

Our study underscores the importance of elastin fragmentation in the vessel wall as an accelerator of atherosclerosis with enhanced inflammation and increased neovascularization, thereby promoting the development of unstable plaques that eventually may rupture. The present mouse model offers the opportunity to further investigate the role of key factors involved in plaque destabilization and potential targets for therapeutic interventions.
Keywords: Plaque rupture, Angiogenesis, Myocardial infarction, Stroke, Animal model
Abstract
Aims
There is a need for animal models of plaque rupture. We previously reported that elastin fragmentation, due to a mutation (C1039G+/−) in the fibrillin-1 (Fbn1) gene, promotes atherogenesis and a highly unstable plaque phenotype in apolipoprotein E deficient (ApoE−/−) mice on a Western-type diet (WD). Here, we investigated whether plaque rupture occurred in ApoE−/−Fbn1C1039G+/− mice and was associated with myocardial infarction, stroke, and sudden death.
Methods and results
Female ApoE−/−Fbn1C1039G+/− and ApoE−/− mice were fed a WD for up to 35 weeks. Compared to ApoE−/− mice, plaques of ApoE−/−Fbn1C1039G+/− mice showed a threefold increase in necrotic core size, augmented T-cell infiltration, a decreased collagen I content (70 ± 10%), extensive neovascularization, intraplaque haemorrhage, and a significant increase in matrix metalloproteinase-2, -9, -12, and -13 expression or activity. Plaque rupture was observed in 70% of ascending aortas and in 50% of brachiocephalic arteries of ApoE−/−Fbn1C1039G+/− mice. In ApoE−/− mice, plaque rupture was not seen in ascending aortas and only in 10% of brachiocephalic arteries. Seventy percent of ApoE−/−Fbn1C1039G+/− mice died suddenly, whereas all ApoE−/− mice survived. ApoE−/−Fbn1C1039G+/− mice showed coronary plaques and myocardial infarction (75% of mice). Furthermore, they displayed head tilt, disorientation, and motor disturbances (66% of cases), disturbed cerebral blood flow (73% of cases; MR angiograms) and brain hypoxia (64% of cases), indicative of stroke.
Conclusions
Elastin fragmentation plays a key role in plaque destabilization and rupture. ApoE−/−Fbn1C1039G+/− mice represent a unique model of acute plaque rupture with human-like complications.
Translational Perspective.
Our study underscores the importance of elastin fragmentation in the vessel wall as an accelerator of atherosclerosis with enhanced inflammation and increased neovascularization, thereby promoting the development of unstable plaques that eventually may rupture. The present mouse model offers the opportunity to further investigate the role of key factors involved in plaque destabilization and potential targets for therapeutic interventions.
Introduction
Atherosclerosis is a chronic inflammatory disease of large- and medium-sized arteries and leads to development of plaques. It is generally accepted that stability rather than size of a plaque is determinant for acute clinical complications. Plaques with an unstable phenotype are prone to rupture, which can lead to myocardial infarction, stroke, and sudden death.1 Unstable plaques are characterized by a large lipid core (usually taking >30–40% of total lesion area), a high number of inflammatory cells (mostly macrophages) and a thin fibrous cap, with few smooth muscle cells and collagen fibres.2–5 Moreover, vulnerable human plaques frequently show intraplaque neovascularization and haemorrhage6 and expansive enlargement of the vessel wall, known as outward remodelling.2,7 Despite major advances in cardio- and cerebrovascular researches, plaque rupture remains the leading cause of acute events.3 Therefore, the need for the development of plaque stabilizing therapies is high. Several research groups have tried to develop suitable models of plaque rupture for the last 15 years, but in these models, rupture occurs only sporadically, after a long period of time, or depends on mechanical injury.8–10 Moreover, reproducibility is low and events as seen in humans are rarely observed.
Recently, we reported the effect of an impaired elastin structure of the vessel wall on the progression of atherosclerosis by cross-breeding apolipoprotein E knock-out (ApoE−/−) mice with mice containing a heterozygous mutation (C1039G+/−) in the fibrillin-1 (Fbn1) gene.11 This mutation led to fragmentation of the elastic fibres in the vessel wall, resulting in increased arterial stiffness and the development of highly unstable plaques with sporadic ruptures.11 Here, we investigated whether plaque ruptures were reproducible and accompanied by ‘human-like’ events, such as myocardial infarction, stroke and/or sudden death. Moreover, we aimed to get better insight into mechanisms involved in plaque rupture in this model by focusing on intraplaque neovascularization, inflammation, and matrix degradation.
Methods
Results
Vulnerable plaque phenotype in ApoE−/−Fbn1C1039G+/− mice on Western-type diet
Compared to ApoE−/− mice on Western-type diet (WD), ApoE−/−Fbn1C1039G+/− mice on WD or normal diet (ND) showed fragmentation of the elastic fibres, thickening of the vessel wall, and vessel dilatation (Supplementary material online, Figure S1). Total plasma cholesterol levels were similar in ApoE−/− and ApoE−/−Fbn1C1039G+/− mice after 20 (609 ± 40 and 645 ± 38 mg/dL) and 35 weeks on WD (664 ± 32 and 675 ± 26 mg/dL, respectively). ApoE−/−Fbn1C1039G+/− mice on ND for 35 weeks showed plasma levels of 153 ± 43 mg/dL. ApoE−/−Fbn1C1039G+/− mice on WD or ND showed larger plaques (twofold) in the proximal ascending aorta compared with plaques in ApoE−/− mice on WD (Supplementary material online, Table S1), in spite of respectively similar or lower plasma cholesterol levels. Because the proximal ascending aorta was significantly dilated in ApoE−/−Fbn1C1039G+/− mice when compared with ApoE−/− mice (2.4-fold increase in cross-sectional area), the increased plaque size did not result in enhanced stenosis. In contrast, in the brachiocephalic artery of ApoE−/−Fbn1C1039G+/− mice on WD (but not on ND), plaque size increased more than arterial calibre, resulting in enhanced stenosis (Supplementary material online, Table S1). Atherosclerotic lesions of ApoE−/−Fbn1C1039G+/− mice on WD showed several features of unstable plaques, such as a threefold increased necrotic core (P < 0.001), 1.5-fold augmented T-cell infiltration (P < 0.05), 40% decrease in total collagen (P < 0.001), 70% decrease in collagen type I (P < 0.01), and 60% loss in elastin content (P < 0.05), resulting in significantly thinner fibrous caps (40–60%), when compared with ApoE−/− mice on WD and ApoE−/−Fbn1C1039G+/− mice on ND (P < 0.001, one-way ANOVA or Kruskal–Wallis test, Figure 1A; Supplementary material online, Table S1). Though the Mac-3-positive area (macrophages) was increased in plaques of ApoE−/−Fbn1C1039G+/− mice, the relative area remained unchanged due to the increased lesion size (Supplementary material online, Table S1).
Figure 1.

Atherosclerotic plaques in brachiocephalic arteries of ApoE−/−Fbn1C1039G+/− mice on Western-type diet show features of a highly unstable phenotype when compared with age-matched ApoE−/− mice on Western-type diet and ApoE−/−Fbn1C1039G+/− mice on normal diet (20 weeks of diet on average). (A) type I collagen (red/arrowheads, sirius red stain under polarized light) was decreased fivefold (for quantification, see Supplementary material online, Table S1). Scale bar = 100 µm. L, lumen. (C) Inducible nitric oxide synthase expression was augmented in plaques of ApoE−/−Fbn1C1039G+/− mice on Western-type diet and normal diet when compared with ApoE−/− mice on Western-type diet, indicating increased macrophage activation. Accordingly, inflammatory cytokines TNF-α, IL-1β, and IL-6 were highly expressed in aortas of ApoE−/−Fbn1C1039G+/− mice on Western-type diet compared to ApoE−/− mice. In addition, an increase in T-cell activation marker interferon γ, vascular endothelial growth factor, transforming growth factor-β, and elastase MMP-12 was observed. (D) Gel zymography showed increased activity of MMP-9 and MMP-2 when compared with aortas of age-matched ApoE−/− mice on Western-type diet. For quantification, see Supplementary material online, Figure S2.
Increased expression of inflammatory markers and matrix degrading enzymes in plaques of ApoE−/−Fbn1C1039G+/− mice on WD
Since the relative area of plaque macrophages was unchanged, we examined whether differences in activation status could point to increased inflammation. When compared with ApoE−/− mice on WD, aortas of ApoE−/−Fbn1C1039G+/− mice showed a twofold increase in inducible nitric oxide synthase (iNOS) and increased expression of inflammatory cytokines tumour necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6. In addition, an augmentation in interferon γ (IFNγ), vascular endothelial growth factor (VEGF), and transforming growth factor-β(TGFβ) was observed (Figure 1B; Supplementary material online, Figure S2A). Furthermore, matrix degrading enzymes MMP-12 (Figure 1B), MMP-2, and MMP-9 (Figure 1C; Supplementary material online, Figure S2B–E) were more than doubled. Immunohistochemistry showed a twofold rise in type I collagenase MMP-13 in plaques of proximal ascending aorta and brachiocephalic artery (not shown) in ApoE−/−Fbn1C1039G+/− mice on WD compared with ApoE−/− mice on WD (Supplementary material online, Figure S2F).
Abundant neovascularization and haemorrhage in plaques of ApoE−/−Fbn1C1039G+/− mice on WD
Because vulnerable plaques in humans are characterized by the presence of intraplaque neovascularization and haemorrhage, we examined whether these features were also present in ApoE−/−Fbn1C1039G+/− mice on WD. Neovascularization and intraplaque haemorrhage were frequently observed in the common carotid arteries (Figure 2A–E) and brachiocephalic artery (Figure 2F–K) of ApoE−/−Fbn1C1039G+/− mice on WD (22 of 24 mice). In contrast, neovascularization was observed in only one ApoE−/− mouse on WD and was absent in ApoE−/−Fbn1C1039G+/− mice on ND. Neovessels were present at the base (Figure 2C and E) and in the centre of plaques and sprouted out of the media (Figure 2C). They contained erythrocytes (Figure 2C, F, G) and a thin layer of endothelial cells (Figure 2D and J). Their number correlated with the degree of medial elastin fragmentation (Supplementary material online, Table S2). Neovascularization occurred in plaques with a high degree of stenosis (94 ± 2%, Figure 2A; carotid arteries) and areas of hypoxia, as shown on a pimonidazole stain (Figure 2E, Table 1; Supplementary material online, Figure S3). Moreover, the vessels were highly leaky, as shown by extravasation of dextran particles (size 70 000 MW). Dextran leakage was also observed in media and adventitia, indicating the presence of vasa vasorum (Figure 2K, Table 1; Supplementary material online, Figure S4). Finally, intraplaque haemorrhages were observed at the base and in the centre of the plaque (Figure 2B, H, I; Table 1).
Figure 2.
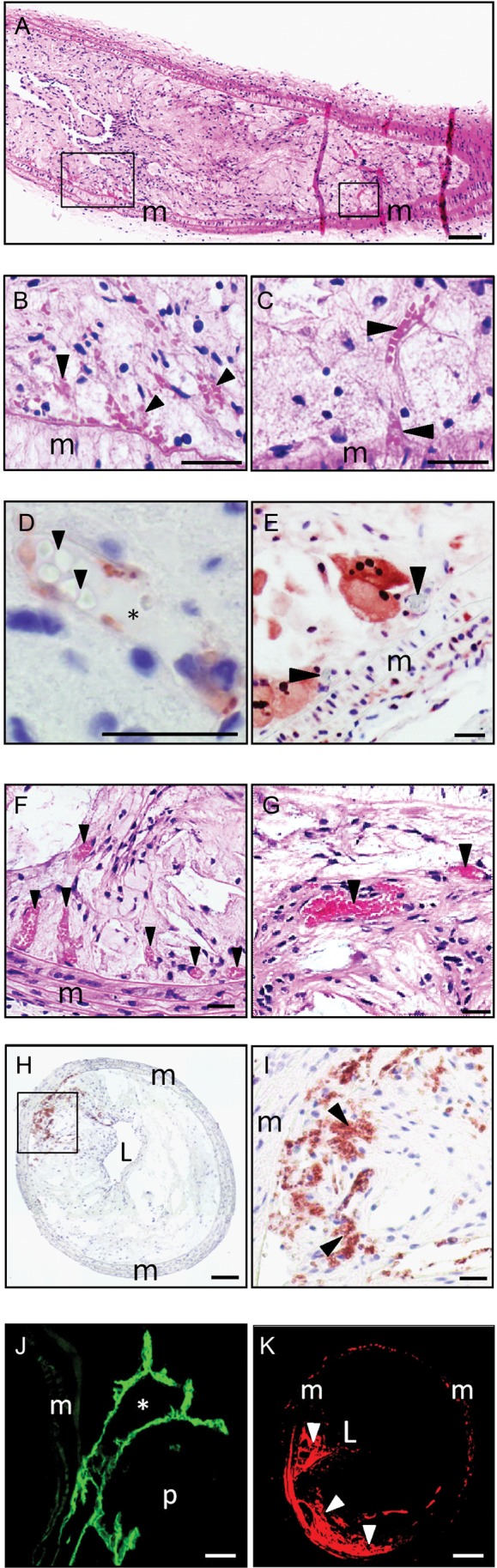
ApoE−/−Fbn1C1039G+/− mice on Western-type diet show intraplaque neovascularization and haemorrhage in common carotid arteries (A–E) and brachiocephalic arteries (F–K). (A) Left common carotid artery of an ApoE−/−Fbn1C1039G+/− mouse that died suddenly after 19 weeks on Western-type diet. Common carotid arteries show pronounced stenosis, intraplaque neovessels, and haemorrhage. (B) Detail of intraplaque haemorrhage at the plaque base (left box in (A)), erythrocytes indicated with arrowheads (H&E). (C) Neovessel (right box in (A)) sprouting out of the media, erythrocytes indicated with arrowheads; H&E. (D) Detail of a neovessel in carotid artery plaque (endothelial cells: von Willebrand stain, red; erythrocytes: arrowheads; lumen of the neovessel: asterisk). (E) Neovessels (arrowheads) are located in hypoxic plaque regions (pimonidazole stain, red). (F) Multiple microvessels in brachiocephalic artery plaques (arrowheads, H&E). (G) Detail of microvessels (arrowheads). (H and I) Intraplaque haemorrhage in the brachiocephalic artery (erythrocytes: TER-119 stain, red, arrowheads). (J) In vivo CD31 stain (endothelial cells, green; lumen of neovessel, asterisk) showing microvessels sprouting out of the media. (K) TMR-dextran leakage in brachiocephalic artery plaques (red, arrowheads). L, lumen; m, media; p, plaque. Scale bar = 100 µm (A, H, K); 50 µm (F, I) and 25 µm (B–E and G, J).
Table 1.
Characteristics of neovessels and intraplaque haemorrhages in ApoE−/− and ApoE−/−Fbn1C1039G+/− mice after 20 weeks (on average) on Western-type diet or normal diet
| ApoE−/− WD | ApoE−/−Fbn1C1039G+/− |
P-value | ||
|---|---|---|---|---|
| WD | ND | |||
| Number of mice with microvessels | ||||
| Left common carotid artery | 1/24 | 22/24 | 0 | P < 0.001 |
| Brachiocephalic artery | 1/24 | 22/24 | 0 | P < 0.001 |
| Number of microvessels/107 µm2a | ||||
| Left common carotid artery | 0 [0–0.2] | 6 [3–12] | n.a. | P < 0.001 |
| TMR-dextran positivity (%)b | ||||
| Left common carotid artery | 1.5 ± 0.8 | 15.0 ± 1.9 | n.d. | P = 0.003 |
| Brachiocephalic artery | 0.8 ± 0.4 | 3.7 ± 0.7 | n.d. | P = 0.026 |
| Pimonidazole positivity (%)c | ||||
| Left common carotid artery | 4 ± 1 | 24 ± 1 | n.d. | P < 0.001 |
| Brachiocephalic artery | 5 ± 1 | 19 ± 1 | n.d. | P < 0.001 |
| Ascending aorta | 5 ± 1 | 9 ± 1c | n.d | P = 0.013 |
| Number of mice with intraplaque haemorrhages | ||||
| Left common carotid artery | 0/24 | 22/24 | 0 | P < 0.001 |
| Brachiocephalic artery | 2/24 | 21/24 | 0 | P < 0.001 |
| Number of intraplaque haemorrhages/107 µm2a | ||||
| Left common carotid artery | 0 [0–0] | 1 [0.4–3] | n.a. | P < 0.001 |
n.d., not determined; n.a., not applicable. P-values vs. ApoE−/−; Fisher's exact test (number of mice with microvessels and intraplaque haemorrhage) or unpaired t-test (TMR-dextran and pimonidazole positivity).
aMedian, minimum–maximum value, n = 15 animals per group, 15 slices per animal.
bMean ± SEM, n = 3 animals per group, 10 slices per artery.
cMean ± SEM, n = 3 animals per group, 5–10 slices per animal.
dP < 0.001 vs. left common carotid artery and brachiocephalic artery (Kruskal–Wallis test followed by Dunn's multiple comparison test).
Plaque rupture in the proximal ascending aorta and brachiocephalic artery of ApoE−/−Fbn1C1039G+/− mice on WD
Plaque rupture was observed in 50% of brachiocephalic arteries (Figure 3Aa and b, 10 of 20 animals) and 70% of proximal ascending aortas of ApoE−/−Fbn1C1039G+/− mice on WD (14 of 20 animals). In contrast, only 1 of 10 ApoE−/− mice on WD (10%) showed plaque rupture in the brachiocephalic artery but none in the ascending aorta (P < 0.001, Fisher's exact test). In ApoE−/−Fbn1C1039G+/− mice on ND, no plaque rupture was observed. Moreover, coronary and carotid artery plaques of ApoE−/−Fbn1C1039G+/− mice on WD showed the presence of fibrin deposits and thrombi, suggestive of plaque rupture (Figure 3Ac–e).
Figure 3.

ApoE−/−Fbn1C1039G+/− mice on Western-type diet show acute plaque rupture and die suddenly. (Aa and b) Plaque rupture with thrombus in the brachiocephalic artery of an ApoE−/−Fbn1C1039G+/− mouse on Western-type diet that died suddenly after 19 weeks. Plaque ruptures were characterized by disruption of the fibrous cap (arrow, dotted line) with intrusion of erythrocytes (arrowheads) into the necrotic core. (a) Low-power micrograph and (b) high-power micrograph of box in (a), showing a fibrin-rich thrombus (purple, T). (c) Coronary artery plaque showing presence of fibrin deposits and thrombus (asterisk). (d) Magnification of boxes in (c). Fibrin deposits indicated with arrowheads. (e) Carotid artery with large thrombus at bifurcation (purple, T). Scale bar = 100 µm (a, e) and 50 µm (b–d). L, lumen; RBC, red blood cells; aBr, brachiocephalic artery; BFC, bifurcation; T, thrombus. (B) Kaplan–Meier survival curves of ApoE−/−Fbn1C1039G+/− and ApoE−/− mice on Western-type diet or normal diet for up to 35 weeks. Seventy percent of ApoE−/−Fbn1C1039G+/− mice on Western-type diet (n = 24) died suddenly within 35 weeks, whereas all ApoE−/− mice on Western-type diet (n = 24) and ApoE−/−Fbn1C1039G+/− mice on normal diet (n = 20) survived. ***P < 0.001 (log-rank test). WD, Western-type diet; ND, normal diet.
Sudden death in ApoE−/−Fbn1C1039G+/− mice on WD
Seventeen of 24 ApoE−/−Fbn1C1039G+/− mice on WD (70%) died suddenly, whereas all ApoE−/− mice on WD (n = 24) and ApoE−/−Fbn1C1039G+/− mice on ND (n = 20) survived (Figure 3B). Sudden death of ApoE−/−Fbn1C1039G+/− mice on WD mainly occurred between 16 and 23 weeks, with 50% mortality after 20 weeks (Figure 3B).
Neurological symptoms, disturbed cerebral flow, and brain hypoxia in ApoE−/−Fbn1C1039G+/− mice on WD
Sixteen of 24 (66%) ApoE−/−Fbn1C1039G+/− mice on WD displayed neurological symptoms such as head tilt, disorientation, and motor disturbances. In most cases, those neurological symptoms preceded sudden death (13 of 24 mice (54%), average age 20 weeks on WD; Supplementary material online, Movie S1). Head tilt was observed in only one ApoE−/− mouse at 32 weeks on WD and never in ApoE−/−Fbn1C1039G+/− mice on ND. In vivo MR angiograms of brains of ApoE−/−Fbn1C1039G+/− and ApoE−/− mice on WD (n = 11 and n = 7, respectively) showed cerebral flow deficits in 73% of ApoE−/−Fbn1C1039G+/− mice (from 16 weeks on WD onward, median: 18 weeks on WD), whereas brains of ApoE−/− mice appeared normal (Figure 4A and B; P < 0.001, Fisher's exact test). Moreover, 2,3,5-triphenyltetrazolium chloride (TTC) stain of the brain showed the presence of hypoxia in 64% of ApoE−/−Fbn1C1039G+/− mice on WD (6 of 7 displaying neurological symptoms), indicative of stroke (P < 0.001, Fisher's exact test) (Figure 4C and D). 2,3,5-Triphenyltetrazolium chloride positivity in the brain was observed in only one ApoE−/− mouse on WD (of eight) that also displayed neurological symptoms.
Figure 4.

Brains of ApoE−/−Fbn1C1039G+/− mice on Western-type diet displayed disturbed cerebral flow and hypoxia, indicative of stroke. (A and B) Representative cerebral MR angiograms of ApoE−/− (A) and ApoE−/−Fbn1C1039G+/− mice (B) on Western-type diet for 25 weeks. ApoE−/−Fbn1C1039G+/− mice showed perfusion deficits in both right and left brain hemispheres (asterisks), in comparison to age-matched ApoE−/− mice (A) (n = 11 and n = 7, respectively, P < 0.001, Fisher's exact test). (C and D) Representative 2,3,5-triphenyltetrazolium chloride stain of brains of an ApoE−/− and ApoE−/−Fbn1C1039G+/− mouse on Western-type diet for 25 weeks. Vital brain tissue is stained red, while hypoxic areas are pale/white. Sixty-four percent of ApoE−/−Fbn1C1039G+/− mice showed hypoxic areas in the brain, indicative of stroke (D; white/pale regions, arrowheads, n = 11), whereas brains of most ApoE−/− mice on Western-type diet (C) were normal (87%, n = 8) (P < 0.001, Fisher's exact test). Scale bar = 200 µm.
Cardiac hypertrophy, decreased left ventricular function, coronary artery plaque, and myocardial infarction in ApoE−/−Fbn1C1039G+/− mice on WD
ApoE−/−Fbn1C1039G+/− mice on WD developed left ventricular dysfunction starting from 5 weeks of diet, as shown by significant increases in end-diastolic diameter (EDD) and end-systolic diameter (ESD), whereas left ventricular diameter of ApoE−/− mice on WD remained unchanged (Supplementary material online, Table S3). In ApoE−/−Fbn1C1039G+/− mice on ND, the EDD and ESD were increased compared to ApoE−/− mice on WD (Supplementary material online, Table S3). ApoE−/−Fbn1C1039G+/− mice on WD that died suddenly showed significantly higher EDD and ESD compared with surviving ApoE−/−Fbn1C1039G+/− mice on WD (Supplementary material online, Table S3). Furthermore, heart weight of ApoE−/−Fbn1C1039G+/− mice on WD was significantly increased when compared with ApoE−/− mice on WD, whereas lung mass remained unchanged (Supplementary material online, Table S3). Post-mortem histological analysis showed coronary artery plaque and myocardial infarction in 96% of ApoE−/−Fbn1C1039G+/− mice on WD (23 of 24), compared with 18% of ApoE−/− mice on WD (2 of 11), respectively (P < 0.001, one-way ANOVA; Figure 5A–I, Supplementary material online, Table S3). Myocardial infarctions were characterized by large areas of collagen and inflammatory cells (Figure 5B, C and H, I) and were mostly seen in the left ventricle. Moreover, a significant increase in coronary artery plaque (Figure 5A–C, D–F) and infarcted area (Figure 5B, C and H, I) was observed in ApoE−/−Fbn1C1039G+/− mice on WD that died suddenly compared to ApoE−/− mice and ApoE−/−Fbn1C1039G+/− mice on WD that survived (number of coronary arteries with >25% stenosis: 8 vs. 0 and 1, P < 0.05, one-way ANOVA and infarcted area (%): 1.0 vs. 0 and 0.3, overview in Supplementary material online, Table S3).
Figure 5.

Sections of hearts of an ApoE−/− mouse at 35 weeks on Western-type diet and two ApoE−/−Fbn1C1039G+/− mice that died suddenly (Masson's trichrome stain). Hearts of ApoE−/−Fbn1C1039G+/− mice showed large infarcted areas, mainly in the left ventricle (green-blue, B, C and H, I) and coronary artery plaque (B, C and E, F), whereas hearts of ApoE−/− mice appeared normal (A, D, G). Scale bar = 500 µm. (E, F) Detail of coronary artery plaque and infarcted area (H and I) in B and C, respectively, in the left ventricle. Infarcted areas showed infiltration of inflammatory cells (arrowheads). The corresponding areas in ApoE−/− mice on Western-type diet appeared normal and coronary arteries were plaque free (D, G). Scale bar = 50 µm.
Discussion
In the present study, we characterized a mouse model of atherosclerotic plaque rupture with endpoints including myocardial infarction, stroke, and sudden death. Importantly, these events did not – or only very occasionally – occur in ApoE−/− mice on WD or in ApoE−/−Fbn1C1039G+/− mice fed an ND. These findings underscore the importance of elastin fragmentation in combination with a WD as prerequisites for atherosclerotic plaque rupture in mice.
Mutations in the Fbn1 gene lead to Marfan syndrome, a genetic disorder characterized by progressive fragmentation of elastic fibres.12 This results in increased arterial stiffening, elevated pulse pressure, and progressive aortic dilatation (especially of the proximal ascending aorta), resembling vascular aging.11,13,14 Fbn1 is essential for proper formation of extracellular matrix (ECM), including the biogenesis and maintenance of elastic fibres. The ECM is not only critical for the structural integrity of connective tissue but also serves as a reservoir for growth factors, such as TGFβ. Deficiency in Fbn1 leads to an increase in unsequestered TGFβ, which may trigger inflammation accompanied by release of proteases that degrade elastin fibres and other ECM components.12 We confirmed that TGFβ was significantly increased in atherosclerotic plaques of ApoE−/−Fbn1C1039G+/− mice on WD.11 Similar to humans, the Fbn1 mutation frequently led to aortic dissection in male ApoE−/−Fbn1C1039G+/− mice, but not in female ApoE−/−Fbn1C1039G+/− mice, as used in the present study.
In ApoE−/− mice on WD, the Fbn1C1039G+/− mutation resulted in significantly larger plaques with a highly unstable phenotype, characterized by a large necrotic core (occupying ∼30% of total plaque area), and a strongly diminished collagen content. Accelerated atherogenesis in these mice was likely the result of enhanced vascular inflammation, leading to increased monocyte attraction, oxidation, and accumulation of lipids.15 Here, we showed that iNOS, a marker for activated macrophages and inflammation, was significantly more expressed in plaques of ApoE−/−Fbn1C1039G+/− mice on either WD or ND when compared with ApoE−/− mice on WD. Accordingly, inflammatory cytokines TNF-α, IL-1β, and IL-6 were highly increased. In addition, we observed a higher infiltration of T-cells and their activation marker IFNγ, the latter playing an important role in collagen turnover by inhibiting smooth muscle cells to synthesize collagen, required to repair and maintain fibrous cap integrity.16,17 Moreover, we observed significant increases in MMP-2, -9, -12, and -13 expression or activity in ApoE−/−Fbn1C1039G+/− mice. MMP-2 and -9 not only contribute to vascular remodelling in Marfan syndrome but are also implicated in atherosclerosis and angiogenesis.18 For example, ApoE−/− mice lacking MMP-2 develop smaller and more stable plaques, whereas macrophages overexpressing active MMP-9 promote neovascularization, intraplaque haemorrhage,19,20 and features of plaque rupture in ApoE−/− mice.20 In the latter case, those features were attributed to elastin degradation, underscoring its role in plaque destabilization and rupture, in line with our findings. MMP-12 and -13 additionally contribute to elastin and (type I) collagen degradation, respectively. Taken together, in ApoE−/−Fbn1C1039G+/− mice on WD, enhanced collagen/ECM breakdown together with decreased synthesis and repair are likely responsible for weakening of the fibrous cap and rendering it more rupture prone.18,17
Interestingly, the present data show extensive neovascularization and intraplaque haemorrhage in brachiocephalic and common carotid arteries of ApoE−/−Fbn1C1039G+/− mice on WD. These features are rarely observed in murine atherosclerosis models but are known to highly affect plaque progression and vulnerability in humans.21 In ApoE−/−Fbn1C1039G+/− mice on WD, intraplaque neovessels, likely arising from adventitial vasa vasorum, clearly sprouted out of the media.22,23 Neovessels were not only present at the base of the plaque but were also frequently observed in its centre, similar to human pathology.21,23 Angiogenesis requires ECM degradation by proteases, including MMPs, to enable endothelial cell migration into the surrounding tissue.18 In addition, degradation of the ECM induces release of sequestered angiogenic factors such as VEGF and TGFβ,18,19 also observed in ApoE−/−Fbn1C1039G+/− mice on WD. In the present study, the extent of neovascularization correlated with the degree of elastin fragmentation in the vessel wall. However, degradation of the ECM alone was not sufficient to induce neovascularization in atherosclerotic plaques, because microvessels were not present in plaques of ApoE−/−Fbn1C1039G+/− mice on ND. This observation indicates that an additional factor is needed to trigger plaque neovascularization. Pimonidazole staining showed that hypoxia, a well-known angiogenesis trigger,24 was strongly increased in plaques of brachiocephalic and carotid arteries in ApoE−/−Fbn1C1039G+/− mice on WD. By contrast, hypoxia in the ascending aorta was minor, which likely explains the absence of neovessels at that site. In conclusion, the highly permeable arterial wall, due to degradation of the ECM, combined with intraplaque hypoxia seems required for neovessel formation in atherosclerotic plaques of ApoE−/−Fbn1C1039G+/− mice on WD.
Importantly, those neovessels were highly leaky, as shown by extravasation of dextran particles. Moreover, the presence of intraplaque erythrocytes near neovessels at the base of the plaque pointed to intraplaque haemorrhages, substantiating ruptured neovessels as source of intraplaque bleeding.19,21 Erythrocytes are important sources of free cholesterol, thereby increasing necrotic core size. Hence, neovascularization, besides supplying plaques with leucocytes and lipoproteins, can promote focal plaque expansion when microvessels rupture or become thrombotic.6,21,24 Taken together, the findings in this mouse model are in line with current concepts of human vulnerable plaques.
In addition to enhanced plaque vulnerability, we showed that plaque rupture was consistently present in ApoE−/−Fbn1C1039G+/− mice on WD, but very rarely in ApoE−/− mice on WD. Moreover, we could identify fibrin-rich mural thrombi in brachiocephalic, carotid and coronary arteries, and ascending aortas. Both intrinsic (i.e. a highly unstable plaque phenotype) and extrinsic factors (i.e. forces acting on the plaque) are elementary for plaque rupture.25,26 In general, rupture occurs when the mechanical stress applied on the fibrous cap exceeds its tensile strength.25 The latter is mainly determined by the collagen content of the plaque, which was significantly decreased in plaques of ApoE−/−Fbn1C1039G+/− mice on WD.17,26 Elevated pulse pressure (as a consequence of arterial stiffening11) leads to repetitive plaque deformation, increasing the tensile stress on the cap.14,27 When applied chronically, this can lead to plaque fatigue, making it prone to rupture.25–27 Moreover, due to the progressive aortic dilatation and outward remodelling (as a result of the large plaques), the collagen and elastin fibres of the cap are stretched and become more rigid, increasing the susceptibility to mechanical stress.25 Aortic dilatation was highly pronounced in the ascending aorta, suggesting that this mechanism was responsible for rupture of unstable plaques at this site. In brachiocephalic and carotid arteries, intraplaque neovascularization and haemorraghe were frequently present, further increasing plaque size and vulnerability to rupture (as illustrated by thrombi in the carotid arteries).
ApoE−/−Fbn1C1039G+/− mice on WD developed left cardiac dysfunction and hypertrophy, as shown by significant increases in EDD, ESD, and heart weight, without affecting lung mass. However, also ApoE−/−Fbn1C1039G+/− mice on ND showed cardiac dysfunction after 25 weeks, indicating an effect of the genotype on the heart. Nevertheless, those mice did not show coronary artery plaque nor died suddenly, suggesting that the presence of coronary artery plaque plays an important role in cardiac death. This was further supported by the fact that ApoE−/−Fbn1C1039G+/− mice on WD that died suddenly showed a significantly higher frequency of coronary stenosis compared with survivors. Moreover, the majority of ApoE−/−Fbn1C1039G+/− mice on WD showed infarcted areas, which compromised cardiac function even more. Indeed, ApoE−/−Fbn1C1039G+/− mice on WD that died suddenly showed larger infarcted areas and a significant higher EDD and ESD when compared with ApoE−/−Fbn1C1039G+/− mice on WD that survived for 25 weeks. Although we do not know whether the increased infarcted area was the result of plaque rupture or due to pronounced plaque formation and coronary artery stenosis, our findings are remarkable, because coronary artery plaque and spontaneous myocardial infarctions almost never develop in ApoE−/− mice on WD. Also in humans, differences in Fbn1 genotype have shown to greatly influence plaque progression and severity of coronary artery disease, underscoring the pathophysiological relevance of Fbn1 mutations in cardiovascular disease.14
Another important feature of ApoE−/−Fbn1C1039G+/− mice on WD is the occurrence of cerebral complications. We confirmed the development of neurological symptoms as shown previously,11 but here we show that this was actually linked with cerebral hypoperfusion and local brain hypoxia (stroke).
Next to emboli, other mechanisms could invoke sudden death in the current model as shown in Figure 6. The severe stenosis of carotid (Figure 2A) and some coronary arteries (Figure 5E) may compromise perfusion to such an extent that it results in local hypoxia, indicated by brain angiograms. Chronically reduced perfusion of heart and brain could lead to permanent damage to these vital organs, and therefore could also cause or contribute to sudden death (Figure 6).
Figure 6.

Schematic overview of mechanisms leading to the formation of vulnerable plaques, plaque rupture, myocardial infarction, stroke, and sudden death in ApoE−/−Fbn1C1039G+/− mice (white boxes indicate the effects of each genotype separately, blue boxes the combination of both). The C1039G+/− mutation in the fibrillin-1 gene causes elastic fibre fragmentation (1), resulting in both increased arterial stiffness (and consequently elevated pulse pressure and progressive aortic dilatation) and cardiac dysfunction (due to the decreased contractility of the cardiomyocytes). In hypercholesterolemical ApoE−/−Fbn1C1039G+/− mice, this elastin fragmentation and arterial stiffness lead to the development of large plaques with a highly unstable phenotype, characterized by enhanced inflammation (2) large necrotic cores (NC) (3) and a thin fibrous cap (FC) (4). Additionally, in the brachiocephalic and carotid arteries intraplaque neovascularisation and haemorrhage (IPH) are abundantly present (5). Due to the elevated pulse pressure and extensive aortic dilatation (especially in the ascending aorta), the mechanical stress on plaques is increased, leading to rupture and subsequent thrombus formation (6). The pronounced atherosclerosis also leads to increased carotid and coronary artery stenosis, resulting in cerebral and/or cardiac hypoperfusion. Since ApoE−/−Fbn1C1039G+/− mice are already sensible to develop cardiac dysfunction as a consequence of the genotype, decreased cardiac perfusion may further impair cardiac function. Plaque rupture with (occlusive) thrombosis as well as hypoperfusion of the heart and brain most likely result in myocardial infarction, stroke, and eventually sudden death.
Conclusion and clinical perspectives
The present study shows that elastin fragmentation in combination with a WD leads to plaque destabilization and rupture in ApoE−/− mice. ApoE−/−Fbn1C1039G+/− mice show many features of human end-stage atherosclerosis, such as an enlarged necrotic core, a thin fibrous cap with an important loss of collagen fibres, outward remodelling, and the presence of intraplaque microvessels and haemorrhage, resulting in plaque rupture, myocardial infarction, stroke, and sudden death. ApoE−/−Fbn1C1039G+/− mice on WD offer the opportunity to further investigate the role of key factors involved in plaque destabilization, including intraplaque neovascularization, which will provide more insight into the mechanisms of plaque disruption and potential targets for therapeutic interventions.
Supplementary Material
Supplementary material is available at European Heart Journal online.
Funding
This work was supported by the Agency for Innovation by Science and Technology (IWT), the Fund for Scientific Research (FWO)—Flanders (G.0126.11), and the University of Antwerp (BOF). C.V.d.D. is a fellow of the IWT. The funding for Open Access was provided by the FWO Flanders and the University of Antwerp.
Conflict of interest: none declared.
Acknowledgements
The authors thank Rita Van den Bossche, Hermine Fret, Anne-Elise Van Hoydonck, Joeri Veiro, Wendy Van Leuven, David De Wilde, Inge Bats, Mark Kockx, Patrick Cras, Katrien Lemmens, and Benedicte De Winter for technical assistance and fruitful discussions.
References
- 1.Davies MJ, Thomas AC. Plaque fissuring – the cause of acute myocardial infarction, sudden ischaemic death, and crescendo angina. Br Heart J 1985;53:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moreno PR. Vulnerable plaque: definition, diagnosis, and treatment. Cardiol Clin 2010;28:1–30. [DOI] [PubMed] [Google Scholar]
- 3.Yla-Herttuala S, Bentzon JF, Daemen M, Falk E, Garcia-Garcia HM, Herrmann J, Hoefer I, Jukema JW, Krams R, Kwak BR, Marx N, Naruszewicz M, Newby A, Pasterkamp G, Serruys PW, Waltenberger J, Weber C, Tokgozoglu L. Stabilisation of atherosclerotic plaques. Position paper of the European Society of Cardiology (ESC) Working Group on atherosclerosis and vascular biology. Thromb Haemost 2011;106:1–19. [DOI] [PubMed] [Google Scholar]
- 4.Alsheikh-Ali AA, Kitsios GD, Balk EM, Lau J, Ip S. The vulnerable atherosclerotic plaque: scope of the literature. Ann Intern Med 2010;153:387–395. [DOI] [PubMed] [Google Scholar]
- 5.Bond AR, Jackson CL. The fat-fed apolipoprotein E knockout mouse brachiocephalic artery in the study of atherosclerotic plaque rupture. J Biomed Biotechnol 2011;2011:379069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kockx MM, Cromheeke KM, Knaapen MW, Bosmans JM, De Meyer GRY, Herman AG, Bult H. Phagocytosis and macrophage activation associated with hemorrhagic microvessels in human atherosclerosis. Arterioscler Thromb Vasc Biol 2003;23:440–446. [DOI] [PubMed] [Google Scholar]
- 7.Varnava AM, Mills PG, Davies MJ. Relationship between coronary artery remodeling and plaque vulnerability. Circulation 2002;105:939–943. [DOI] [PubMed] [Google Scholar]
- 8.Ni M, Chen WQ, Zhang Y. Animal models and potential mechanisms of plaque destabilisation and disruption. Heart 2009;95:1393–1398. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz SM, Galis ZS, Rosenfeld ME, Falk E. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol 2007;27:705–713. [DOI] [PubMed] [Google Scholar]
- 10.Chen YC, Bui AV, Diesch J, Manasseh R, Hausding C, Rivera J, Haviv I, Agrotis A, Htun NM, Jowett J, Hagemeyer CE, Hannan RD, Bobik A, Peter K. A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microRNA expression profiling. Circ Res 2013;113:252–265. [DOI] [PubMed] [Google Scholar]
- 11.Van Herck JL, De Meyer GRY, Martinet W, Van Hove CE, Foubert K, Theunis MH, Apers S, Bult H, Vrints CJ, Herman AG. Impaired fibrillin-1 function promotes features of plaque instability in apolipoprotein E-deficient mice. Circulation 2009;120:2478–2487. [DOI] [PubMed] [Google Scholar]
- 12.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest 2004;114:172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mariko B, Pezet M, Escoubet B, Bouillot S, Andrieu JP, Starcher B, Quaglino D, Jacob MP, Huber P, Ramirez F, Faury G. Fibrillin-1 genetic deficiency leads to pathological ageing of arteries in mice. J Pathol 2011;224:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medley TL, Cole TJ, Gatzka CD, Wang WY, Dart AM, Kingwell BA. Fibrillin-1 genotype is associated with aortic stiffness and disease severity in patients with coronary artery disease. Circulation 2002;105:810–815. [DOI] [PubMed] [Google Scholar]
- 15.Fulop T, Jr., Larbi A, Fortun A, Robert L, Khalil A. Elastin peptides induced oxidation of LDL by phagocytic cells. Pathol Biol (Paris) 2005;53:416–423. [DOI] [PubMed] [Google Scholar]
- 16.Koenig W, Khuseyinova N. Biomarkers of atherosclerotic plaque instability and rupture. Arterioscler Thromb Vasc Biol 2007;27:15–26. [DOI] [PubMed] [Google Scholar]
- 17.Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med 2013;368:2004–2013. [DOI] [PubMed] [Google Scholar]
- 18.Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol 2008;75:346–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Nooijer R, Verkleij CJ, von der Thusen JH, Jukema JW, van der Wall EE, van Berkel TJ, Baker AH, Biessen EA. Lesional overexpression of matrix metalloproteinase-9 promotes intraplaque hemorrhage in advanced lesions but not at earlier stages of atherogenesis. Arterioscler Thromb Vasc Biol 2006;26:340–346. [DOI] [PubMed] [Google Scholar]
- 20.Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest 2006;116:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, Wrenn SP, Narula J. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol 2005;25:2054–2061. [DOI] [PubMed] [Google Scholar]
- 22.Moulton KS, Olsen BR, Sonn S, Fukai N, Zurakowski D, Zeng X. Loss of collagen XVIII enhances neovascularisation and vascular permeability in atherosclerosis. Circulation 2004;110:1330–1336. [DOI] [PubMed] [Google Scholar]
- 23.Rademakers T, Douma K, Hackeng TM, Post MJ, Sluimer JC, Daemen MJ, Biessen EA, Heeneman S, van Zandvoort MA. Plaque-associated vasa vasorum in aged apolipoprotein E-deficient mice exhibit proatherogenic functional features in vivo. Arterioscler Thromb Vasc Biol 2013;33:249–256. [DOI] [PubMed] [Google Scholar]
- 24.Sluimer JC, Kolodgie FD, Bijnens AP, Maxfield K, Pacheco E, Kutys B, Duimel H, Frederik PM, van Hinsbergh VW, Virmani R, Daemen MJ. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J Am Coll Cardiol 2009;53:1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pasterkamp G, Falk E. Atherosclerotic plaque rupture: an overview. J Clin Basic Cardiol 2000;3:81–86. [Google Scholar]
- 26.Slager CJ, Wentzel JJ, Gijsen FJ, Thury A, van der Wal AC, Schaar JA, Serruys PW. The role of shear stress in the destabilization of vulnerable plaques and related therapeutic implications. Nat Clin Pract Cardiovasc Med 2005;2:456–464. [DOI] [PubMed] [Google Scholar]
- 27.Huang Y, Teng Z, Sadat U, He J, Graves MJ, Gillard JH. In vivo MRI-based simulation of fatigue process: a possible trigger for human carotid atherosclerotic plaque rupture. Biomed Eng Online 2013;12:36. [DOI] [PMC free article] [PubMed] [Google Scholar]