

Abstract
Oxidation of alcohols and amines is catalyzed by multiple families of flavin-and pyridine nucleotide-dependent enzymes. Measurement of solvent isotope effects provides a unique mechanistic probe of the timing of the cleavage of the OH and NH bonds, necessary information for a complete description of the catalytic mechanism. The inherent ambiguities in interpretation of solvent isotope effects can be significantly decreased if isotope effects arising from isotopically labeled substrates are measured in combination with solvent isotope effects. The application of combined solvent and substrate (mainly deuterium) isotope effects to multiple enzymes is described here to illustrate the range of mechanistic insights that such an approach can provide.
Keywords: Solvent isotope effect, flavoprotein, oxidase, dehydrogenase, enzyme kinetics, kinetic isotope effect
1. Introduction
Solvent isotope effects, in which one determines the effect(s) on a reaction of replacing water as the solvent with deuterium oxide, can have advantages over other kinds of isotope effects. There is no need to synthesize an isotopically labeled substrate, often the limiting factor in measurement of a kinetic isotope effect; instead one simply makes up solutions in D2O instead of H2O. As a result, solvent isotope effects can be simple to measure experimentally. In addition, in the case of exchangeable protons such as those on carbon, oxygen, or sulfur, use of D2O as solvent is the only way to incorporate the heavier isotope. Conversely, interpretation of solvent isotope effects can be complex. Because all exchangeable protons are replaced in D2O, multiple protons in the substrate can be replaced. In addition, many protons in the enzyme are exchanged in D2O; this can have subtle effects on activity and structure. The properties of D2O as solvent are not identical to those of H2O; the former is ∼24% more viscous than H2O at 25 °C and 20% more viscous at 37 °C [1, 2]. More critically, pKa values are altered in D2O and proper controls must be carried out to compensate for this. Despite these complications, when properly measured solvent isotope effects are an indispensable probe of reactions involving solvent-exchangeable protons, such as those in alcohols or amines.
The measurement of isotope effects arising from substrates that are isotopically substituted at non-exchangeable positions can be highly effective in addressing the inherent ambiguities in interpreting solvent isotope effects. The present review focuses on the combination of solvent isotope effects with other isotope effects to probe the mechanisms of amine and alcohol oxidations by enzymes. It is not meant to be a comprehensive review of the mechanisms of enzyme-catalyzed alcohol and amine oxidation or of the application of solvent isotope effects to study enzyme-catalyzed reactions.
2. Measurement of solvent isotope effects
There have been a number of comprehensive treatments of the theory and analysis of solvent isotope effects [3-5], and the reader is directed to those for a more comprehensive treatment. Typically, solvent kies arising from transfer of a single proton from nitrogen or oxygen are in the range 1.5-3, while transfer of a proton from sulfur yields an inverse solvent isotope effect that can be as low as 0.5 [4]. Inverse solvent isotope effects of a similar magnitude have also been associated with low-barrier hydrogen bonds and metal-bound hydroxide or alkoxide [6]. Normal and inverse solvent kies can also arise from the combined effects of a large number of small isotope effects; one possible origin for such effects is a change in the relative amounts of different conformational substates when a protein is transferred to D2O.
For most enzymes key active-site residues must be properly protonated for tight binding and/or catalysis, so that the activity of most enzymes is sensitive to solution pH. The pKa values of these residues will exhibit solvent isotope effects, in that the pKa value will shift in D2O, increasing by 0.3-0.7 [3]. Similar shifts will occur in the pKa values for buffers. There is also a solvent isotope effect on the glass electrode commonly used to measure pH, so that 0.4 must be added to the pH meter reading to obtain the correct pD for a buffer in D2O [7]. Because of the likelihood of a shift in the pH dependence of an enzyme in D2O, measurement of a solvent isotope effects requires that the pH/D dependence of the kinetic parameter(s) of interest, usually kcat and kcat/Km, be determined in both H2O and D2O. One can then determine the solvent isotope effect on the kinetic parameter in a pH/D-insensitive region of the profile.
A key question in interpretation of a solvent isotope effect is knowledge of the number of protons contributing to the observed effect. This is typically addressed by carrying out a proton inventory, in which one determines the solvent isotope effect in mixtures of D2O and H2O [5, 8]. The data are then fit to a version of the Kresge-Gross-Butler equation (eq 1), which describes changes in the state of the proton as it goes from the reactant (R) state to the transition (T) state. Here, k0 is the kinetic parameter of interest in H2O, kn is the kinetic parameter of interest in a solution containing a mole fraction of D2O of n, x is the number of protons in the reactant or transition state, ϕ is the respective fractionation factor, and (Z)n reflects a medium effect. The convention in treating solvent isotope effects has been to describe the properties of the proton in the R and T states in terms of fractionation factors, which are equilibria for isotope exchange reactions. For many reactions, the reactant has the same fractionation factor as solvent, so that the solvent isotope effect can be attributed only to a change in the state of the proton(s) in going to the transition state. The data can then be fit to the much simpler eq 2, where the isotope effect (kie) is simply the inverse of the transition state fractionation factor. A reaction in which a single exchangeable proton is in flight in the transition state (x = 1) will yield a linear relationship between the observed isotope effect and the fraction of D2O. If more than one exchangeable proton is involved (x>1), the inventory is bowl-shaped. It can be very difficult to measure the kinetics with sufficient precision to distinguish whether a curved inventory arises from 2 or more than 2 protons. A solvent isotope effect due to a combination of a large number of small effects, referred to as a medium effect, will yield the greatest curvature in the solvent inventory plot. While one could simply use eq 1 with a large and arbitrary value of a, such data should instead be fit to eq 3. Here Z is a fractionation factor, the inverse of the isotope effect. In some cases, curved proton inventories can occur if the solvent isotope effect being measured is suppressed from the intrinsic value due the presence of another solvent-insensitive step of comparable magnitude, so that there is a change in the commitments [9].
| (1) |
| (2) |
| (3) |
3. Utility of multiple isotope effects
A complete description of the structure of the transition state for a reaction requires knowledge of the changes in all of the bonds in the substrate when the transition state is formed. This is seldom done due to the amount of labor involved. However, at a minimum one would want to determine the effect of isotopic substitution of all atoms undergoing bond cleavage or formation. This is simplest if the reaction is concerted, with a single transition state. However, it is not uncommon for more than one step in an enzyme-catalyzed reaction to involve breaking of a bond. An important application of the use of multiple isotope effects is discrimination between concerted and stepwise reaction [10]. If a reaction is concerted, the isotope effect due to substitution of one reacting atom with a heavier isotope will be unaffected or increase if the isotope effect is measured using substrate in which another reacting atom is substituted with a heavier isotope. The former occurs if the isotope effect being measured is the intrinsic value, while the latter occurs if the measured isotope effect is decreased by kinetic complexity. Conversely, if a reaction is stepwise and the two steps exhibit isotope effects arising from substitution of different atoms, the isotope effect measured for one isotope will be smaller when the substrate is also substituted with the other heavier isotope.
4. Isotope effects on alcohol oxidation
4.1 Flavoenzymes
The application of isotope effects to study the mechanisms of flavoproteins that catalyze oxidation of alcohols or amines is often simplified by their steady-state kinetic mechanisms. Typically, oxidation of the substrate and concomitant reduction of the flavin occurs in the absence of any interaction with the second substrate; in addition, this reductive half-reaction is often irreversible. The subsequent oxidation of the reduced flavin then occurs by electron transfer to oxygen to form hydrogen peroxide in the oxidases or transfer to another redox cofactor in the dehydrogenases. A result of an irreversible reductive half-reaction is that the kcat/Km value of alcohol or amine substrate is independent of the concentration of the oxidizing substrate. The separate reductive and oxidative half-reactions and the sensitivity of the flavin visible absorbance spectrum to redox state also makes these enzymes very amenable to single turnover methods, in that the reductive half-reaction can usually be analyzed in the absence of the oxidizing substrate, allowing the rate constant for flavin reduction by the substrate to be measured directly.
A number of flavoproteins that catalyze the oxidation of alcohols to aldehydes belong to the GMC oxidoreductase family. These enzymes have a common fold that was first identified from the sequences of glucose dehydrogenase, glucose oxidase, methanol oxidase, and choline dehydrogenase [11] and subsequently confirmed by three-dimensional structures [12]. Methanol oxidase (also known as alcohol oxidase) appears to be the first GMC oxidoreductase for which solvent isotope effects were used as a mechanistic probe. Sherry and Abeles reported that the enzyme from Hansenula polymorpha was inactivated by cyclopropanol, and that the inactivated enzyme contained an adduct of the flavin with a ring-opened form of the inhibitor [13]. Two mechanisms were considered for the inactivation (Figure 1). In both the reaction is initiated by abstraction of the hydroxyl proton by an active-site base. In path i opening of the cyclopropyl ring in the alkoxide generates a transient carbanion that attacks the flavin. In path ii an electron is transferred from the alkoxide to the flavin to generate the cyclopropoxy and flavin radicals. Rapid opening of the ring in the former to yield a 3-propanol radical would be followed by recombination of the two radicals to form the adduct. The radical mechanism was favored because no adduct was detected when the native FAD was replaced with 5-deazaFAD as the cofactor. This synthetic flavin is generally considered incompetent in radical reactions [14].
Figure 1.
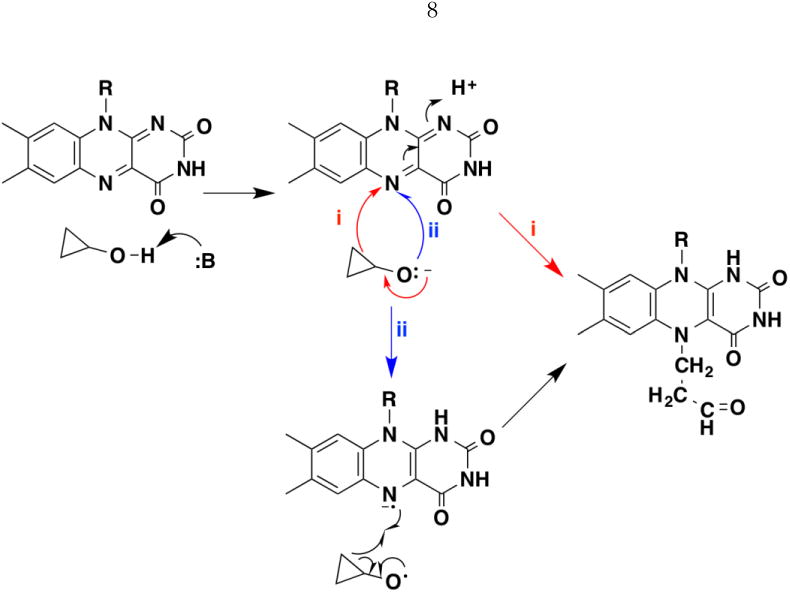
Proposed mechanisms for inactivation of methanol oxidase by cyclopropyl alcohol.
The mechanism of methanol oxidase was re-examined by Menon et al. [15] using isotope effects on the oxidation of 2-substituted ethanols and p-substituted benzyl alcohols. The kcat/Km values for substituted benzyl alcohols showed a good correlation with the σ- value of the substrate yielding a ρ value of 1.9, consistent with an electron-rich transition state as in alkoxide formation. With benzyl alcohol as substrate, the D(kcat/Km) value was 1.2 and the D2O(kcat/Km) value 2.0, consistent with little change in the CH bond during cleavage of the OH bond. The solvent inventory with benzyl alcohol was linear, consistent with a single proton being responsible for the solvent isotope effect, as expected if the isotope effects reflect formation of the alkoxide. In contrast, the kcat/Km values for ethyl alcohols showed the best correlation with the σI value of the substrate and a ρ value of -1.2, suggesting a slightly electron-deficient transition state. The isotope effects for these substrates varied with the substituent. With ethanol the D(kcat/Km) value was 1.5 and the D2O(kcat/Km) value was 1.9, while 2-Cl- and 2-Br-ethanol both had D(kcat/Km) values of 5 and D2O(kcat/Km) values of 1.1. These data were interpreted as evidence for cleavage of the OH and CH bonds in separate steps. With ethanol as substrate the energetics of the two steps are comparable, while with the more electron-rich 2-Cl- and 2-Br-substituents cleavage of the CH bond has become rate-limiting. The mechanism proposed to account for these results is shown in Figure 2. In line with the suggestion of Sherry and Abeles [13], the reaction is initiated by abstraction of a proton from the alcohol by an active-site base to form the alkoxide. Transfer of a hydride from the alkoxide to the flavin then generates the aldehyde and reduced flavin.
Figure 2.
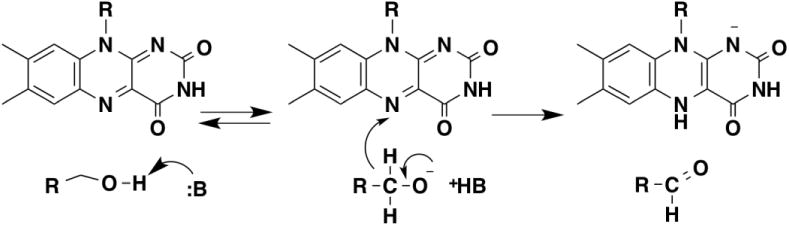
Mechanism of methanol oxidase.
Similar studies have been carried out with other GMC oxidoreductases. Choline oxidase catalyzes the oxidation of choline to betaine aldehyde and the subsequent oxidation of that compound to glycine betaine (Figure 3). With fully deuterated choline, the D(kcat/Km) value is 10.7 and the D2O(kcat/Km) value is not significantly different from unity, establishing that the CH and OH bonds are cleaved in different steps [16]. A mechanism analogous to that of Figure 3 was proposed to explain these results, with rapid and reversible formation of the alkoxide followed by hydride transfer. When the active-site residue His466 was mutated to alanine, the kcat/Km value decreased three orders of magnitude [17]. The D(kcat/Km) value for the mutant enzyme was 6.3, but, more critically, the D2O(kcat/Km) value was 2.2 and was the same with both deuterated and non-deuterated choline. The lack of a change in the D2O(kcat/Km) value with the deuterated substrate established that both isotope effects arise from a single rate-limiting transition state. The authors proposed that the role of His466 was to stabilize the alkoxide. The loss of the interaction between the positively-charged His466 and the alkoxide would delay cleavage of the OH bond until the transfer of the hydride to the flavin began. A similar result had previously been reported for a mutant of flavocytochrome b2, leading to a similar mechanistic proposal (vide infra) [18].
Figure 3.
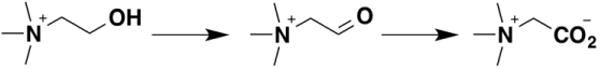
The reactions catalyzed by choline oxidase.
Pyranose 2-oxidase, another member of the GMC oxidoreductase family, catalyzes the oxidation of the C2 hydroxyl of aldopyranoses to the corresponding aldehyde (Figure 4). The FAD cofactor is attached covalently to the enzyme through His167. Kinetic isotope effects were measured for the oxidation of glucose by the H167A enzyme using single-turnover methods [19]. Use of 2-[2H]-glucose as substrate decreased the rate constant for flavin reduction by 3.9-fold, but this rate constant was unchanged when the reaction was carried out in D2O. A stepwise reaction involving hydride transfer from the alkoxide similar to that of Figure 2 was proposed to explain these results.
Figure 4.
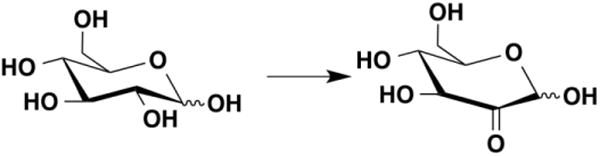
The reaction catalyzed by pyranose 2-oxidase.
Similar studies of a fungal aryl-alcohol oxidase came to a somewhat different mechanistic conclusion, even though this enzyme is also member of the GMC oxidoreductase family. With either p-methoxybenzyl alcohol or 2,4-hexadiene-1-ol as substrate, the dideuterated substrate gave an isotope effect of ~9 and a solvent isotope effect of 1.4 on the rate constant for flavin reduction [20]. The deuterium isotope effect was unchanged in D2O and the solvent isotope effect was unchanged when deuterated substrate was used, consistent with a concerted reaction. Based on QM/MM calculations, Hernandez-Ortega et al. [21] concluded that the reaction is concerted but asynchronous, with proton transfer from the oxygen preceding hydride transfer. These authors also carried out a similar QM/MM analysis for choline oxidase; the results in that case were also consistent with proton transfer preceding hydride transfer but with the alkoxide being an actual intermediate in the reaction so that the reaction is stepwise.
The reported isotope effects for members of the GMC oxidoreductase family are fully consistent with the mechanism of Figure 2, in which the proton is removed from the alcohol oxygen before a hydride is transferred to the flavin. Different members of this family appear to differ in the extent to which the alkoxide is stabilized. With choline oxidase and pyranose 2-oxidase, the alkoxide is a stable intermediate preceding rate-limiting hydride transfer. With p-methoxybenzyl alcohol and 2,4-hexadiene-1-ol as substrates for aryl-alcohol oxidase, the alkoxide is not an intermediate, but proton transfer from the oxygen still precedes hydride transfer in an asynchronous transition state. Indeed, the results with different substrates for methanol oxidase suggest that the relative energetics of OH and CH bond cleavage are quite sensitive to the identity of the substrate, such that the barrier for OH bond cleavage can be higher than that for hydride transfer from the alkoxide in some cases.
Oxidation of α-hydroxy acids to the corresponding keto acids is catalyzed by a family of flavoproteins with a TIM barrel fold. This family includes both oxidases, e. g., lactate oxidase, glycolate oxidase, and long chain hydroxy acid oxidase, and dehydrogenases, e. g., lactate dehydrogenase and mandelate dehydrogenase; the dehydrogenases also contain a heme domain [22]. Some members of this family, such as lactate monooxygenase, will catalyze an oxidative decarboxylation of the enzyme-bound keto acid product during the oxidative half reaction [23, 24].
The mechanism of α-hydroxy acid oxidation by members of this enzyme family has been controversial. Based on the structure of the yeast lactate dehydrogenase flavocytochrome b2 with pyruvate bound, two possible mechanisms were proposed for oxidation of lactate (Figure 5) [25]. In one (path i) the active-site base His373 abstracts a proton from the α-carbon of the substrate to form a carbanion. This is followed by transfer of the proton from the oxygen to the phenolate of Tyr254 as electrons are transferred to the FMN cofactor. In the second (path ii) His373 abstracts a proton from the substrate hydroxyl as a hydride is transferred to the FMN. Solvent and deuterium kinetic isotope effects were measured with wild-type flavocytochrome b2 and mutant enzymes to distinguish between the two proposed mechanisms. With the wild-type enzyme, there was a kinetic isotope effect of 5.4 on the rate constant for flavin reduction when 2-[2H]-lactate was the substrate; this same step showed a solvent isotope effect of 1.0, establishing that OH and CH bond cleavage are not concerted [26]. While the result is consistent with mechanism i, the pH profiles of the enzyme were consistent with a need for His373 to be protonated but provided no evidence for the involvement of the phenolate of Tyr254. With the Y254F enzyme, the primary deuterium kinetic isotope effects on the kcat and kcat/Klactate values were both 5.0, and both kinetic parameters exhibited identical solvent isotope effects of 1.5 [18]. The former value decreased slight to 4.5 in D2O, while the latter value decreased to 1.4 with deuterated lactate. These results are consistent with a slightly asynchronous concerted mechanism in the mutant protein due to loss of the interaction with Tyr254. In the wild-type enzyme Tyr254 is able to stabilize the alkoxide, allowing it to form before hydride transfer to the FMN. This stepwise mechanism in which alkoxide formation precedes hydride transfer is essentially the same mechanism as is described above for oxidation of simple alcohols by flavoproteins.
Figure 5.
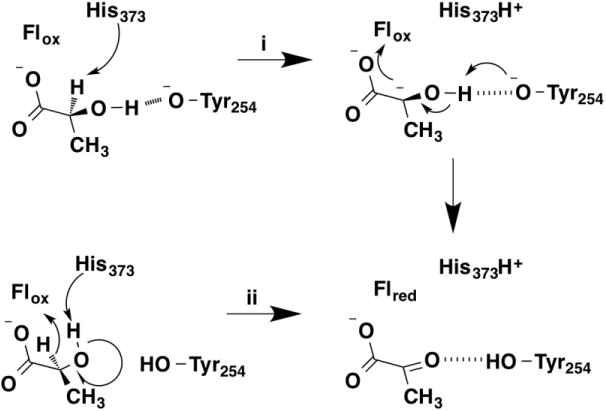
Proposed mechanisms for oxidation of lactate by flavocytochrome b2.
4.2 Pyridine nucleotide-dependent enzymes
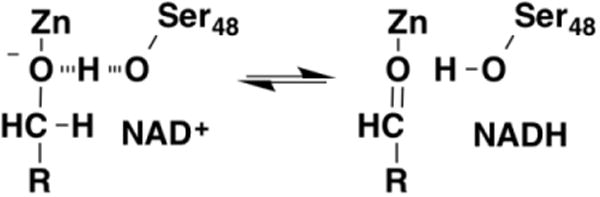
A large number of pyridine-nucleotide dependent enzymes will catalyze oxidation of alcohols. These enzymes typically utilize ternary complex kinetic mechanisms that, together with the lack of a bound chromophore, can make their kinetics more complex than is the case for the flavoproteins discussed above. The pyridine nucleotide-dependent alcohol dehydrogenases can be divided into three structural families: the metal-independent short-chain dehydrogenase/reductase (SDR) superfamily; the medium-chain dehydrogenase/reductase (MDR) family, which contains metal-dependent and independent members; and the metal-independent polyol-specific long-chain dehydrogenase/reductase (PSLDR) family [27-29]. Within the MDR superfamily the alcohol dehydrogenase from horse liver has long served as a model system for understanding fundamentals of enzyme-catalyzed reactions and for development of kinetic methodology [30-34]. Structures of the enzyme with NAD+ and 2,2,2-trifluoroethanol or 2,3,4,5,6-F5-benzylalcohol bound have been obtained at very high resolution (1.1 Å) [35]. These show a distance from the active-site zinc to the substrate oxygen of 1.96 Å, consistent with a zinc-alkoxide, and a distance from the oxygen of Ser48 to the substrate oxygen of 2.48 Å, consistent with a low-barrier hydrogen bond (Figure 6). While solvent and deuterium kinetic isotope effects on the steady state kinetic parameters for oxidation of cyclohexanol by horse liver alcohol dehydrogenase were reported separately early on, interpretation of these is complex due to the pH dependence of the steady-state kinetic parameters and to their kinetic complexity [36, 37]. Isotope effects obtained from rapid-reaction studies, where it is possible to monitor the hydride transfer to NAD directly, are simpler to interpret. With benzyl alcohol as substrate, the deuterium and solvent isotope effects were 4.6 and 0.57, respectively; the substrate deuterium isotope effect was unchanged in D2O, and the solvent isotope effect was unchanged with the deuterated substrate, establishing that both isotope effects arise from the same step [38]. Oxidation of ethanol similarly showed a normal deuterium isotope effect of 3.8 and an inverse solvent isotope effect of 0.5; the solvent inventory determined with this substrate was curved [39]. These data are consistent with the mechanism of Figure 6 [38]. Here, hydride transfer occurs from the metal-bound alkoxide, and the low-barrier hydrogen bond is lost as the alcohol is oxidized to the aldehyde [30, 39]. This mechanism resembles that proposed in Figure 2 for flavin-dependent oxidation of alcohols in that hydride transfer occurs from an enzyme-stabilized alkoxide. There is a solvent isotope effect because a low-barrier hydrogen bond is used to stabilize the zinc-bound alkoxide, and the proton involved undergoes a change in its fractionation factor during the reaction. The curvature of the proton inventory can be explained by a combination of a ground-state effect fractionation factor of 0.37 due to the low-barrier hydrogen bond and a transition state fractionation factor of 0.73.
Figure 6. Alcohol oxidation by zinc-dependent alcohol dehydrogenase [38].
Similar analyses were carried out with Pseudomonas fluorescens mannitol dehydrogenase, a member of the metal-independent PSLDR family [40, 41]. With this enzyme the deuterium and solvent isotope effects changed differently with pH. The primary deuterium kinetic isotope effect on the kcat/Km value for mannitol had a value 1.8 at pH 9 and below and decreased to 1 above a pKa value of ∼11; the deuterium isotope effect at pH 10 value did not change significantly in D2O. At pH > 10, where the kcat/Kmannitol value was pH-independent, there was a solvent isotope effect of 2.4 that was associated with a linear proton inventory. In the reverse direction, fructose reduction, the primary deuterium isotope effect on the kcat/Kfructose value was maximal at pH 7 with a value of 2.5, decreasing to 0.83 at pH 10. The solvent isotope effect was 2.6 at pH 7. These data were interpreted as involving a stepwise mechanism of alkoxide formation prior to mannitol oxidation, with a pH-sensitive conformational change preceding hydride transfer.
NADP+-Dependent isocitrate dehydrogenase is a member of a superfamily of metal-dependent decarboxylases [42, 43]. The mechanism involves oxidation of isocitrate to oxalosuccinate followed by decarboxylation to yield 2-ketoglutarate (Figure 7) [44, 45]. There are two proton transfers in this reaction, the deprotonation of the alcohol, analogous to the reactions discussed above, and the protonation of the 2-ketoglutarate enolate formed by decarboxylation. Combined solvent and primary deuterium kinetic isotope effects have been used to study the mechanism of the enzyme from Mycobacterium tuberculosis [46]. Both the kcat and kcat/Kisocitrate values showed modest primary deuterium isotope effects of ∼1.4; these decreased to ∼1.2 in D2O. The solvent isotope effects on kcat and kcat/Kisocitrate were 3.0 and 1.5, respectively; these decreased to 1.7 and ∼1.0 with deuterated isocitrate. These data clearly establish that the solvent and primary isotope effects arise from separate steps. The authors concluded that the solvent isotope effect was more likely due to the protonation of the enolate than deprotonation of the alcohol, which was expected to be coupled to hydride transfer.
Figure 7.
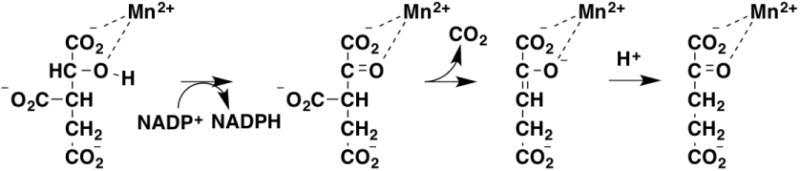
The mechanism of isocitrate dehydrogenase.
4.3 Other cofactors
Galactose oxidase is the best-characterized member of the small family of radical copper oxidases that contain a stable tyrosyl radical coordinated to a single copper atom. With galactose as substrate, there was a deuterium isotope effect on the rate constant for reduction of the enzyme of 21, and a solvent isotope effect of 0.99 [47]. The results with a series of benzyl alcohols as substrates were more complex. With 4-NO2-benzyl alcohol, the deuterium isotope effect was 12 and the solvent isotope effect 1.07, while with 4-CH3O-benzyl alcohol the deuterium isotope effect was 4.3 and the solvent isotope effect was 1.2 [48]. With the latter substrate the deuterium isotope effect decreased slightly in D2O. These results are similar to those for methanol oxidase described above [15]. Overall the results for galactose oxidase are consistent with proton transfer preceding the proton-coupled electron transfer in either an asynchronous concerted reaction or in a stepwise reaction in which the alkoxide is a short-lived intermediate, depending on the substrate.
5. Isotope effects on amine oxidation
5.1 Flavoproteins
Several families of flavoproteins will catalyze the oxidation of amines and amino acids. The initial oxidation product is released from the enzyme before it is hydrolyzed nonenzymatically to free amine and aldehyde or ketone [49] (Figure 8). The mechanisms proposed for oxidation of amines by flavoproteins include CH bond cleavage by abstraction of a proton, a hydrogen atom, or a hydride as well as mechanisms in which CH bond cleavage occurs concomitant with or after formation of an amine-flavin adduct [50]. Solvent isotope effects in combination with other isotope effects have been used to distinguish among these possibilities. The presence of nitrogen in the reacting bond has allowed secondary nitrogen kinetic isotope effects to be utilized as mechanistic probes in addition to primary deuterium and solvent isotope effects [51].
Figure 8.
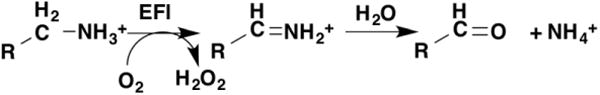
The reaction catalyzed by flavin amine oxidases. The second step is nonenzymatic.
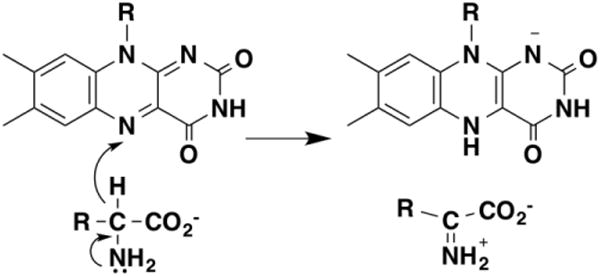
The flavoproteins that catalyze oxidation of amines belong to several different structural families. Flavoenzymes with the same overall structure as D-amino acid oxidase will catalyze the oxidation of D-amino acids, glycine, and some N-methylated amino acids [52-56]. D-Amino acid oxidase from pig kidney has been the most-studied member of this family. The primary deuterium kinetic isotope effect with D-serine, a substrate for which the isotope effect on the kcat/Km value equals the intrinsic isotope effect, is 4.5, and the solvent isotope effect is unity [57, 58], establishing that the amino proton is not in flight in the transition state for CH bond cleavage. The observed 15N isotope effect was affected by both pH and solvent, decreasing from 1.0128 at pH 7.5 to 0.999 at pH 10.1 in H2O and increasing to 1.0175 at pD 7.5 in D2O [59]. Both effects can be explained by the 15N equilibrium isotope effect of 1.026 on the pKa of the amine in an amino acid; this effect is larger in D2O [60]. This equilibrium effect results in a larger fraction of the 15N-amino acid being the zwitterion than is the case for the 14N-amino acid; the effect becomes smaller as the pH increases so that an increasing fraction of the amine is in the reactive neutral form. Thus the observed decrease in the 15N isotope effect kcat/Km value with increasing pH and the increase in D2O provide evidence for the oxidation requiring a form of the substrate in which the nitrogen is uncharged. Correction of the measured 15N isotope effects at both high and low pH and in both H2O and D2O for the equilibrium 15N effect gave identical 15N isotope effects under all four conditions, 0.9963 ± 0.0016, establishing that the amine nitrogen is undergoing rehybridization in the transition state for CH bond cleavage. This value is consistent with values predicted for mechanisms involving hydride or hydrogen atom transfer, but not with carbanion formation by proton abstraction or the involvement of an amino acid-flavin adduct [61]. The energetic barrier for amine oxidation by transfer of a hydrogen atom or a single electron to the flavin is much higher than that for hydride transfer, and no flavin radical has been detected in stopped-flow studies of these enzymes, so that a mechanism involving a flavin radical is unlikely. The mechanism most consistent with all the kinetic isotope effects is binding of the anionic amino acid to the enzyme followed by hydride transfer to the flavin (Figure 9).
Figure 9. The mechanism of D-amino acid oxidase.
Similar results were found with N-methyltryptophan oxidase, an enzyme with the same fold as D-amino acid oxidase that catalyzes demethylation of N-methyl amino acids by oxidizing the bond between the amino acid nitrogen and the methyl group (Figure 10). The deuterium isotope on both the kcat/Ksarcosine and kcat value was 7.2 with N-(2H3-methyl)-glycine, and there was no solvent isotope effect on the kcat/Ksarcosine value [62]. The 15N isotope effect on the kcat/Ksarcosine value was pH-sensitive, decreasing above the pKa for sarcosine to a limiting value of 0.9940 ± 0.0004 [61]. These results are again consistent with the anionic amino acid as the substrate and a hydride transfer mechanism.
Figure 10.
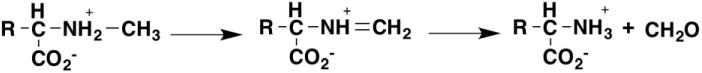
The reaction catalyzed by N-methyltryptophan oxidase and sarcosine oxidase. The second step is nonenzymatic.
D-Arginine dehydrogenase from Pseudomonas aeruginosa has an overall structure similar to that of D-amino acid oxidase, although the sequence identity is low [63]. The enzyme catalyzes a similar reaction, oxidation of D-amino acids to the imino acids, but differs in substrate specificity and reactivity with oxygen [64]. Kinetic isotope effects and pH studies were used to determine the mechanism of the enzyme with the slow substrate D-leucine [65]. The rate constant for flavin reduction exhibited a solvent isotope effect of 1.8 and a primary deuterium isotope effect of 5.1; the former decreased to 1.6 with the deuterated substrate and the latter decreased to 4.7 in D2O. The authors concluded that the enzyme binds the zwitterionic form of the amino acid substrate, with loss of the amino proton occuring after a conformational change and preceding hydride transfer in a stepwise reaction. This proposal differs from those above for other amine-oxidizing flavoproteins in that loss of the amine proton occurs in the enzyme active site, but agrees that the amine oxidation step involves hydride transfer from the anionic amino acid.
A separate family of flavin-dependent amine oxidases has structures similar to that of monoamine oxidase [66], but pH-independent solvent isotope effects do not appear to have been reported for these enzymes. Still, the pH and kinetic isotope effects for these enzymes are consistent with the mechanism proposed for the D-amino acid oxidase family. Tryptophan 2-monooxygenase is an amino acid oxidase/decarboxylase in the monoamine oxidase structural family [67]. The effects of pH on the kcat/Km values for the amino acid substrate established that nitrogen must be neutral for productive binding [68, 69]. With the slow substrate alanine, the primary deuterium kinetic isotope effect was 6.0 and the pH-corrected 15N isotope effect 0.992, consistent with hydride transfer form the anionic amino acid [60]. In the case of monoamine oxidase A, the pH dependence similarly established the neutral amine as the substrate [70]. With monoamine oxidase B, there was a deuterium kinetic isotope effect on the kcat/Km value of 5.2, and a pH-corrected 15N isotope effect of 0.985; the latter increased to 0.990 with deuterated benzylamine [71]. These data suggest that CH bond cleavage and nitrogen rehybridization are asynchrous with this enzyme. However, quantitative analyses of these isotope effects are complicated by the evidence that there is a significant forward external commitment with benzylamine as a substrate at the pH of the measurements, in that the deuterium isotope effect on the kcat/Km value is only about one-half the isotope effects on kcat and on the rate constant for flavin reduction [72].
The flavoprotein proline dehydrogenase catalyzes the oxidation of proline to 3,4-dehydroproline in both eukaryotes and bacteria. The enzyme is found both as a monofunctional protein and as part of a bifunctional enzyme that also contains Δ1-pyrroline-5-carboxylate dehydrogenase. In both cases the enzyme has a TIM barrel fold, in contrast to the other amino acid-oxidizing flavoproteins [73, 74]. Solvent and deuterium kinetic isotope effects have been carried out with the monofunctional enzyme from Mycobacterium tuberculosis [75]. There was a deuterium kinetic isotope effect of 5.6 and a solvent isotope effect of 2.1 on the kcat/Km value for proline. The former value decreased to 3.4 in D2O, consistent with a stepwise mechanism in which deprotonation precedes hydride transfer. The deuterium isotope effect on the rate constant for flavin reduction exhibited a primary isotope effect of 5.2 and a solvent isotope effect of close to unity, establishing that hydride transfer occurs from the anionic amino acid.
Berberine bridge enzyme catalyzes a ring closure reaction in the biosynthesis of plant alkaloids (Figure 11) [76]. The reaction is proposed to involve oxidation of the bond between the nitrogen and the methyl group by hydride transfer to the flavin followed by (path i) or concerted with (path ii) nucleophilic attack by C2' of the phenolic ring on the iminium ion. The latter would be facilitated by abstraction by an active-site base of the phenolic hydrogen, further activating the ring for nucleophilic attack. Solvent and primary deuterium kinetic isotope effects were carried out with berberine bridge enzyme using methyl-deuterated berberine [77]. The rate constant for flavin reduction showed a deuterium kie of 3.5 and a solvent isotope effect of 1.0; the former was unchanged in D2O. Similar results were obtained when the active-site base Glu417, was mutated to glutamine. These results establish that the phenolic proton is not in flight in the transition state for amine oxidation, so that the reaction is either stepwise, with rapid ring closure after the rate-limiting amine oxidation, or formation of the phenoxide does not occur.
Figure 11.
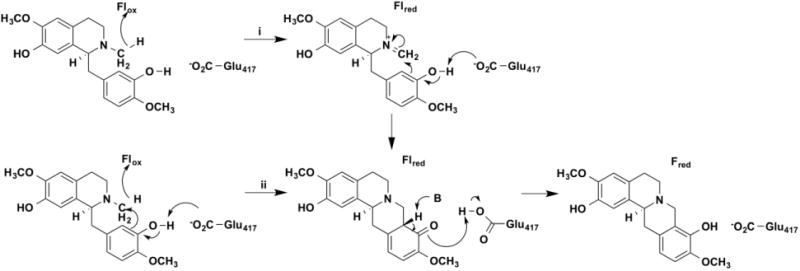
Proposed mechanisms for berberine bridge enzyme.
5.2 Pyridine-nucleotide dependent enzymes
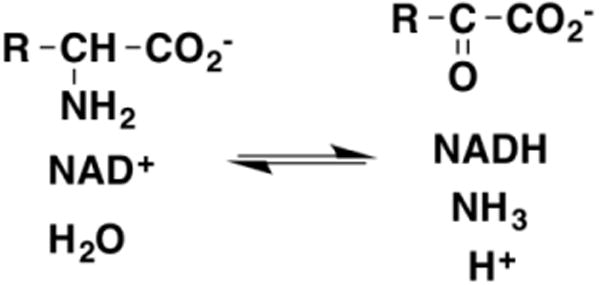
A number of pyridine-nucleotide dependent enzymes catalyze the reversible oxidation of amino acids to the respective keto acids and ammonium ion (Figure 12). These enzymes catalyze both oxidation of the amino acid by hydride transfer to the pyridine nucleotide and hydrolysis of the Schiff base intermediate, in contrast to the flavoproteins described above. Detailed mechanistic studies of glutamate and alanine dehydrogenase using pH and both deuterium and 15N kinetic isotope effects [51, 78, 79] support the anionic amino acid as the active form of the amine substrate in the forward direction and ammonia as the active form in the reverse direction. While the kcat values in both directions show solvent isotope effects, the rate constant for oxidation of the amino acid does not [80], consistent with hydride transfer involving the anionic amino acid.
Figure 12. The reaction catalyzed by glutamate and alanine dehydrogenase.
Combined primary deuterium and solvent isotope effects have been used to determine the catalytic mechanism of yeast saccharopine dehydrogenase, the enzyme catalyzing the final step in lysine biosynthesis. These were done in the direction of saccharopine formation, using NADD to determine the deuterium isotope effect. The deuterium isotope effects on both the kcat and the kcat/Klysine values were 1.5-1.6 in both H2O and D2O, consistent with concerted transfer of a hydride and a proton from the positively charged nitrogen in the reactive CN bond. The solvent isotope effect on the kcat/Klysine value was 1.9 with both NADH and NADD, while the solvent isotope effect on the kcat value decreased from 2.2 with lysine to 1.8 with deuterated lysine; this suggested that there was an additional step that was sensitive to D2O, likely loss of water from the carbinolamine intermediate (Figure 13). Consistent with that conclusion, the solvent inventories for both kcat/Klysine and kcat were bowed upwards, with the kcat value exhibiting substantially more curvature [81]. Based on the crystal structure, Lys77 was identified as the base that removed the proton from lysine and His 96 as the residue involved in loss of water from the carbinolamine [82]. With the H96Q mutant, the deuterium isotope effects on both kcat and kcat/Klysine were not significantly different form 1.0 in H2O or D2O, and the solvent isotope effects of 2.4 on kcat and 2.2 on kcat/Klysine were unchanged with the deuterated substrate; these values suggest that interconversion of the carbinolamine and the imine is fully rate-limiting in the mutant protein, as expected for the proposed role of His96.
Figure 13.
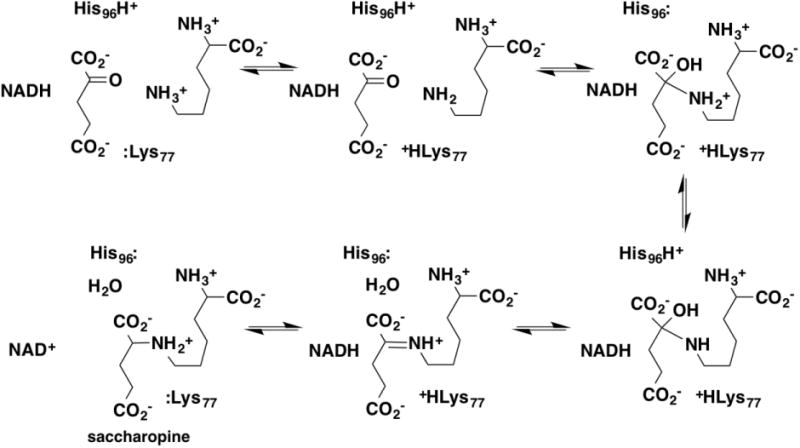
Proposed mechanism for saccharopine dehydrogenase, based on Kumar et al. [82, 83].
6. Applications of combined solvent and primary kinetic isotope effects to other reactions
In the reaction catalyzed by glyceraldehyde-3-phosphate dehydrogenase, attack of an active-site thiol on the substrate aldehyde moiety forms a thiohemiacetal (Figure 14). Oxidation of the thiohemiacetal by hydride transfer to an oxidized pyridine nucleotide generates a thioester for nucleophilic attack by phosphate to form the 1,3-diphosphoglycerate product; this step also involves proton transfer from the oxygen of the thiohemiacetal. Wolfson-Stofko et al. [84] measured solvent and deuterium isotope effects with 1-[2H]-glyceraldehyde-3-phosphate to examine the relative order of alkoxide formation and hydride transfer for the M. tuberculosis enzyme. The solvent isotope effect on the kcat/Km value for glyceraldehyde-3-phosphate was significantly smaller with the deuterated substrate, consistent with a stepwise reaction in which the alkoxide is formed before hydride transfer.
Figure 14.
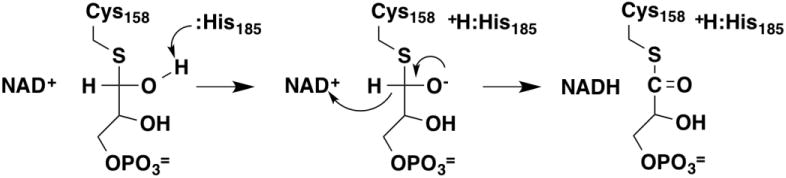
Mechanism of thiohemiacetal oxidation by glyceraldehyde 3-phosphate dehydrogenase [84].
Heme oxygenase catalyzes the hydroxylation of its own cytochrome P450 heme moiety in the catabolism of heme. Davydov et al. [85] measured secondary deuterium and solvent isotope effects of 0.7 and 2.3 on the rate constant for the formation of the hydroxylated heme from a hydroperoxy precursor at 215K. These values are consistent with concerted protonation of the hydroperoxy-ferri-heme and oxygen addition to the heme ring.
7. Conclusion
For oxidations of both alcohols and amines by a variety of enzymes, the use of multiple isotope effects has established that proton transfer precedes hydride transfer. For amine oxidation, the much lower pKa values of amines than alcohols can result in there being a sufficiently high fraction of the amine in the reactive neutral form at physiological pH that the enzyme does not absolutely require an active-site base to remove a proton from the amine. For alcohol oxidation, a variety of enzymes stabilize the alkoxide sufficiently that its formation does not contribute to the rate of catalysis. Still, the exact timing of the two bond cleavages appears to vary with different enzymes and even with different substrates for the same enzyme. Methanol oxidase and galactose oxidase provide examples of how the use of alternate, typically nonphysiological, substrates can alter the relative energetics of alkoxide formation and the subsequent CH bond cleavage; aryl-alcohol oxidase may be another example where the selection of substrate for mechanistic studies influences the results. The suggestion that the flavoprotein D-arginine dehydrogenase binds the zwitterionic form of the amino acid substrate productively, in contrast to the binding of the anionic substrate by other amine-oxidizing enzymes, implies that there may be some diversity even among structurally similar enzymes.
The examples cited here illustrate the utility of combining solvent isotope effects with other kinetic isotope effects. Measurement of isotope effects involving bond order changes for atoms that do not readily exchange, whether deuterium, 15N, or other isotopes not discussed here, can identify the specific transition state(s) that contribute to the kinetic parameter being measured. This knowledge can substantially decrease the ambiguity inherent in interpretation of solvent isotope effects. The advantage of such an approach is most obvious in cases where there is an isotope effect upon substitution of a non-exchanging atom in the substrate but none upon replacing H2O as the solvent with D2O, since such a result rules out transfer of a solvent-exchangeable proton in the transition state. For many of the flavoproteins catalyzing alcohol or amine oxidation, such an absence of an effect of D2O on the kinetics established the alkoxide or neutral amine, respectively, as the form of the substrate from which hydride transfer occurs. In more complicated cases, as frequently occurs with pyridine nucleotide dependent enzymes, measurement of combined isotope effects, in which one determines if the solvent isotope effect changes when there is isotopic substitution in the substrate or vice versa, has been critical in distinguishing stepwise and concerted reactions and in identifying the individual step exhibiting the solvent isotope effect.
Highlights.
Oxidation of an amine or alcohol requires removal of a solvent-exchangeable proton.
Solvent isotope effects can be used to probe the timing of NH and OH bond cleavages.
Combining solvent and substrate isotope effects helps define the origin of the solvent effect.
Alcohol oxidation typically involves hydride transfer from an alkoxide.
Amine oxidation typically involves hydride transfer from the neutral amine.
Acknowledgments
The work from the author's laboratory described here would not have been possible without the contributions of talented graduate students, postdoctoral fellows, and collaborators or financial support from the NIH (GM058698) and The Welch Foundation (AQ-1256). I also thank Drs. John Blanchard and Bryce Plapp for helpful comments on the manuscript.
Footnotes
This manuscript is dedicated to the memory of the late W. W. Cleland.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hardy RC, Cottington RL. Viscosity of deuterium oxide and water in the range 5° to 125° C. J Res Nat Bur Stand. 1949;42:573–578. [Google Scholar]
- 2.Millero FJ, Dexter R, Hoff E. Density and viscosity of deuterium oxide solutions from 5-70°C. J Chem Eng Data. 1971;16:85–87. [Google Scholar]
- 3.Schowen KB, Schowen RL. Solvent isotope effects on enzyme systems. Methods Enzymol. 1982;87:551–606. [PubMed] [Google Scholar]
- 4.Quinn DM, Sutton LD. Theoretical basis and mechanistic utility of solvent isotope effects. In: Cook PF, editor. Enzyme mechanism from isotope effects. CRC Press, Place Published; 1991. pp. 73–126. [Google Scholar]
- 5.Quinn DM. Theory and practice of solvent isotope effects. In: Kohen A, Limbach HH, editors. Isotope Effects in Chemistry and Biology. Taylor and Francis, Place Published; 2006. pp. 995–1018. [Google Scholar]
- 6.Cleland WW. Low-barrier hydrogen bonds and enzymatic catalysis. Arch Biochem Biophys. 2000;382:1–5. doi: 10.1006/abbi.2000.2011. [DOI] [PubMed] [Google Scholar]
- 7.Salomaa P, Schaleger LL, Long FA. Solvent deuterium isotope effects on acid-base equilibria. J Am Chem Soc. 1964;86:1–7. [Google Scholar]
- 8.Venkatasubban KS, Schowen RL. The proton inventory technique. 1. Introduction to proton inventories. CRC Critical Rev Biochem. 1984;17:1–44. doi: 10.3109/10409238409110268. [DOI] [PubMed] [Google Scholar]
- 9.Kiick DM. Effect of commitments to catalysis on the degree of curvature in proton inventories of the kinetic parameters for enzyme-catalyzed reactions: Application to tryptophan indole-lyase. J Am Chem Soc. 1991;113:8499–8504. [Google Scholar]
- 10.O'Leary MH. Multiple isotope effects on enzyme-catalyzed reactions. Ann Rev Biochem. 1989;58:377–401. doi: 10.1146/annurev.bi.58.070189.002113. [DOI] [PubMed] [Google Scholar]
- 11.Cavener DR. GMC oxidoreductases a newly defined family of homologous proteins with diverse catalytic activities. J Mol Biol. 1992;223:811–814. doi: 10.1016/0022-2836(92)90992-s. [DOI] [PubMed] [Google Scholar]
- 12.Wongnate T, Chaiyen P. The substrate oxidation mechanism of pyranose 2-oxidase and other related enzymes in the glucose-methanol-choline superfamily. FEBS J. 2013;280:3009–3027. doi: 10.1111/febs.12280. [DOI] [PubMed] [Google Scholar]
- 13.Sherry B, Abeles RH. Mechanism of action of methanol oxidase, reconstitution of methanol oxidase with 5-deazaflavin, and inactivation of methanol oxidase by cyclopropanol. Biochemistry. 1985;24:2594–2605. doi: 10.1021/bi00332a002. [DOI] [PubMed] [Google Scholar]
- 14.Hemmerich P, Massey V, Fenner H. Flavin and 5-deazaflavin: A chemical evaluation of ‘modified’ flavoproteins with respect to the mechanisms of redox biocatalysis. FEBS Lett. 1977;84:5–21. doi: 10.1016/0014-5793(77)81047-8. [DOI] [PubMed] [Google Scholar]
- 15.Menon V, Hsieh CT, Fitzpatrick PF. Substituted alcohols as mechanistic probes of alcohol oxidase. Bioorg Chem. 1995;23:42–53. [Google Scholar]
- 16.Fan F, Gadda G. On the catalytic mechanism of choline oxidase. J Am Chem Soc. 2005;127:2067–2074. doi: 10.1021/ja044541q. [DOI] [PubMed] [Google Scholar]
- 17.Ghanem M, Gadda G. On the catalytic role of the conserved active site residue His466 of choline oxidase. Biochemistry. 2005;44:893–904. doi: 10.1021/bi048056j. [DOI] [PubMed] [Google Scholar]
- 18.Sobrado P, Fitzpatrick PF. Solvent and primary deuterium isotope effects show that lactate CH and OH bond cleavages are concerted in Y254F flavocytochrome b2, consistent with a hydride transfer mechanism. Biochemistry. 2003;42:15208–15214. doi: 10.1021/bi035546n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sucharitakul J, Wongnate T, Chaiyen P. Kinetic isotope effects on the noncovalent flavin mutant protein of pyranose 2-oxidase reveal insights into the flavin reduction mechanism. Biochemistry. 2010;49:3753–3765. doi: 10.1021/bi100187b. [DOI] [PubMed] [Google Scholar]
- 20.Ferreira P, Hernandez-Ortega A, Herguedas B, Martínez ÁT, Medina M. Aryl-alcohol oxidase involved in lignin degradation: A mechanistic study based on steady and pre-steady state kinetics and primary and solvent isotope effects with two alcohol substrates. J Biol Chem. 2009;284:24840–24847. doi: 10.1074/jbc.M109.011593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernández-Ortega A, Lucas F, Ferreira P, Medina M, Guallar V, Martínez AT. Role of active site histidines in the two half-reactions of the aryl-alcohol oxidase catalytic cycle. Biochemistry. 2012;51:6595–6608. doi: 10.1021/bi300505z. [DOI] [PubMed] [Google Scholar]
- 22.Cunane LM, Barton JD, Chen ZW, Le KH, Amar D, Lederer F, Mathews FS. Crystal structure analysis of recombinant rat kidney long chain hydroxy acid oxidase. Biochemistry. 2005;44:1521–1531. doi: 10.1021/bi048616e. [DOI] [PubMed] [Google Scholar]
- 23.Emanuele JJ, Jr, Fitzpatrick PF. Mechanistic studies of the flavoprotein tryptophan 2-monooxygenase. 1. Kinetic mechanism. Biochemistry. 1995;34:3710–3715. doi: 10.1021/bi00011a028. [DOI] [PubMed] [Google Scholar]
- 24.Maeda-Yorita K, Aki K, Sagai H, Misaki H, Massey V. L-Lactate oxidase and L-lactate monooxygenase: mechanistic variations on a common structural theme. Biochimie. 1995;77:631–642. doi: 10.1016/0300-9084(96)88178-8. [DOI] [PubMed] [Google Scholar]
- 25.Dubois J, Chapman SK, Mathews FS, Reid GA, Lederer F. Substitution of Tyr254 with Phe at the active site of flavocytochrome b2: Consequences on catalysis of lactate dehydrogenation. Biochemistry. 1990;29:6393–6400. doi: 10.1021/bi00479a008. [DOI] [PubMed] [Google Scholar]
- 26.Sobrado P, Daubner SC, Fitzpatrick PF. Probing the relative timing of hydrogen abstraction steps in the flavocytochrome b2 reaction with primary and solvent deuterium isotope effects and mutant enzymes. Biochemistry. 2001;40:994–1001. doi: 10.1021/bi002283d. [DOI] [PubMed] [Google Scholar]
- 27.Jörnvall H, Hedlund J, Bergman T, Oppermann U, Persson B. Superfamilies SDR and MDR: From early ancestry to present forms. Emergence of three lines, a Zn-metalloenzyme, and distinct variabilities. Biochem Biophys Res Commun. 2010;396:125–130. doi: 10.1016/j.bbrc.2010.03.094. [DOI] [PubMed] [Google Scholar]
- 28.Klimacek M, Kavanagh KL, Wilson DK, Nidetzky B. Pseudomonas fluorescens mannitol 2-dehydrogenase and the family of polyol-specific long-chain dehydrogenases/reductases: sequence-based classification and analysis of structure–function relationships. Chem Biol Interact. 2003;143–144:559–582. doi: 10.1016/s0009-2797(02)00219-3. [DOI] [PubMed] [Google Scholar]
- 29.Persson B, Kallberg Y. Classification and nomenclature of the superfamily of short-chain dehydrogenases/reductases (SDRs) Chem Biol Interact. 2013;202:111–115. doi: 10.1016/j.cbi.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 30.Plapp BV. Catalysis by alcohol dehydrogenases. In: Kohen A, Limbach HH, editors. Isotope Effects in Chemistry and Biology. CRC Press, Place Published; 2006. pp. 811–836. [Google Scholar]
- 31.Nagel ZD, Klinman JP. Update 1 of: Tunneling and dynamics in enzymatic hydride transfer. Chem Rev. 2010;110:PR41–PR67. doi: 10.1021/cr1001035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plapp BV. Conformational changes and catalysis by alcohol dehydrogenase. Arch Biochem Biophys. 2010;493:3–12. doi: 10.1016/j.abb.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Billeter SR, Webb SP, Agarwal PK, Iordanov T, Hammes-Schiffer S. Hydride transfer in liver alcohol dehydrogenase: quantum dynamics, kinetic isotope effects, and role of enzyme motion. J Am Chem Soc. 2001;123:11262–11272. doi: 10.1021/ja011384b. [DOI] [PubMed] [Google Scholar]
- 34.Cui Q, Elstner M, Karplus M. A theoretical analysis of the proton and hydride transfer in liver alcohol dehydrogenase (LADH) J Phys Chem B. 2002;106:2721–2740. [Google Scholar]
- 35.Plapp BV, Ramaswamy S. Atomic-resolution structures of horse liver alcohol dehydrogenase with NAD+ and fluoroalcohols define strained Michaelis complexes. Biochemistry. 2012;51:4035–4048. doi: 10.1021/bi300378n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor KB. Solvent isotope effects on the reaction catalyzed by alcohol dehydrogenase from equine liver. Biochemistry. 1983;22:1040–1045. doi: 10.1021/bi00274a007. [DOI] [PubMed] [Google Scholar]
- 37.Cook PF, Cleland WW. pH variation of isotope effects in enzyme-catalyzed reactions. 2. Isotope-dependent step not pH dependent. Kinetic mechanism of alcohol dehydrogenase. Biochemistry. 1981;20:1805–1816. doi: 10.1021/bi00510a015. [DOI] [PubMed] [Google Scholar]
- 38.Ramaswamy S, Park DH, Plapp BV. Substitutions in a flexible loop of horse liver alcohol dehydrogenase hinder the conformational change and unmask hydrogen transfer. Biochemistry. 1999;38:13951–13959. doi: 10.1021/bi991731i. [DOI] [PubMed] [Google Scholar]
- 39.Sekhar VC, Plapp BV. Rate constants for a mechanism including intermediates in the interconversion of ternary complexes by horse liver alcohol dehydrogenase. Biochemistry. 1990;29:4289–4295. doi: 10.1021/bi00470a005. [DOI] [PubMed] [Google Scholar]
- 40.Slatner M, Nidetzky B, Kulbe KD. Kinetic study of the catalytic mechanism of mannitol dehydrogenase from Pseudomonas fluorescens. Biochemistry. 1999;38:10489–10498. doi: 10.1021/bi990327g. [DOI] [PubMed] [Google Scholar]
- 41.Klimacek M, Nidetzky B. Examining the relative timing of hydrogen abstraction steps during NAD+-dependent oxidation of secondary alcohols catalyzed by long-chain D-mannitol dehydrogenase from Pseudomonas fluorescens using pH and kinetic isotope effects. Biochemistry. 2002;41:10158–10165. doi: 10.1021/bi025517x. [DOI] [PubMed] [Google Scholar]
- 42.Tipton PA, Beecher BS. Tartrate dehydrogenase, a new member of the family of metal-dependent decarboxylating R-hydroxyacid dehydrogenases. Arch Biochem Biophys. 1994;313:15–21. doi: 10.1006/abbi.1994.1352. [DOI] [PubMed] [Google Scholar]
- 43.Dean AM, Golding GB. Protein engineering reveals ancient adaptive replacements in isocitrate dehydrogenase. Proc Natl Acad Sci USA. 1997;94:3104–3109. doi: 10.1073/pnas.94.7.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hermes JD, Roeske CA, O'Leary MH, Cleland WW. Use of multiple isotope effects to determine enzyme mechanisms and intrinsic isotope effects. Malic enzyme and glucose-6-phosphate dehydrogenase. Biochemistry. 1982;21:5106–5114. doi: 10.1021/bi00263a040. [DOI] [PubMed] [Google Scholar]
- 45.Grissom CB, Cleland WW. Isotope effect studies of the chemical mechanism of pig heart NADP isocitrate dehydrogenase. Biochemistry. 1988;27:2934–2943. doi: 10.1021/bi00408a040. [DOI] [PubMed] [Google Scholar]
- 46.Quartararo CE, Hazra S, Hadi T, Blanchard JS. Structural, kinetic and chemical mechanism of isocitrate dehydrogenase-1 from Mycobacterium tuberculosis. Biochemistry. 2013;52:1765–1775. doi: 10.1021/bi400037w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whittaker MM, Ballou DP, Whittaker JW. Kinetic isotope effects as probes of the mechanism of galactose oxidase. Biochemistry. 1998;37:8426–8436. doi: 10.1021/bi980328t. [DOI] [PubMed] [Google Scholar]
- 48.Minasian SG, Whittaker MM, Whittaker JW. Stereoselective hydrogen abstraction by galactose oxidase. Biochemistry. 2004;43:13683–13693. doi: 10.1021/bi048554s. [DOI] [PubMed] [Google Scholar]
- 49.Roberts KM, Tormos JR, Fitzpatrick PF. Characterization of unstable products of flavin-and pterin-dependent enzymes by continuous-flow mass spectrometry. Biochemistry. 2014;53:2672–2679. doi: 10.1021/bi500267c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fitzpatrick PF. Oxidation of amines by flavoproteins. Arch Biochem Biophys. 2010;493:13–25. doi: 10.1016/j.abb.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weiss PM, Chen CY, Cleland WW, Cook PF. Use of primary deuterium and 15N isotope effects to deduce the relative rates of steps in the mechanisms of alanine and glutamate dehydrogenases. Biochemistry. 1988;27:4814–4822. doi: 10.1021/bi00413a035. [DOI] [PubMed] [Google Scholar]
- 52.Mattevi A, Vanoni MA, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B. Crystal structure of D-amino acid oxidase: a case of active site mirror-image convergent evolution with flavocytochrome b2. Proc Natl Acad Sci USA. 1996;93:7496–7501. doi: 10.1073/pnas.93.15.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trickey P, Wagner MA, Jorns MS, Mathews FS. Monomeric sarcosine oxidase: structure of a covalently flavinylated amine oxidizing enzyme. Structure. 1999;7:331–345. doi: 10.1016/s0969-2126(99)80043-4. [DOI] [PubMed] [Google Scholar]
- 54.Settembre EC, Dorrestein PC, Park J, Augustine AM, Begley TP, Ealick SE. Structural and mechanistic studies on thiO, a glycine oxidase essential for thiamin biosynthesis in Bacillus subtilis. Biochemistry. 2003;42:2971–2981. doi: 10.1021/bi026916v. [DOI] [PubMed] [Google Scholar]
- 55.Ilari A, Bonamore A, Franceschini S, Fiorillo A, Boffi A, Colotti G. The X-ray structure of N-methyltryptophan oxidase reveals the structural determinants of substrate specificity. Proteins: Struct Funct Bioinform. 2008;71:2065–2075. doi: 10.1002/prot.21898. [DOI] [PubMed] [Google Scholar]
- 56.Monaghan PJ, Leys D, Scrutton NS. Mechanistic aspects and redox properties of hyperthermophilic L-proline dehydrogenase from Pyrococcus furiosus related to dimethylglycine dehydrogenase/oxidase. FEBS Journal. 2007;274:2070–2087. doi: 10.1111/j.1742-4658.2007.05750.x. [DOI] [PubMed] [Google Scholar]
- 57.Denu JM, Fitzpatrick PF. pH and kinetic isotope effects on the reductive half-reaction of D-amino acid oxidase. Biochemistry. 1992;31:8207–8215. doi: 10.1021/bi00150a013. [DOI] [PubMed] [Google Scholar]
- 58.Denu JM, Fitzpatrick PF. Intrinsic primary, secondary, and solvent kinetic isotope effects on the reductive half-reaction of D-amino acid oxidase: evidence against a concerted mechanism. Biochemistry. 1994;33:4001–4007. doi: 10.1021/bi00179a029. [DOI] [PubMed] [Google Scholar]
- 59.Kurtz KA, Rishavy MA, Cleland WW, Fitzpatrick PF. Nitrogen isotope effects as probes of the mechanism of D-amino acid oxidase. J Am Chem Soc. 2000;122:12896–12897. [Google Scholar]
- 60.Ralph EC, Anderson MA, Cleland WW, Fitzpatrick PF. Mechanistic studies of the flavoenzyme tryptophan 2-monooxygenase: Deuterium and 15N kinetic isotope effects on alanine oxidation by an L-amino acid oxidase. Biochemistry. 2006;45:15844–15852. doi: 10.1021/bi061894o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ralph EC, Hirschi JS, Anderson MA, Cleland WW, Singleton DA, Fitzpatrick PF. Insights into the mechanism of flavoprotein-catalyzed amine oxidation from nitrogen isotope effects on the reaction of N-methyltryptophan oxidase. Biochemistry. 2007;46:7655–7664. doi: 10.1021/bi700482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ralph EC, Fitzpatrick PF. pH and kinetic isotope effects on sarcosine oxidation by N-methyltryptophan oxidase. Biochemistry. 2005;44:3074–3081. doi: 10.1021/bi047716h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu G, Yuan H, Li C, Lu CD, Gadda G, Weber IT. Conformational changes and substrate recognition in Pseudomonas aeruginosa D-arginine dehydrogenase. Biochemistry. 2010;49:8535–8545. doi: 10.1021/bi1005865. [DOI] [PubMed] [Google Scholar]
- 64.Yuan H, Fu G, Brooks PT, Weber I, Gadda G. Steady-state kinetic mechanism and reductive half-reaction of D-arginine dehydrogenase from Pseudomonas aeruginosa. Biochemistry. 2010;49:9542–9550. doi: 10.1021/bi101420w. [DOI] [PubMed] [Google Scholar]
- 65.Yuan H, Xin Y, Hamelberg D, Gadda G. Insights on the mechanism of amine oxidation catalyzed by D-arginine dehydrogenase through pH and kinetic isotope effects. J Am Chem Soc. 2011;133:18957–18965. doi: 10.1021/ja2082729. [DOI] [PubMed] [Google Scholar]
- 66.Gaweska H, Fitzpatrick PF. Structures and mechanism of the monoamine oxidase family. BioMol Concepts. 2011;2:365–377. doi: 10.1515/BMC.2011.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gaweska HM, Taylor AB, Hart PJ, Fitzpatrick PF. Structure of the flavoprotein tryptophan 2-monooxygenase, a key enzyme in the formation of galls in plants. Biochemistry. 2013;52:2620–2626. doi: 10.1021/bi4001563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sobrado P, Fitzpatrick P. Analysis of the role of the active site residue Arg98 in the flavoprotein tryptophan 2-monooxygenase, a member of the L-amino oxidase family. Biochemistry. 2003;42:13826–13832. doi: 10.1021/bi035299n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emanuele JJ, Jr, Fitzpatrick PF. Mechanistic studies of the flavoprotein tryptophan 2-monooxygenase. 2. pH and kinetic isotope effects. Biochemistry. 1995;34:3716–3723. doi: 10.1021/bi00011a029. [DOI] [PubMed] [Google Scholar]
- 70.Dunn RV, Marshall KR, Munro AW, Scrutton NS. The pH dependence of kinetic isotope effects in monoamine oxidase A indicates stabilization of the neutral amine in the enzyme-substrate complex. FEBS Journal. 2008;275:3850–3858. doi: 10.1111/j.1742-4658.2008.06532.x. [DOI] [PubMed] [Google Scholar]
- 71.MacMillar S, Edmondson DE, Matsson O. Nitrogen kinetic isotope effects for the monoamine oxidase B-catalyzed oxidation of benzylamine and (1,1-2H2)benzylamine: Nitrogen rehybridization and CH bond cleavage are not concerted. J Am Chem Soc. 2011;133:12319–12321. doi: 10.1021/ja205629b. [DOI] [PubMed] [Google Scholar]
- 72.Walker MC, Edmondson DE. Structure-activity relationships in the oxidation of benzylamine analogues by bovine liver mitochondrial monoamine oxidase B. Biochemistry. 1994;33:7088–7098. doi: 10.1021/bi00189a011. [DOI] [PubMed] [Google Scholar]
- 73.White TA, Krishnan N, Becker DF, Tanner JJ. Structure and kinetics of monofunctional proline dehydrogenase from Thermus thermophilus. J Biol Chem. 2007;282:14316–14327. doi: 10.1074/jbc.M700912200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang M, White TA, Schuermann JP, Baban BA, Becker DF, Tanner JJ. Structures of the Escherichia coli PutA proline dehydrogenase domain in complex with competitive inhibitors. Biochemistry. 2004;43:12539–12548. doi: 10.1021/bi048737e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Serrano H, Blanchard JS. Kinetic and isotopic characterization of L-proline dehydrogenase from Mycobacterium tuberculosis. Biochemistry. 2013;52:5009–5015. doi: 10.1021/bi400338f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kutchan TMD, Heinz Characterization and mechanism of the berberine bridge enzyme, a covalently flavinylated oxidase of benzophenanthridine alkaloid biosynthesis in plants. J Biol Chem. 1995;270:24475–24481. doi: 10.1074/jbc.270.41.24475. [DOI] [PubMed] [Google Scholar]
- 77.Gaweska HM, Roberts KM, Fitzpatrick PF. Isotope effects suggest a stepwise mechanism for berberine bridge enzyme. Biochemistry. 2012;51:7342–7347. doi: 10.1021/bi300887m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rife JE, Cleland WW. Determination of the chemical mechanism of glutamate dehydrogenase from pH studies. Biochemistry. 1980;19:2328–2333. doi: 10.1021/bi00552a008. [DOI] [PubMed] [Google Scholar]
- 79.Grimshaw CE, Cook PF, Cleland WW. Use of isotope effects and pH studies to determine the chemical mechanism of Bacillus subtilis L-alanine dehydrogenase. Biochemistry. 1981;20:5655–5661. doi: 10.1021/bi00523a003. [DOI] [PubMed] [Google Scholar]
- 80.Colen AH, Wilkinson RR, Fisher HF. Location of deuterium oxide solvent isotope effects in the glutamate dehydrogenase reaction. J Biol Chem. 1975;250:5243–5246. [PubMed] [Google Scholar]
- 81.Xu H, Alguindigue SS, West AH, Cook PF. A proposed proton shuttle mechanism for saccharopine dehydrogenase from Saccharomyces cerevisiae. Biochemistry. 2006;46:871–882. doi: 10.1021/bi061980o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kumar VP, Thomas LM, Bobyk KD, Andi B, Cook PF, West AH. Evidence in support of lysine 77 and histidine 96 as acid–base catalytic residues in saccharopine dehydrogenase from Saccharomyces cerevisiae. Biochemistry. 2012;51:857–866. doi: 10.1021/bi201808u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kumar VP, West AH, Cook PF. Supporting role of lysine 13 and glutamate 16 in the acid– base mechanism of saccharopine dehydrogenase from Saccharomyces cerevisiae. Arch Biochem Biophys. 2012;522:57–61. doi: 10.1016/j.abb.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 84.Wolfson-Stofko B, Hadi T, Blanchard JS. Kinetic and mechanistic characterization of the glyceraldehyde 3-phosphate dehydrogenase from Mycobacterium tuberculosis. Arch Biochem Biophys. 2013;540:53–61. doi: 10.1016/j.abb.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Davydov R, Matsui T, Fujii H, Ikeda-Saito M, Hoffman BM. Kinetic isotope effects on the rate-limiting step of heme oxygenase catalysis indicate concerted proton transfer/heme hydroxylation. J Am Chem Soc. 2003;125:16208–16209. doi: 10.1021/ja038923s. [DOI] [PubMed] [Google Scholar]