

Abstract
Mulberry is an important cultivated plant that has received the attention of biologists interested in sericulture and plant–insect interaction. Morus notabilis, a wild mulberry species with a minimal chromosome number is an ideal material for whole-genome sequencing and assembly. The genome and transcriptome of M. notabilis were sequenced and analyzed. In this article, a web-based and open-access database, the Morus Genome Database (MorusDB), was developed to enable easy-to-access and data mining. The MorusDB provides an integrated data source and an easy accession of mulberry large-scale genomic sequencing and assembly, predicted genes and functional annotations, expressed sequence tags (ESTs), transposable elements (TEs), Gene Ontology (GO) terms, horizontal gene transfers between mulberry and silkworm and ortholog and paralog groups. Transcriptome sequencing data for M. notabilis root, leaf, bark, winter bud and male flower can also be searched and downloaded. Furthermore, MorusDB provides an analytical workbench with some built-in tools and pipelines, such as BLAST, Search GO, Mulberry GO and Mulberry GBrowse, to facilitate genomic studies and comparative genomics. The MorusDB provides important genomic resources for scientists working with mulberry and other Moraceae species, which include many important fruit crops. Designed as a basic platform and accompanied by the SilkDB, MorusDB strives to be a comprehensive platform for the silkworm–mulberry interaction studies.
Database URL: http://morus.swu.edu.cn/morusdb.
Introduction
Morus (mulberry) is a genus of flowering plants from the family Moraceae. The deciduous mulberry tree is an economically important food crop for the domesticated silkworm. Human beings have used the mulberry–silkworm interaction for at least 5000 years in the sericulture industry, which greatly changed world history. More than 150 mulberry species were registered, among which Morus notabilis C.K.Schneid was first recorded by Schneide in 1916. It became well known as a naturally available mulberry species, which had 14 chromosomes according to the cytological and morphological studies (1). Morus notabilis was discovered in Sichuan province, Southwest of China, and is named Chuansang, has seven chromosomal pairs as determined by cytological studies (2). Chuansang is a dioecious tree. It is 9–15 m tall and has grayish brown bark, orbicular leaves with triangular serrated and paired male flower, which is 4–5 cm long. As Chuansang has a minimal chromosomal number, it was selected for genome sequencing in the Morus Genome Project (MGP). The MGP was performed on Illumina Hiseq 2000 platform using a whole-genome shotgun sequencing strategy. Twelve libraries were constructed and sequenced to produce 78.34 billion high-quality reads (236-fold genome coverage), from which we assembled a 330.79-Mb genome using SOAPdenovo 2 (3). The MGP also included transcriptomic sequencing of the Chuansang root, branch bark, winter bud, male flower and leaf and expressed sequence tag (EST) sequencing of a combined cDNA library from the five tissues mentioned above (2).
Combined with data of silkworm and other closely related plants, the high-throughput mulberry genomic data provide bases and references for researchers to carry out studies on mulberry genomics, comparative genomics, biology and interactions between silkworm and mulberry. However, it is difficult to use this data efficiently without a platform and tools. Based on the MGP data, we constructed the MorusDB, a database and platform for researchers to search, analyze, collect and share the mulberry genomic and related data.
Data sets and methods
System implementation
The server that MorusDB depends on was built with Linux Ubuntu Sever 12.04, Apache 2, MySQL Server 5.5 and PHP 5.3. The framework of MorusDB is composed of three layers (Figure 1). A relational database, morusdb, is the core layer and implemented in the MySQL relational database management system. All mulberry data and information were stored in MySQL tables so that they can be managed, searched and displayed efficiently. Common gateway interface (CGI) programs and content management system (CMS) constitute the intermediate layer. The CGIs were mainly developed using Perl, PHP, JavaScript and C programming languages, with which we developed scripts for fetching and cutting sequences, searching genes, performing analyses on gene expression, transposable elements and homologous genes, as well as tools and pipelines for aligning sequence and searching gene ontology. The front end is managed by the Drupal (https://drupal.org), an open source CMS that is distributed under the terms of the GNU's Not Unix (GNU) General Public License. Results of search and analyses will be submitted to the Drupal CMS and displayed to user end. The mulberry genome browser, Mulberry GBrowse, is driven by the Generic Genome Browser (4, 5), one of the Generic Model Organism Database (http://gmod.org) components for manipulating and displaying annotations on genomes. The Mulberry GBrowse was configured following instructions so that it can access mulberry data in the morusdb database.
Figure 1.

The framework of MorusDB. MorusDB core is implemented in MySQL database for storing data and contents. CGI programs and CMS were used to constitute the intermediate layer, which was used to carry out analysis and manage and display data and contents to the outer layer, web pages.
Data and processing
The M. notabilis genomic assembly is composed of 110 760 scaffolds, from which 27 085 protein-coding genes consisting of 124 550 exons, 81 rRNA genes, 560 tRNA genes, 311 snRNA genes, 233 microRNA genes and 324 736 repetitive sequences were predicted using de novo and homologous methods (Table 1). Translated proteins were aligned against the latest nonredundant protein database (nr) from the GenBank and UniProtKB/SwissProt from the UniProt release 4.0 (6) using BLASTP 2.2.26 (7, 8) under E-value threshold of 1e-5 to obtain functional references, which were then manually curated. Protein domains and Gene Ontologies (GO) were predicted by searching the SMART (9) and InterPro (10) databases using a local batch script and InterProScan5 (10), respectively. The repetitive elements include 53 725 classified transposable elements (TE) and 271 011 other repeats. The scaffolds accompanied by all the predicted genes, repeats and annotations were processed and imported into the morusdb. Meanwhile, the M. notabilis genome data have been deposited in National Center for Biotechnology Information (NCBI) under BioProject no. PRJNA202089 and GenBank no. ATGF00000000.1.
Table 1.
Data set of mulberry M. notabilis deposited in MorusDB (1)
| No. | Size (base pairs / amino acids) | |
|---|---|---|
| Genome | ||
| Scaffold | 110 760 | 330 791 087 |
| Gene | 27 085 | 77 647 891 |
| CDS | 124 550 | 29 437 365 |
| Protein | 27 085 | 9 785 370 |
| rRNA | 81 | 12 174 |
| tRNA | 560 | 43 692 |
| snRNA | 311 | 34 162 |
| microRNA | 233 | 26 520 |
| Repeats | ||
| Known transposable element | 53 725 | 74 910 659 |
| Others | 271 011 | 53 073 173 |
| Transcriptome | ||
| Libraries for mRNA sequencing | ||
| Leaf | 25 853 672 | 2 326 830 480 |
| Root | 25 583 558 | 2 302 520 220 |
| Branch bark | 25 969 690 | 2 337 272 100 |
| Winter bud | 26 774 498 | 2 409 704 820 |
| Male flower | 25 688 148 | 2 311 933 320 |
| cDNA library | ||
| EST of above tissues | 9573 | 4 595 661 |
To identify orthologous and paralogous groups (OPGs) of M. notabilis genes (MOPGs), the latest version of protein sequences of relative plants were downloaded. Arabidopsis thaliana proteins were downloaded from the FTP server (ftp://ftp.arabidopsis.org/home/tair/Proteins/TAIR10_protein_ lists) of TAIR database (http://www.ara bidopsis.org). Proteins of Populus trichocarpa, Malus domestica and Fragaria vesca were downloaded from the FTP server (ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v9.0) of DOE JGI (http://www.jgi.doe.gov). Thereafter, the OPGs of the aforementioned five plants were predicted using OrthoMCL v2.0.9. Proteins were all-against-all aligned using BLASTP 2.2.26 (7, 8) under E-value cutoff of 1e-6. The OrthoMCL (11) pipeline was then run under the percentMatchCutoff of 50. Finally, 24 065 MOPGs, including 19 858 M. notabilis genes, were obtained from a collection of 231 832 proteins and imported into the morusdb.
The Illumina sequencing of M. notabilis transcriptome from the five tissues generated 11.7 Gb sequences, which were deposited in the NCBI SRA under accession number of SRP040752. The transcriptome sequences were mapped to the genome sequences using SOAPaligner 2.20 (http://soap.genomics.org.cn/soapaligner.html) to determine gene expression levels using number of reads per kilobase per million mapped reads (RPKM) (12). Features of gene expression in the five tissues were then analyzed based on the RPKMs. In addition to screen out the tissue-specific expressed genes and housekeeping genes (2), we also found 8814 genes that encoded two or more isoforms and 4.97 Mb of untranslated region (UTR) from 16 513 mulberry genes. Meanwhile, the 9573 ESTs sequenced from the combined cDNA library of the five tissues were aligned against the scaffolds using BLAT (13).
Results
We intended to provide users with an efficient and direct way to access MorusDB data. Therefore, a clean and simple home page (Figure 2) was built to enable users to search for the mulberry data and information, perform analyses and download all data just by clicking hyperlinks on a navigation menu on top of the page. MorusDB is also a responsive Web site and friendly to both mobile and desktop devices.
Figure 2.

A snapshot of the MorusDB home page. The home page describes the mulberry genome sequencing project and provides links to all other parts of the website.
Search database
MorusDB contents, including mulberry knowledge, news, tools, etc., can be quickly searched with top-right search engine. Using Fetch Data tool on the left side of pages, users can easily cut a sequence region, retrieve sequences in batches and search genes with keywords (Figure 3). Homologous sequence search can be carried out using the NCBI-BLAST 2.2.26 (7, 8) and the faster and more sensitive AB-BLAST 3.0 (http://blast.advbiocomp.com) against genome data of mulberry, silkworm and the public nr and UniProt databases. The search results can be printed out as a standard output with alignment figures or parsed into a tabular format under a given number of best matches and best hits.
Figure 3.

Searching and browsing M. notabilis genes. Genes of M. notabilis can be browsed or searched (A) using tools ‘Index of Chuansang Genes’ and ‘Search by Keywords’. Gene structures can be bird-eye viewed by mouse-hovering gene IDs (A and B). Users can click on these IDs to open the Mulberry GBrowse to see the detailed information (C and D).
Mulberry GO
Morus notabilis gene ontologies can be analyzed using the Mulberry GO (Figure 4) tool package. GO can be retrieved using Fetch GO with given gene IDs and searched using Search GO, which will align given sequence against the InterPro database by running the InterProScan 5 locally. Both tools will list GO IDs with explanation hyperlinks to AmiGO (14), QuickGO (15), GONUTS (16) and External2GO (17). Browse GO provides a birds-eye view of the M. notabilis GO. The Browse GO classified M. notabilis genes into three GO categories, cellular component, molecular function and biological process. Morus notabilis genes under each subcategory were listed and linked to detailed gene information pages.
Figure 4.

Snapshots of searching M. notabilis gene ontologies (GOs). Researchers can fetch GOs by gene IDs using Fetch GO (A and B) and search GOs by sequences or sequence IDs using Search GO (C and D). All M. notabilis GOs can also be birds-eye viewed with Browse GO (E and F).
Mulberry Transcriptome, TEs and MOPGs
To manage large data efficiently, M. notabilis gene expression patterns in the five tissues, classified transposable elements and orthologous and paralogous groups were stored in MySQL tables, which can be exported to the JavaScript Object Notation (JSON, http://www.json.org) data. The JSON data can be easily sorted, filtered and exported using the jqxGrid (http://www.jqwidgets.com), which is a powerful datagrid widget built entirely with open web standards and offers rich functionality. Therefore, researchers can search each column by keywords or numbers (Figure 5). Hyperlinks were added to IDs and numbers for a quick accession of detailed information and sequence.
Figure 5.
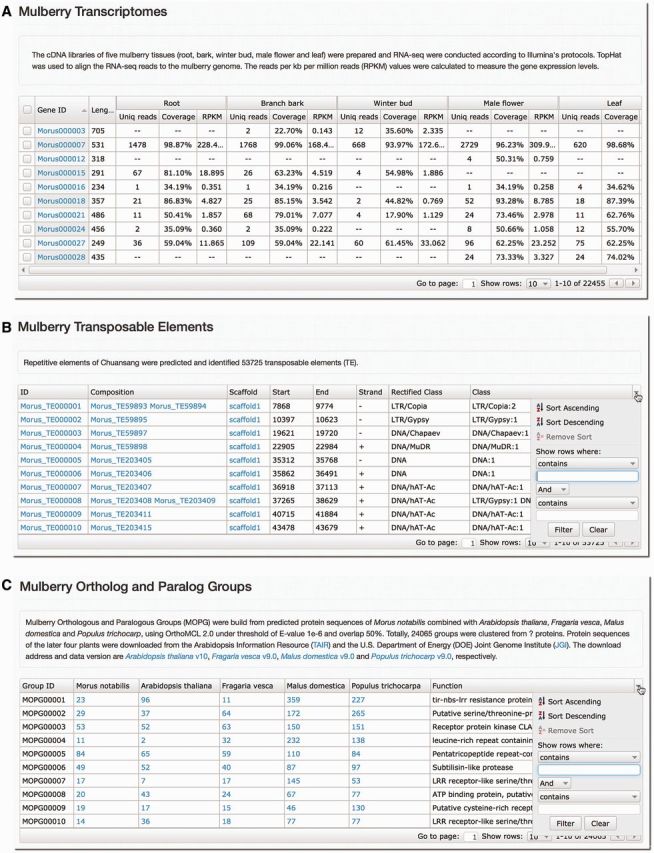
Screenshots of analyses on M. notabilis transcriptome, TEs and OPGs. Gene expressions in the five tissues (A), TEs (B) and OPGs (C) of the M. notabilis can be searched and filtered by keyword or number in each column of the tables.
Mulberry GBrowse
The Mulberry GBrowse is a comprehensive and powerful analysis tool, which has integrated the M. notabilis genes, CDSs, proteins and TEs, as well as the transcriptome data. Using this tool, users can easily browse and search M. notabilis data on a large scale and with a graphic interface, and conveniently view and fetch detailed gene information including location, annotation, GO, sequences, etc. (Figure 3).
Conclusion and future perspective
The mulberry genomic information provides a foundation for many kinds of studies: revealing the global organization of the mulberry genome, enabling studies of comparative genomics among mulberry and other eudicot species, accelerating gene identification and characterization and applying ‘omics’ technologies to better understand the biological phenomena of mulberry. The MorusDB is a comprehensive resource and platform that provides researchers with not only the present mulberry genome data but also tools for carrying out data analysis. Mulberry and silkworm were both sequenced as a plant–herbivore pair so that they are ideal objects and materials for interactional studies. In this regard, the MorusDB and SilkDB (18, 19), both maintained in our laboratory, not only provide kinds of data, but also form an associated platform for related studies in the future.
Beyond its initial release, MorusDB is a continuous effort to follow advances in studies on mulberry and expand, revise and improve the mulberry data of genome, transcriptome and proteome, as well as biological information. Furthermore, we will keep making efforts to develop MorusDB built-in tools and pipelines to facilitate and promote studies on mulberry.
Acknowledgements
The authors thank all people’s contribution on this work. They would like to thank the anonymous reviewers for their valuable comments and suggestions.
Funding
The National Hi-Tech research and Development Program of China (2013AA100605-3); the ‘111’ Project (B12006); the Fundamental Research Funds for the Central Universities (2362014xk05); the Science Fund for Distinguished Young Scholarship of Chongqing (cstc 2011jjjq0010). Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest. None declared.
References
- 1.Janaki Ammal E. (1948) The origin of black mulberry. J.R. Hortic. Soc., 73, 117–120. [Google Scholar]
- 2.He N., Zhang C., Qi X., et al. (2013) Draft genome sequence of the mulberry tree Morus notabilis. Nat. Commun., 4, 2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo R., Liu B., Xie Y., et al. (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience, 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stein L.D., Mungall C., Shu S., et al. (2002) The generic genome browser: a building block for a model organism system database. Genome Res., 12, 1599–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stein L.D. (2013) Using GBrowse 2.0 to visualize and share next-generation sequence data. Brief. Bioinform., 14, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Apweiler R., Bairoch A., Wu C.H., et al. (2004) UniProt: the universal protein knowledgebase. Nucleic Acids Res., 32, D115–D119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camacho C., Coulouris G., Avagyan V., et al. (2009) BLAST+: architecture and applications. BMC Bioinform., 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altschul S.F., Madden T.L., Schaffer A.A., et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Letunic I., Doerks T., Bork P. (2012) SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res., 40, D302–D305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mulder N., Apweiler R. (2007) InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol. Biol., 396, 59–70. [DOI] [PubMed] [Google Scholar]
- 11.Li L., Stoeckert C.J., Jr, Roos D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res., 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mortazavi A., Williams B.A., McCue K., et al. (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods, 5, 621–628. [DOI] [PubMed] [Google Scholar]
- 13.Kent W.J. (2002) BLAT–the BLAST-like alignment tool. Genome Res., 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carbon S., Ireland A., Mungall C.J., et al. (2009) AmiGO: online access to ontology and annotation data. Bioinformatics, 25, 288–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Binns D., Dimmer E., Huntley R., et al. (2009) QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics, 25, 3045–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renfro D.P., McIntosh B.K., Venkatraman A., et al. (2012) GONUTS: the Gene Ontology normal usage tracking system. Nucleic Acids Res., 40, D1262–D1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye J., Fang L., Zheng H., et al. (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res., 34, W293–W297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J., Xia Q., He X., et al. (2005) SilkDB: a knowledgebase for silkworm biology and genomics. Nucleic Acids Res., 33, D399–D402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan J., Li R., Cheng D., et al. (2010) SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res., 38, D453–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]