

Abstract
Self-amplifying messenger RNA (mRNA) of positive-strand RNA viruses are effective vectors for in situ expression of vaccine antigens and have potential as a new vaccine technology platform well suited for global health applications. The SAM vaccine platform is based on a synthetic, self-amplifying mRNA delivered by a nonviral delivery system. The safety and immunogenicity of an HIV SAM vaccine encoding a clade C envelope glycoprotein formulated with a cationic nanoemulsion (CNE) delivery system was evaluated in rhesus macaques. The HIV SAM vaccine induced potent cellular immune responses that were greater in magnitude than those induced by self-amplifying mRNA packaged in a viral replicon particle (VRP) or by a recombinant HIV envelope protein formulated with MF59 adjuvant, anti-envelope binding (including anti-V1V2), and neutralizing antibody responses that exceeded those induced by the VRP vaccine. These studies provide the first evidence in nonhuman primates that HIV vaccination with a relatively low dose (50 µg) of formulated self-amplifying mRNA is safe and immunogenic.
Keywords: self-amplifying mRNA, HIV, vaccine, antibodies, T cells
Vaccination is one of the most important tools to combat infectious diseases such as human immunodeficiency virus (HIV) infection. Nucleic acid vaccines (eg, plasmid DNA, viral vector, and messenger RNA [mRNA] vaccines) have been evaluated in a variety of experimental clinical settings against cancer, malaria parasites, hepatitis B virus, and HIV type 1 [1–3]. RNA approaches in particular have been studied for cancer [4], allergy [5], and influenza vaccines [6], as well as for gene therapies [7, 8]. So far, DNA vaccines have shown low potency in humans during clinical trials, in contrast to the robust cellular and humoral immunogenicity elicited by standard intramuscularly injected vaccines in small animals [9]. As a consequence, the focus of many DNA vaccine strategies has shifted to their ability to prime the immune response in heterologous prime-boost regimens, using various viral vectors, such as adenovirus or modified vaccinia Ankara [10–12], or recombinant subunit proteins [13] as boosts.
Recombinant viral vector technologies have the advantage of efficient delivery of the nucleic acid payload, but their usefulness is often hampered by preexisting antivector immunity, production limitations, and safety concerns, as in the case with adenovirus serotype 5–based vaccines [14, 15]. Also, antivector immunity develops rapidly after vaccination with recombinant viral vectors, thereby interfering with subsequent immunizations [16]. DNA/RNA vaccines are not limited by such constraints and can be safely and effectively administered repeatedly to humans [17, 18].
We have previously described the SAM vaccine platform [8, 19–21]. This platform, now in preclinical development, is based on a synthetic, self-amplifying mRNA delivered by a synthetic lipid nanoparticle [19]. We used a self-amplifying RNA based on an alphavirus genome [22], which contains the genes encoding the alphavirus RNA replication machinery but lacks the genes encoding the viral structural proteins required to make an infectious alphavirus particle. The structural protein genes are replaced with genes encoding protein antigens, which are abundantly expressed from a subgenomic mRNA in the cytoplasm of cells transfected with these self-amplifying RNAs [19]. It was shown that nonviral delivery of a 9-kb self-amplifying RNA encapsulated within a lipid nanoparticle substantially increased immunogenicity, compared with delivery of unformulated RNA, and that this novel vaccine technology was able to elicit broad, potent, and protective immune responses in rodents, comparable to a viral delivery technology and to a 200-fold higher dose of pDNA delivered using electroporation. In the current study, we evaluated the usefulness of an alternative nonviral delivery system. This newer formulation is based on a cationic nanoemulsion (CNE) that binds to the self-amplifying mRNA or “SAM” vector and enhances the delivery and potency of the vaccine [23]. CNE is based on the oil-in-water emulsion adjuvant MF59, which has been extensively tested in clinical trials, is licensed in 30 countries, and has an established safety profile in children, adults, and elderly individuals [24, 25]. Here we evaluated CNE delivery of an HIV SAM vaccine in rhesus macaques and compared its immunogenicity to that of 2 vaccine modalities known to be immunogenic in humans: viral delivery of self-amplifying RNA using viral replicon particles (VRPs) [26] and recombinant envelope protein formulated with the potent adjuvant MF59 [27].
METHODS
Preparation of CNE
CNE was prepared as described elsewhere [23, 28]. Briefly, squalene, DOTAP, and sorbitan trioleate were combined and heated to 37°C. The resulting oil phase was then combined with an aqueous phase consisting of polysorbate 80 in 10 mM citrate buffer at pH 6.5. The final weight by weight percentages of squalene, DOTAP, sorbitan trioleate, and polysorbate 80 were 4.3%, 0.4%, 0.5%, and 0.5%, respectively. This mixture was homogenized using a T25 homogenizer with a 13.4 mm diameter rotor (IKA, Wilmington, North Carolina) at 24 000 RPM to produce a primary emulsion. This was then passed through a M-110P Microfluidizer (Microfluidics, Newton, Massachusetts) with an ice bath cooling coil at a homogenization pressure of 137 Mpa approximately 8 times. The formulation was stored at 4°C before use. The 100-nm CNE had a positive surface charge, which was used to adsorb the RNA to the surface of the oil droplet through an electrostatic interaction with the negatively charge phosphate backbone. RNA was diluted to 200 µg/mL and was added to an equal volume of CNE, mixed, and allowed to equilibrate on ice for 30 minutes to 2 hours. Endotoxin levels were measured by the gel clot LAL assay per the manufacturer's instructions (Cape Cod Associates, Massachusetts) and found to be <2.48 EU/mL (or 0.62 EU/dose).
Production of VRPs
To compare RNA vaccines to traditional RNA-vectored approaches for achieving in vivo expression of antigens, we used VRP produced from baby hamster kidney cells and characterized as previously described [19, 22]. The replicon used was derived from the genome of Venezuelan equine encephalitis virus engineered to contain the 3′ terminal sequences (3′ untranslated region) of Sindbis virus and a Sindbis virus packaging signal. These replicons were packaged into VRPs by co-electroporating them into baby hamster kidney cells along with defective helper RNAs encoding the Sindbis virus capsid and glycoprotein genes.
Production and Formulation of Recombinant TV1 gp140 Protein
The recombinant trimeric TV1 gp140 protein used in this study was expressed, purified, and further analyzed for purity, homogeneity, soluble CD4, monoclonal antibody binding, and endotoxin levels before immunization studies, as previously described [29, 30]. Shortly before immunization, the TV1 gp140 protein was mixed with an equal volume of MF59, which was prepared as described elsewhere [31].
Animals, Immunization, and Safety Evaluations
Twenty-four mature outbred male rhesus macaques of Chinese origin were housed at the Biomedical Primate Research Centre (BPRC; Rijswijk, the Netherlands), according to international guidelines for nonhuman primate care and use (European Council Directive 86/609/EEC, and Convention ETS 123, including the revised Appendix A). The institutional animal care and use committee (DEC-BPRC) approved the study. Four groups of 6 animals each were primed via intramuscular injection in the upper left arm at weeks 0, 4, and 12 (volume, 0.5 mL). Group 1 received 1 × 108 infectious units (IU) of VRPs encoding HIV-1 TV1 Env gp140. Group 2 received 50 µg of CNE-formulated SAM RNA encoding HIV-1 TV1 Env gp140. Group 3 received 100 µg of HIV-1 TV1 Env gp140 protein with MF59. All experimental groups received a boost dose intramuscularly in the upper left arm at weeks 24 and 36 with 100 µg of HIV-1 TV1 Env gp140 with MF59 (volume, 0.5 mL). One control group of 6 animals was divided in 2 subgroups of 3 animals each. Both subgroups received a prime dose at weeks 0, 4, and 12. One subgroup received 1 × 108 IU of VRPs encoding a control viral antigen, whereas the other subgroup received 50 µg of SAM vaccine expressing a control viral glycoprotein. Both subgroups received a boost dose at weeks 24 and 36 with control protein in MF59. Blood specimens were obtained 2 weeks before, 2 weeks after, and on the day of every immunization. To investigate possible adverse effects, sites of immunization were observed for redness, swelling, and/or inflammation. Furthermore, body weight, rectal temperature, routine hematologic variables, and clinical chemistry variables were monitored.
Humoral Immune Responses
Enzyme-Linked Immunosorbent Assay (ELISA) for Antigen-Specific Antibody
Antibodies specific for gp140 envelope were measured in serum using an ELISA [32]. Binding antibody levels targeting HIV-1 gp120 variable regions 1 and 2 (V1V2) were determined using customized multiplex binding assays as described elsewhere [33–36]. The gp70-CaseA V1V2 scaffold antigen was provided by Drs Liao and Haynes [35, 37]. Each sample was tested at six 5-fold serial dilutions, starting at 1:80. Areas under the curve (AUCs) were calculated using GraphPad Prism 5.
Antigen-Specific B-Cell Enzyme-Linked Immunosorbent Spot (ELISPOT) Assay
Antigen-specific B-cell counts were measured as described previously [38]. This assay uses an ex vivo 5-day stimulation of peripheral blood mononuclear cells (PBMCs) to induce quiescent memory B cells to differentiate into plasma cells/antibody-secreting cells (ASCs) that can then be measured by an ELISPOT assay, using plates coated with the antigen. Data are shown as the number of ASCs/1 × 106 PBMCs.
Virus Neutralization
For the standardized neutralization assays, the TZM-bl cell line was used [39–41]; the cells were obtained through the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program in the Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH, from Dr John C. Kappes, Dr Xiaoyun Wu, and Tranzyme. Sera were diluted 1:20 initially and, subsequently, in a 3-fold series to achieve a final dilution of 1:43 740. Each dilution was mixed with pseudoviruses to give 500 000 counts per second in a Perkin-Elmer Victor 6016971 luminometer. Neutralizing titers are expressed as the serum dilution required to reduce the luciferase activity in cultures exposed to pseudovirus alone by 50% [42–44]. The virus neutralization activity of immune sera was measured against pseudoviruses (constructed at the BPRC) derived from clade C MW965.26, SHIV1157ipEL-p, SHIV1157ipd3N4, and HIV-1TV1 and the clade B SHIVSF162P4.
Cellular Immunology: T-Cell ELISPOT Assay
Quantification of antigen-specific cytokine-secreting cells was performed on freshly isolated PBMCs by interferon γ (IFN-γ), interleukin 2 (IL-2), and interleukin 4 (IL-4) ELISPOT assays, as described elsewhere [45]. Separate peptide pools, consisting of 15mers with an 11–amino acid overlap, that covered the entire gp120 of ConS, gp41 ConS, or gp120 TV1 protein were used to measure antigen-specific immune responses after each immunization.
Statistical Analyses
Comparisons among multiple groups were performed using analysis of variance. A 2-sided Wilcoxon rank sum analysis was used to test for differences between immunization groups. For all comparisons, a 2-sided P value of < .05 was considered statistically significant. All analyses were performed using the analysis software within the GraphPad Prism package.
RESULTS
Assessment of Safety of HIV SAM Vaccines
None of the vaccines evaluated in this study appeared to cause any notable adverse events. No swelling, redness, or inflammation at the injection sites was observed after immunization, and no differences in behavior were seen. To assess possible systemic off-target effects caused by the vaccines, biochemical and hematological parameters were recorded from every blood sampling time point. As examples, body mass, proportion of systemic lymphocytes, and alanine aminotransferase (ALT) levels in the blood are shown (Figure 1). None of the vaccines resulted in changes in body mass or deviations outside of the normal ranges for lymphocyte counts, ALT levels, or other biochemical and hematological parameters (data not shown).
Figure 1.

Assessment of safety. Safety evaluations were performed by measuring standard hematological and biochemical parameters throughout the study (see Materials and Methods). Mean body weights (in kg) ± standard error of the mean (SEM) are shown in the left panel. A representative of hematological parameters is given by the mean lymphocyte percentage ( ± SEM; upper right panel). As a representative of biochemical analysis, the mean alanine aminotransferase (ALT) level (± SEM) is shown in the lower right panel. The gray area in the hematology and clinical chemistry panels represents the normal range of the corresponding parameters, as measured in the colony of Rhesus macaques (n = 1200) from the Biomedical Primate Research Centre (Rijswijk, the Netherlands). Groups were primed 3 times (weeks 0, 4, and 12) with either 1 × 108 IU of viral replicon particles (VRPs) encoding human immunodeficiency virus type 1 (HIV-1) TV1 Env gp140 (black symbols), 50 µg cationic nanoemulsion (CNE)–formulated SAM RNA encoding HIV-1 TV1 Env gp140 (red symbols), 100 µg HIV-1 TV1 Env gp140 protein with MF59 as adjuvant (green symbols), or sham VRP/SAM vaccines (black open symbols and dotted line). All experimental groups received a boost dose at weeks 24 and 36 with 100 µg HIV-1 TV1 Env gp140 with MF59 as adjuvant and control groups. Blue arrowheads indicate the 3 prime immunizations, and the 2 brown arrowheads indicate the 2 Env/MF59 booster immunizations.
Elicitation of Humoral Immune Responses by HIV SAM Vaccine
Anti-envelope humoral immune responses were assessed in 4 ways. First, anti–Env-specific binding antibody titers were measured by ELISA (Figure 2). Rapid and strong responses were induced in all experimental animals, with the highest levels seen in the Env/MF59 group, in which titers ranged between 103.5 and 105.5 after 3 immunizations (geometric mean titer [GMT] at peak, 105.00). Animals receiving the HIV SAM and HIV-VRP vaccines reached anti-Env levels of 103–104.5, with GMTs of 103.94 and 103.41, respectively. Anti-Env antibody titers increased significantly after boosting with Env/MF59, reaching levels 10–100-fold higher than those found after priming (Figure 2), with peak GMTs of 106.14, 106.25, and 104.79 for Env/MF59, HIV SAM, and HIV-VRP, respectively.
Figure 2.
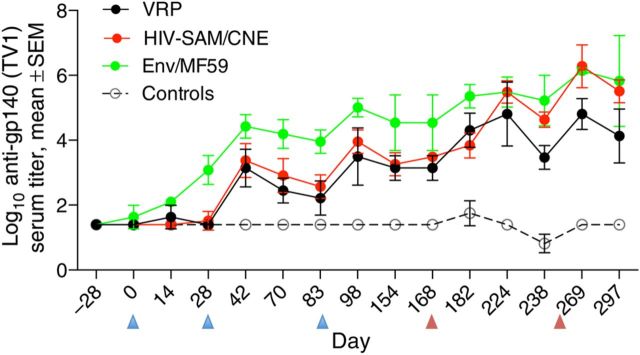
Kinetics of antibody responses following immunizations. TV1 envelope-specific enzyme-linked immunosorbent assay end point titers are given as means (±standard error of the mean) for 6 animals per group. The various groups of animals are immunized as described in the legend of Figure 1. Blue arrowheads represent the 3 prime immunizations, and the 2 brown arrowheads represent the 2 Env/MF59 booster immunizations. Abbreviations: CNE, cationic nanoemulsion; HIV, human immunodeficiency virus; VRP, viral replicon particle.
Second, to enumerate the number of antigen-specific B-cells, ELISPOT analysis was performed. The number of ASCs in the HIV-VRP–immunized animals remained between approximately 300 and 650 cells per 106 PBMCs during both the priming and boosting phases of the study (Figure 3). In contrast, the HIV SAM and Env/MF59 vaccines induced ASCs ranging from 350 to 1450 and 250 to 1550 per 106 PBMCs, respectively, which increased over time with subsequent immunizations. Very low numbers of Env-specific ASCs (<150 per 106 PBMCs) were detected in the control group (Figure 3). No significant differences between HIV SAM and Env/MF59-immunized groups were observed (P > .05), but both groups generated higher numbers of ASCs per 106 PBMCs than the VRP and control immunized groups (P < .05).
Figure 3.

Dynamic of TV1 Env-specific B-cells in peripheral blood. The frequency of Env-specific memory B cells were determined by B-cell enzyme-linked immunosorbent spot assays in the 4 different groups. The values (numbers of antibody-secreting cells (ASCs) per 106 peripheral blood mononuclear cells) are means (±standard error of the mean) for 6 animals per group. Blue arrowheads represent the 3 prime immunizations, and the 2 brown arrowheads represent the 2 Env/MF59 booster immunizations. Abbreviations: CNE, cationic nanoemulsion; HIV, human immunodeficiency virus; VRP, viral replicon particle.
Third, the antibody response directed against the V1V2 of HIV-1 gp120 was evaluated by customized binding antibody multiplex assays, using the gp70-CaseA V1V2 scaffold. At week 14, 2 weeks after the last prime dose for the VRP and the HIV SAM group or the third immunization for the Env/MF59 group, 4 of 6 animals in the VRP group, 6 of 6 animals in the HIV SAM group, and 4 of 5 animals in the Env/MF59 group (1 animal in this group was excluded because of significant binding to gp70 backbone in the assays) developed an anti-V1V2 IgG response. The magnitude of the response for the HIV SAM group was slightly lower but not significantly different from that of the Env/MF59 group (P > .05) and was significantly higher than that of the VRP group (P = .013; Figure 4). The median AUCs for the VRP, HIV SAM, Env/MF59, and control groups were 860, 14 865, 21 966, and 50, respectively (data not shown). After 2 protein boosts, the anti-V1V2 response was enhanced for all 3 vaccine groups. No significant difference in magnitude was observed among the groups, although the VRP group remained lower, compared with the other 2 groups (Figure 4). The median AUCs for the VRP, HIV SAM, Env/MF59, and control groups were 16 501, 42 129, 32 233, and 50, respectively (data not shown).
Figure 4.

Multiplex assays for variable regions 1 and 2 (V1V2) immunoglobulin G binding antibodies. Week 14 (left panel) and week 38 (right panel) serum samples were serially diluted and tested for binding to gp70 CaseA2 V1V2. Antibody titers are expressed as areas under the curve (AUCs) for each group. The mean AUCs and standard error of the mean for each group are shown. P values are from a Mann–Whitney U test of the median values. One animal in the Env/MF59 group was excluded from the analysis because of high background binding to gp70 backbone. Abbreviations: HIV, human immunodeficiency virus; VRP, viral replicon particle.
Fourth, the virus-neutralizing capacity of sera was measured in the standardized TZM-bl luciferase reporter assay after a single round of infection against tier 1 and tier 2 pseudoviruses. After the 3 priming immunizations, the highest levels of neutralizing antibodies were detected in the Env/MF59 group against the homologous clade C TV-1, the heterologous clade C tier 1 SHIV1157ipEL-p, and the clade B tier 1 SHIVSF162p4 (Figure 5). These neutralizing antibody titers were boosted after 2 booster immunizations. None of the HIV-VRP–immunized animals showed neutralizing activity against the panel tested during the priming phase, and only low levels were seen after Env/MF59 boosting. Three of the 6 HIV SAM–immunized animals had very low but detectable levels of neutralizing antibodies during the priming phase against the SHIV1157ipEL-p, which could also cross-neutralize the clade B strain. The levels of neutralizing antibody titers against the same pseudoviruses increased significantly after Env/MF59 boosting to the same levels as those in animals that received Env/MF59 5 times (Figure 5). Importantly, the SAM prime immunization group showed high neutralization responses against a tier 1 clade C strain MW965.26 (Figure 5).
Figure 5.

Kinetics of neutralizing antibody responses following immunizations. Virus-neutralizing activities of sera from immunized macaques were measured against pseudoviruses derived from clade C MW965.26, HIV-1TV1, SHIV1157ipEL-p, and SHIV1157ipd3N4 and clade B SHIVSF162P4. Antibody titers are expressed as the dilution of serum required to reduce the luciferase activity in cultures exposed to pseudovirus alone by 50%. The values are means (±standard error of the mean) for 6 animals per group. Dotted lines indicate the lowest dilution tested. Pre: sera from week −2; 2wp3, 2 weeks after the third immunization; 2wp4, 2 weeks after the fourth immunization; 2wp5, 2 weeks after the fifth immunization. Abbreviations: CNE, cationic nanoemulsion; HIV, human immunodeficiency virus; VRP, viral replicon particle.
Elicitation of Cellular Immune Responses to HIV SAM Vaccine
The induction of cellular immune responses was evaluated by quantification of antigen-specific cytokine-secreting T-cells (IFN-γ, IL-2, and IL-4) performed on freshly isolated PBMCs by ELISPOT analysis (Figure 6). Two HIV SAM prime immunizations resulted in clearly detectable cellular immune responses at week 6 by the IFN-γ ELISPOT assay, with mean values (±standard error of the mean [SEM]) of 362 ± 46 spot-forming units (SFUs)/106 PBMCs after cells were stimulated with gp120 ConS pp, 375 ± 117 SFUs/106 PBMCs after gp41 ConS pp stimulation, and 533 ± 93 SFUs/106 PBMCs after gp120 TV1 protein stimulation. Mean responses (±SEM) further increased after the third immunization to levels up to 947 ± 99 SFUs/106 PBMCs (gp120 pp ConS stimulation), 705 ± 155 SFUs/106 PBMCs (gp41 ConS pp stimulation), and 1135 ± 122 SFUs/106 PBMCs (TV1 stimulation). Responses were relatively stable for 10 weeks. Booster immunizations had only a marginal effect on the number of IFN-γ–secreting cell populations. In contrast, priming with the HIV-VRP vaccine elicited very low levels of IFN-γ–producing cells (<30 SFUs/106 PBMCs) between weeks 0 and 22. Env/MF59 boosting gave slightly higher responses in this group, with mean values (±SEM) ranging between 132 ± 39 and 366 ± 57 SFUs/106 PBMCs (gp120 ConS), 37 ± 10 and 197 ± 32 SFUs/106 PBMCs (gp41 ConS), and 98 ± 18 and 480 ± 31 SFUs/106 PBMCs (TV1 stimulation). Five immunizations with Env/MF59 induced moderate responses when the cells were stimulated with gp120 pp, gp41 pp, or gp120 TV1 antigens (Figure 6). Overall, the HIV SAM–immunized group showed significantly higher IFN-γ responses, compared with the VRP group (P < .0003), the Env/MF59 group (P < .001), and the control group (P < .0004).
Figure 6.

Quantification of antigen-specific cytokine-secreting cells was performed on freshly isolated peripheral blood mononuclear cells (PBMCs) by interferon γ (IFN-γ; left column), interleukin 2 (IL-2; middle column), and interleukin 4 (IL-4; right column) enzyme-linked immunospot assays. Separate peptide pools, consisting of 15mers with an 11–amino acid overlap, which covered the entire gp120 of ConS (upper row), gp41 ConS (middle row), or gp120 TV1 protein (bottom row), were used to measure antigen-specific immune responses after each immunization. Responses after antigen stimulation are shown as the mean numbers of spot-forming cells per 106 PBMCs ( ± standard error of the mean) for 6 animals per group. Groups of animals are color coded as described in the legend of Figure 1. Blue arrows represent the 3 prime immunizations, and the 2 brown arrows represent the 2 Env/MF59 booster immunizations. Abbreviations: CNE, cationic nanoemulsion; HIV, human immunodeficiency virus; SFU, spot forming unit; VRP, viral replicon particle.
Similar patterns of responses were observed with IL-2 secretion (Figure 6). As before, the highest levels of IL-2–producing cell populations were seen in the HIV SAM group, with only low levels in most of the animals in the HIV-VRP and Env/MF59 groups (HIV SAM vs VRP, P < .002 with TV1 antigen; HIV SAM vs Env/MF59, P < .012 with gp120 ConS; HIV SAM vs control, P < .006). In animals from all groups, IL-2–secreting cell levels diminished with time.
IL-4 responses were observed in HIV SAM and Env/MF59 recipients, with similar levels (P > .05). Three HIV SAM prime doses yielded levels of IL-4 similar to those achieved after 3 Env/MF59 immunizations. Env/MF59 boosting did not result in higher IL-4 levels (Figure 6).
It is noteworthy that although the HIV SAM CNE and Env/MF59 vaccines were delivered using similar formulations, the SAM vaccine induced overall higher levels of T-helper type 1 (Th1) cytokine responses (IFN-γ and IL-2) to both gp120 and gp41, whereas the Env vaccine induced higher IL-4 responses to these antigens (albeit at similar levels to those induced by the SAM CNE vaccine). This highlights the potential immune stimulatory effects of the SAM component of the vaccine.
DISCUSSION
This study provides the first evidence in a nonhuman primate species that HIV vaccination with self-amplifying RNA formulated with CNE is safe and immunogenic, eliciting both humoral and cellular immune responses. It also demonstrates the first evidence of the induction of potent anti-HIV–directed immune responses from a nucleic acid vaccine delivered with a nonviral delivery system at submilligram doses (50 µg) in any primate species. The HIV SAM vaccine evaluated here induced potent and broad-based immune responses, including levels of Th1-type T cells that were greater in magnitude than those induced by the VRP or Env/MF59 vaccines, and anti-envelope antibody responses (including neutralizing antibodies) that were greater in magnitude than those induced by the VRP vaccine. The reasons for the higher potency of the SAM vaccine, compared with the VRP vaccine, remain to be determined but may be related to the higher amount of immunostimulatory RNA contained in SAM vaccine versus VRP vaccine and/or a differential antigen expression profile in situ [19]. The highest levels of both antibody and T-cell responses were seen in animals primed with HIV SAM and boosted with Env/MF59. No adverse events or safety signals were observed in any of the animals. These results corroborate recent findings obtained by using a similarly formulated CMV SAM vaccine in rhesus macaques, in which potent antigen-specific antibody and T-cell immune responses were also observed [23].
SAM vaccines, based on the nonviral delivery of self-amplifying mRNA encoding antigenic viral proteins, offer the potential to combine the positive attributes of live-attenuated, inactivated, and recombinant subunit vaccines, while avoiding many of their limitations. First, SAM vaccines are synthetic, produced from an enzymatic transcription reaction, and do not require cell culture for their production. Second, the dose of SAM vaccine needed for induction of potent immune responses in primates will likely be <100 µg, as judged by the data presented in this study. Based on current early-stage production methods developed so far, even a small-scale reaction vessel of 1L is capable of producing up to 100 000 doses of highly pure RNA drug substance in 6 hours [46]. Finally, The SAM vector, once delivered into the cytoplasm of a cell, launches the replication of many copies of itself and mRNA encoding the antigen of interest and stimulates various elements of the innate immune system, thereby providing a built-in adjuvant effect [47].
The aim of the current study was to evaluate the SAM technology for its potential use as an HIV vaccine and compare its potency directly to that of 2 vaccine technologies that have already achieved proof of concept in humans for induction of antibody and T-cell responses. Here we have demonstrated that a SAM vaccine encoding HIV envelope can elicit a broad spectrum of immune responses, including Th1-type T-cell responses, that compared favorably with both the corresponding VRP-based and adjuvanted subunit vaccines at doses comparable to those previously tested in human clinical trials [48]. The quantity and quality of anti-Env antibodies induced by the SAM vaccine are higher than those observed with the VRP vaccine. And although the levels of the SAM vaccine–induced neutralizing antibodies are not as high in this study as those elicited by the adjuvanted subunit protein vaccine during the priming phase, equivalent responses were seen after the protein boost. In addition, the HIV SAM vaccine alone elicited significant anti-V1V2 IgG responses before administration of the protein boosts. V1V2 IgG responses are of particular interest since they were correlated with decreased HIV-1 infection risk in the RV144 trial [35]. In summary, these observations provide an early proof of concept for this vaccine approach for the elicitation of anti-HIV immune responses in nonhuman primates. Future studies in relevant vaccine challenge models and human subjects will be needed to identify optimal regimens for achieving protective immunity against HIV.
For pDNA vaccines, immune responses in larger species have been generally lower than those in small animals, with the amount of pDNA required for effective immunization of larger animals being 1000-fold higher than for small species (milligrams vs micrograms) [9]. The relatively low HIV SAM vaccine dose of 50 µg required for potency in this report is therefore significant, being 1–2 orders of magnitude lower in dose than that needed for plasmid DNA vaccines [9], and indicates that large-scale production is feasible. The current costs of clinical manufacturing of pDNA are on the order of $50–100/mg (1–10 g scale) [49]. The current projection for the cost of mRNA manufacture is comparable [50], which makes a very compelling commercial case for evaluation of nonviral delivery of self-amplifying RNA in human clinical trials.
These data, taken together, indicate that the prospects for a SAM-based HIV vaccine are promising, particularly when combined in a prime-boost regimen with recombinant protein antigens. With continuous improvements of the SAM technology that potentially can be achieved through both antigen engineering and more-efficient RNA delivery systems, a single-modality HIV vaccine may be possible.
Notes
Acknowledgments. We thank I. Nieuwenhuis, M. Hemelop, N. Beenhakker, W. Koornstra, and D. Mortier for their technical support.
Financial support. This work was supported by the National Institutes of Health (grant 5 PO1 AI066287-02).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Rice J, Ottensmeier CH, Stevenson FK. DNA vaccines: precision tools for activating effective immunity against cancer. Nat Rev Cancer. 2008;8:108–20. doi: 10.1038/nrc2326. [DOI] [PubMed] [Google Scholar]
- 2.Wang R, Doolan DL, Le TP, et al. Induction of antigen-specific cytotoxic T lymphocytes in humans by a malaria DNA vaccine. Science. 1998;282:476–80. doi: 10.1126/science.282.5388.476. [DOI] [PubMed] [Google Scholar]
- 3.Rottinghaus ST, Poland GA, Jacobson RM, Barr LJ, Roy MJ. Hepatitis B DNA vaccine induces protective antibody responses in human non-responders to conventional vaccination. Vaccine. 2003;21:4604–8. doi: 10.1016/s0264-410x(03)00447-x. [DOI] [PubMed] [Google Scholar]
- 4.Pascolo S. Vaccination with messenger RNA (mRNA) Handb Exp Pharmacol. 2008;183:221–35. doi: 10.1007/978-3-540-72167-3_11. [DOI] [PubMed] [Google Scholar]
- 5.Weiss R, Scheiblhofer S, Roesler E, Weinberger E, Thalhamer J. mRNA vaccination as a safe approach for specific protection from type I allergy. Expert Rev Vaccines. 2012;11:55–67. doi: 10.1586/erv.11.168. [DOI] [PubMed] [Google Scholar]
- 6.Kallen KJ, Heidenreich R, Schnee M, et al. A novel, disruptive vaccination technology: Self-adjuvanted RNActive vaccines. Hum Vaccin Immunother. 2013;9:2263–76. doi: 10.4161/hv.25181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tavernier G, Andries O, Demeester J, et al. mRNA as gene therapeutic: how to control protein expression. J Control Release. 2011;150:238–47. doi: 10.1016/j.jconrel.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 8.Geall AJ, Mandl CW, Ulmer JB. RNA: the new revolution in nucleic acid vaccines. Semin Immunol. 2013;25:152–9. doi: 10.1016/j.smim.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet. 2008;9:776–88. doi: 10.1038/nrg2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McConkey SJ, Reece WH, Moorthy VS, et al. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat Med. 2003;9:729–35. doi: 10.1038/nm881. [DOI] [PubMed] [Google Scholar]
- 11.Harari A, Bart PA, Stohr W, et al. An HIV-1 clade C DNA prime, NYVAC boost vaccine regimen induces reliable, polyfunctional, and long-lasting T cell responses. J Exp Med. 2008;205:63–77. doi: 10.1084/jem.20071331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goonetilleke N, Moore S, Dally L, et al. Induction of multifunctional human immunodeficiency virus type 1 (HIV-1)-specific T cells capable of proliferation in healthy subjects by using a prime-boost regimen of DNA- and modified vaccinia virus Ankara-vectored vaccines expressing HIV-1 Gag coupled to CD8+ T-cell epitopes. J Virol. 2006;80:4717–28. doi: 10.1128/JVI.80.10.4717-4728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S, Kennedy JS, West K, et al. Cross-subtype antibody and cellular immune responses induced by a polyvalent DNA prime-protein boost HIV-1 vaccine in healthy human volunteers. Vaccine. 2008;26:3947–57. doi: 10.1016/j.vaccine.2007.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nwanegbo E, Vardas E, Gao W, et al. Prevalence of neutralizing antibodies to adenoviral serotypes 5 and 35 in the adult populations of The Gambia, South Africa, and the United States. Clin Diagn Lab Immunol. 2004;11:351–7. doi: 10.1128/CDLI.11.2.351-357.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mast TC, Kierstead L, Gupta SB, et al. International epidemiology of human pre-existing adenovirus (Ad) type-5, type-6, type-26 and type-36 neutralizing antibodies: correlates of high Ad5 titers and implications for potential HIV vaccine trials. Vaccine. 2010;28:950–7. doi: 10.1016/j.vaccine.2009.10.145. [DOI] [PubMed] [Google Scholar]
- 16.Santra S, Seaman MS, Xu L, et al. Replication-defective adenovirus serotype 5 vectors elicit durable cellular and humoral immune responses in nonhuman primates. J Virol. 2005;79:6516–22. doi: 10.1128/JVI.79.10.6516-6522.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolff JA, Malone RW, Williams P, et al. Direct gene transfer into mouse muscle in vivo. Science. 1990;247:1465–8. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- 18.Schalk JA, Mooi FR, Berbers GA, et al. Preclinical and clinical safety studies on DNA vaccines. Hum Vaccin. 2006;2:45–53. doi: 10.4161/hv.2.2.2620. [DOI] [PubMed] [Google Scholar]
- 19.Geall AJ, Verma A, Otten GR, et al. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci U S A. 2012;109:14604–9. doi: 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulmer JB, Mason PW, Geall A, Mandl CW. RNA-based vaccines. Vaccine. 2012;30:4414–8. doi: 10.1016/j.vaccine.2012.04.060. [DOI] [PubMed] [Google Scholar]
- 21.Hekele A, Bertholet S, Archer J, et al. Rapidly produced SAM vaccine against H7N9 influenza is immunogenic in mice. Emerging Microbes Infections. 2013;2:1–7. doi: 10.1038/emi.2013.54. e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perri S, Greer CE, Thudium K, et al. An alphavirus replicon particle chimera derived from Venezuelan equine encephalitis and sindbis viruses is a potent gene-based vaccine delivery vector. J Virol. 2003;77:10394–403. doi: 10.1128/JVI.77.19.10394-10403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brito L, Chan M, Shaw C, et al. A cationic nanoemulsion for the delivery of next generation RNA vaccines. Mol Ther. 2014 doi: 10.1038/mt.2014.133. doi:10.1038/mt.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta RK, Siber GR. Adjuvants for human vaccines--current status, problems and future prospects. Vaccine. 1995;13:1263–76. doi: 10.1016/0264-410x(95)00011-o. [DOI] [PubMed] [Google Scholar]
- 25.Ansaldi F, Canepa P, Parodi V, et al. Adjuvanted seasonal influenza vaccines and perpetual viral metamorphosis: the importance of cross-protection. Vaccine. 2009;27:3345–8. doi: 10.1016/j.vaccine.2009.01.081. [DOI] [PubMed] [Google Scholar]
- 26.Smerdou C, Liljestrom P. Non-viral amplification systems for gene transfer: vectors based on alphaviruses. Curr Opin Mol Ther. 1999;1:244–51. [PubMed] [Google Scholar]
- 27.Spearman P, Lally MA, Elizaga M, et al. A trimeric, V2-deleted HIV-1 envelope glycoprotein vaccine elicits potent neutralizing antibodies but limited breadth of neutralization in human volunteers. J Infect Dis. 2011;203:1165–73. doi: 10.1093/infdis/jiq175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ott G, Singh M, Kazzaz J, et al. A cationic sub-micron emulsion (MF59/DOTAP) is an effective delivery system for DNA vaccines. J Control Release. 2002;79:1–5. doi: 10.1016/s0168-3659(01)00545-4. [DOI] [PubMed] [Google Scholar]
- 29.Srivastava IK, Stamatatos L, Legg H, et al. Purification and characterization of oligomeric envelope glycoprotein from a primary R5 subtype B human immunodeficiency virus. J Virol. 2002;76:2835–47. doi: 10.1128/JVI.76.6.2835-2847.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srivastava IK, Kan E, Sun Y, et al. Comparative evaluation of trimeric envelope glycoproteins derived from subtype C and B HIV-1 R5 isolates. Virology. 2008;372:273–90. doi: 10.1016/j.virol.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 31.Calabro S, Tortoli M, Baudner BC, et al. Vaccine adjuvants alum and MF59 induce rapid recruitment of neutrophils and monocytes that participate in antigen transport to draining lymph nodes. Vaccine. 2011;29:1812–23. doi: 10.1016/j.vaccine.2010.12.090. [DOI] [PubMed] [Google Scholar]
- 32.Mooij P, Bogers WM, Oostermeijer H, et al. Evidence for viral virulence as a predominant factor limiting human immunodeficiency virus vaccine efficacy. J Virol. 2000;74:4017–27. doi: 10.1128/jvi.74.9.4017-4027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomaras GD, Yates NL, Liu P, et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J Virol. 2008;82:12449–63. doi: 10.1128/JVI.01708-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hammer SM, Sobieszczyk ME, Janes H, et al. Efficacy trial of a DNA/rAd5 HIV-1 preventive vaccine. N Engl J Med. 2013;369:2083–92. doi: 10.1056/NEJMoa1310566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haynes BF, Gilbert PB, McElrath MJ, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med. 2012;366:1275–86. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zolla-Pazner S, deCamp A, Gilbert PB, et al. Vaccine-induced IgG antibodies to V1V2 regions of multiple HIV-1 subtypes correlate with decreased risk of HIV-1 infection. PLoS One. 2014;9:e87572. doi: 10.1371/journal.pone.0087572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao HX, Bonsignori M, Alam SM, et al. Vaccine induction of antibodies against a structurally heterogeneous site of immune pressure within HIV-1 envelope protein variable regions 1 and 2. Immunity. 2013;38:176–86. doi: 10.1016/j.immuni.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crotty S, Aubert RD, Glidewell J, Ahmed R. Tracking human antigen-specific memory B cells: a sensitive and generalized ELISPOT system. J Immunol Methods. 2004;286:111–22. doi: 10.1016/j.jim.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 39.Derdeyn CA, Decker JM, Sfakianos JN, et al. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol. 2000;74:8358–67. doi: 10.1128/jvi.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei X, Decker JM, Liu H, et al. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002;46:1896–905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis D, Koornstra W, Mortier D, et al. Protection in macaques immunized with HIV-1 candidate vaccines can be predicted using the kinetics of their neutralizing antibodies. PLoS One. 2011;6:e28974. doi: 10.1371/journal.pone.0028974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li M, Gao F, Mascola JR, et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79:10108–25. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Montefiori DC. Measuring HIV neutralization in a luciferase reporter gene assay. Methods Mol Biol. 2009;485:395–405. doi: 10.1007/978-1-59745-170-3_26. [DOI] [PubMed] [Google Scholar]
- 44.Seaman MS, Janes H, Hawkins N, et al. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol. 2010;84:1439–52. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koopman G, Mortier D, Hofman S, et al. Acute-phase CD4+ T-cell proliferation and CD152 upregulation predict set-point virus replication in vaccinated simian-human immunodeficiency virus strain 89.6p-infected macaques. J Gen Virol. 2009;90:915–26. doi: 10.1099/vir.2008.006148-0. [DOI] [PubMed] [Google Scholar]
- 46.Ulmer JB, Sztein MB. Promising cutting-edge technologies and tools to accelerate the discovery and development of new vaccines. Curr Opin Immunol. 2011;23:374–6. doi: 10.1016/j.coi.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deering RP, Kommareddy S, Ulmer JB, Brito LA, Geall AJ. Nucleic acid vaccines: prospects for non-viral delivery of mRNA vaccines. Expert Opin Drug Deliv. 2014;11:885–99. doi: 10.1517/17425247.2014.901308. [DOI] [PubMed] [Google Scholar]
- 48.Bernstein DI, Reap EA, Katen K, et al. Randomized, double-blind, Phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine. 2009;28:484–93. doi: 10.1016/j.vaccine.2009.09.135. [DOI] [PubMed] [Google Scholar]
- 49.Sardesai NY, Weiner DB. Electroporation delivery of DNA vaccines: prospects for success. Curr Opin Immunol. 2011;23:421–9. doi: 10.1016/j.coi.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pascolo S. Messenger RNA-based vaccines. Expert Opin Biol Ther. 2004;4:1285–94. doi: 10.1517/14712598.4.8.1285. [DOI] [PubMed] [Google Scholar]