google Scholar]
- 270.Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300(5622):1142–5. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- 271.Tatar M, Khazaeli AA, Curtsinger JW. Chaperoning extended life. Nature. 1997;390(6655):30. doi: 10.1038/36237. [DOI] [PubMed] [Google Scholar]
- 272.Yokoyama K, et al. Extended longevity of Caenorhabditis elegans by knocking in extra copies of hsp70F, a homolog of mot-2 (mortalin)/mthsp70/Grp75. FEBS Lett. 2002;516(1–3):53–7. doi: 10.1016/s0014-5793(02)02470-5. [DOI] [PubMed] [Google Scholar]
- 273.Morrow G, et al. Overexpression of the small mitochondrial Hsp22 extends Drosophila life span and increases resistance to oxidative stress. Faseb J. 2004;18(3):598–9. doi: 10.1096/fj.03-0860fje. [DOI] [PubMed] [Google Scholar]
- 274.Erjavec N, et al. Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 2007;21(19):2410–21. doi: 10.1101/gad.439307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Cummings CJ, et al. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10(14):1511–8. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- 276.Niedzwiecki A, Fleming JE. Changes in protein turnover after heat shock are related to accumulation of abnormal proteins in aging Drosophila melanogaster. Mech Ageing Dev. 1990;52(2–3):295–304. doi: 10.1016/0047-6374(90)90133-z. [DOI] [PubMed] [Google Scholar]
- 277.Tang D, et al. Nuclear heat shock protein 72 as a negative regulator of oxidative stress (hydrogen peroxide)-induced HMGB1 cytoplasmic translocation and release. J Immunol. 2007;178(11):7376–84. doi: 10.4049/jimmunol.178.11.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Tang D, et al. The anti-inflammatory effects of heat shock protein 72 involve inhibition of high-mobility-group box 1 release and proinflammatory function in macrophages. J Immunol. 2007;179(2):1236–44. doi: 10.4049/jimmunol.179.2.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Seo K, et al. Heat shock factor 1 mediates the longevity conferred by inhibition of TOR and insulin/IGF-1 signaling pathways in C. elegans. Aging Cell. 2013;12(6):1073–81. doi: 10.1111/acel.12140. [DOI] [PubMed] [Google Scholar]
- 280.Carranco R, et al. Repression by an auxin/indole acetic acid protein connects auxin signaling with heat shock factor-mediated seed longevity. Proc Natl Acad Sci U S A. 2010;107(50):21908–13. doi: 10.1073/pnas.1014856107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell. 2004;15(2):657–64. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 283.Arrigo AP. Editorial: heat shock proteins in cancer. Curr Mol Med. 2012;12 (9):1099–101. doi: 10.2174/156652412803306738. [DOI] [PubMed] [Google Scholar]
- 284.Sherman M, Multhoff G. Heat shock proteins in cancer. Ann N Y Acad Sci. 2007;1113:192–201. doi: 10.1196/annals.1391.030. [DOI] [PubMed] [Google Scholar]
- 285.Dai C, et al. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130(6):1005–18. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Melcher A, et al. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat Med. 1998;4(5):581–7. doi: 10.1038/nm0598-581. [DOI] [PubMed] [Google Scholar]
- 287.Meng L, et al. Heat shock protein Hsp72 plays an essential role in Her2-induced mammary tumorigenesis. Oncogene. 2011;30(25):2836–45. doi: 10.1038/onc.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Shin BK, et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 2003;278(9):7607–16. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- 289.Multhoff G, et al. A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int J Cancer. 1995;61(2):272–9. doi: 10.1002/ijc.2910610222. [DOI] [PubMed] [Google Scholar]
- 290.Garg AD, et al. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother. 2012;61(2):215–21. doi: 10.1007/s00262-011-1184-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Saito K, Dai Y, Ohtsuka K. Enhanced expression of heat shock proteins in gradually dying cells and their release from necrotically dead cells. Exp Cell Res. 2005;310(1):229–36. doi: 10.1016/j.yexcr.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 292.Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science. 1995;269(5230):1585–8. doi: 10.1126/science.7545313. [DOI] [PubMed] [Google Scholar]
- 293.Asea A, et al. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6(4):435–42. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 294.Calderwood SK, et al. Extracellular heat shock proteins in cell signaling. FEBS Lett. 2007;581(19):3689–94. doi: 10.1016/j.febslet.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 295.Binder RJ. Hsp receptors: the cases of identity and mistaken identity. Curr Opin Mol Ther. 2009;11(1):62–71. [PubMed] [Google Scholar]
- 296.Gabai VL, et al. Heat shock transcription factor Hsf1 is involved in tumor progression via regulation of hypoxia-inducible factor 1 and RNA-binding protein HuR. Mol Cell Biol. 2012;32(5):929–40. doi: 10.1128/MCB.05921-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Meng L, V, Gabai L, Sherman MY. Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene. 2010;29(37):5204–13. doi: 10.1038/onc.2010.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Kang R, et al. HMGB1 in health and disease. Mol Aspects Med. 2014 doi: 10.1016/j.mam.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Kim YS, et al. SIRT1 modulates high-mobility group box 1-induced osteoclastogenic cytokines in human periodontal ligament cells. J Cell Biochem. 2010;111(5):1310–20. doi: 10.1002/jcb.22858. [DOI] [PubMed] [Google Scholar]