


Abstract
The program RECON has been designed for constructing profiles of nucleosome potential, characterizing the probability of nucleosome formation along DNA sequences. The program used for recognition of nucleosome formation sites in genomic DNA sequences. It was developed using discriminant analysis based on a genetic algorithm method utilizing statistics for dinucleotide location within local regions of nucleosome formation sites. The program RECON is available at http://wwwmgs.bionet.nsc.ru/mgs/programs/recon/.
INTRODUCTION
Nucleosomes are the major structural element of chromatin. Each nucleosome is formed by a fragment with a length of 147 bp wrapped around an 8mer comprising pairs of four types of histones. Neighboring nucleosomes are connected with linker DNA ranging from 20 to 80 bp (1,2). Sequence-directed nucleosome positioning, providing for proper interaction of functional DNA sites with nonhistone proteins, plays an important functional role among the factors determining the regularity of nucleosome location. Thus, along with DNA compacting, nucleosomes play the most important role in providing access for regulatory transcription factors to gene regulatory regions, which is essential for activation of gene expression (3). The mechanisms of sequence-directed nucleosome positioning have been studied in numerous in vivo and in vitro experiments that suggested the existence of specialized nucleosome code determining this positioning as a result of multiple histone–DNA interactions (4,5).
In this work, we describe an Internet-accessible program, RECON, allowing for calculation of a function that characterizes the ability of DNA to form nucleosomes (hereinafter referred to as nucleosome potential). To develop the program, we used a method based on discriminant analysis and accounting for the frequencies of dinucleotides in local regions of nucleosome sites (6). This method is based on detection of the block structure of the site of nucleosome formation during its partition into local regions with a specific dinucleotide context. Testing this method using control samples with experimentally confirmed increased (7) and decreased (8) affinity for nucleosomes demonstrated its efficiency and potency for localizing nucleosome formation sites (6).
We used the software package RECON for computer analysis of several classes of genomic DNA sequences, namely, promoters with various expression patterns (6), exons, introns, splicing site regions (9), locus control regions (10), NotI flanking sequences (11), alpha-satellite DNA and mobile element insertion regions (12). The goal of this study was to detect interrelations between the nucleosome potential profile and functional characteristics of sequences with certain structure–function significance. It was demonstrated that the nucleosome potential of promoter regions of housekeeping genes and the genes expressed in many tissues is considerably lower than that of the promoter regions of tissue-specific genes. A pronounced increase in nucleosome potential from exons to introns in splicing donor sites was shown, as well as a marked drop from introns to exons in splicing acceptor sites.
METHODS AND ALGORITHMS
The method for calculating nucleosome potential has been described in detail elsewhere (6). Let us consider two samples of sequences—nucleosome formation sites and random sequences with equal nucleotide frequencies. As the sample of nucleosome formation sites, we used 141 DNA sequences from the database Nucleosomal DNA (13). To calculate nucleosome potential, a studied region of the nucleosome formation site is partitioned into fragments. As the partition Ω(b1, b2,…,bp−1) of the site [a, b] we understand a set P of nonoverlapping local fragments [ap, bp] (p = 1, …, P), meeting the following conditions: a1 = a; ap+1 = bp, for p = 1,…, P−1; bP = b. The program accepts a number of regions P up to 13. The search for an optimal partition is intended to provide minimal errors during recognition. The Mahalanobis distance R2 between distributions over two samples is used as the parameter for assessing the quality of a partition:
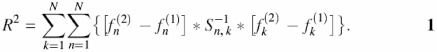
Here, ![]() is the mean frequency of the i-th dinucleotide in the p-th partition fragment for the sample of promoter sequences;
is the mean frequency of the i-th dinucleotide in the p-th partition fragment for the sample of promoter sequences; ![]() , the corresponding frequency for the sample of random sequences [n = (p − 1) × 16 + i, p = 1,…, 12, i = 1,…, 16, n = 1,…, N]; matrix S−1, an inverse matrix for the consolidated covariance matrix S = S(1) + S(2); S(1) and S(2), covariation matrices for the positive and negative samples of sequences of vectors of the dinucleotide frequencies
, the corresponding frequency for the sample of random sequences [n = (p − 1) × 16 + i, p = 1,…, 12, i = 1,…, 16, n = 1,…, N]; matrix S−1, an inverse matrix for the consolidated covariance matrix S = S(1) + S(2); S(1) and S(2), covariation matrices for the positive and negative samples of sequences of vectors of the dinucleotide frequencies ![]() and
and ![]() . The value of R2 depends on N = 16 × P variables, dinucleotide frequencies in the partition fragments (16 is the number of dinucleotides). Growth in R2 corresponds to mutual distances of the centers of distributions over the two samples.
. The value of R2 depends on N = 16 × P variables, dinucleotide frequencies in the partition fragments (16 is the number of dinucleotides). Growth in R2 corresponds to mutual distances of the centers of distributions over the two samples.
When analyzing a random DNA sequence, the value of function ϕ(X) was calculated at each position of a sliding window (fragment X, 160 bp):
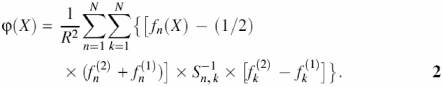
Here, fn(X) is the vector of dinucleotide frequencies constructed taking into account the partitioning of the fragment X into local fragments. The nucleosome potential ϕ(X) (Equation 2) is constructed so that its mean value over the sample of nucleosome formation site sequences equals +1; over the sample of random sequences, −1. This means that a higher probability of nucleosome formation corresponds to values of nucleosome potential ϕ(X) close to +1. When predicting nucleosome formation sites using the potential ϕ(X), it is possible to use the following rule:
![]()
Selection of the value of Δϕ is determined by the specificity of the data analysis. If we use the distribution of ϕ(X) values over the learning sample of nucleosome formation sites, then the value of Δϕ may be selected as
![]()
Here, Pα is the α-quantile of the standard normal distribution; and σϕ, the standard deviation of the values of the recognition function ϕ(X) (Equation 2) over the sample of nucleosome formation sites (σϕ = 0.79). Thus, Δϕ = 1.55 for a 95% confidence interval (P0.95 = 1.96).
Compared with the first version of this program (6), in the current version we use a new normalization of the output profile, namely, the maximal value of the nucleosome potential equals +1 and positive values correspond to reliable predictions of nucleosome formation sites at a certain significance level. To find the values of nucleosome potential ϕ(X) with a specified significance level α, it was transformed as follows:
![]()
Designations here are similar to those in (Equation 4); α is selected to equal 0.95.
Representation of the nucleosome potential as in (Equation 5) is used to bring into correlation larger values of nucleosome potential with larger probability of nucleosome formation. Thus, values of ϕα(X) > 0 (Equation 5) correspond to reliable prediction of nucleosome formation sites.
The program RECON is constructed based on analysis of the statistical distribution of dinucleotide frequencies within local regions of nucleosome formation sites. This is because analysis of DNA nucleotide sequences with essentially nonuniform dinucleotide composition may be incorrect and fail to reflect the actual ability of sequences to form nucleosomes. The nonuniformity of dinucleotide composition may stem from the fact that certain types of dinucleotides may be numerous in a sequence analyzed, while other types may be absent. To exclude this type of sequence we used the following measure—the distance of dinucleotide relative abundance δ(S1, S2) (14):
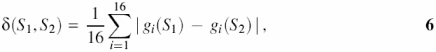
where S1, S2 is a pair of sequences analyzed;
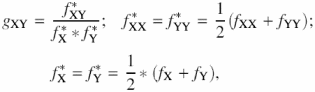
where X and Y are complementary nucleotides. In our case, S1 is a random sequence, and S2 is an integrated sequence of 141 nucleosome sites. Comparison of each nucleosome site S1 with the integrated sequence S2 demonstrated that δ(S1, S2) < 0.5. Hence, we use the value δ0 = 0.5 as a threshold for elimination of the sequences with abnormal dinucleotide content. Thus, the analyzed sequence S1 should meet the following condition in accordance with the definition of measure Equation 6:
![]()
IMPLEMENTATION AND DISCUSSION
The WWW interface http://wwwmgs.bionet.nsc.ru/mgs/programs/recon/ of the program RECON (Figure 1) allows the user to calculate the nucleosome formation potential [ϕα(X), ϕ(X)] (Equations 5 and 2) profile for a sequence of interest. Output data are either in the from of a graphical representation or in numerical form (Figure 1, option ‘Graphic mode’). The default mode transforms original nucleosome formation potential ϕ(X) (Equation 2) according to Equation 5 to the interval ϕα(X) ≤ +1 (Figure 1, option ‘Standardization by dispersion’) so that the value ϕα(X) = +1 corresponds to the best prediction; the interval ϕα(X) > 0 corresponds to reliable predictions with a confidence level P < 0.05 (α = 0.95); and the interval ϕα(X) ≤ 0 corresponds to unreliable predictions (P ≥ 0.05).
Figure 1.

Interface of the program RECON.
Presentation of the results of the program in graphical mode, or numerically in the case when the sequence analyzed does not meet the condition of distance of dinucleotide relative abundance (Equation 7), is shown in Figure 2. In this situation, the numerical output to uses the symbol (*) for the positions excluded from analysis (Figure 2A); the graphical mode shows these positions by changed color (Figure 2B).
Figure 2.

Representation of the results of an analysis in (A) numerical format and (B) graphical mode when a region of the sequence does not meet the condition on dinucleotide composition (Equation 7).
Let us consider the application of various RECON options by the example of the promoter region of GM-CSF (granulocyte–macrophage colony stimulating factor, AC X03020). It was experimentally demonstrated for this gene (15) that centers of nucleosome formation sites are localized to positions −100 and −400 relative to the transcription start (1129). The results of analysis in graphical mode using the option ‘Standardization by dispersion’ and without it are shown in Figure 3. Note that the rather high values of nucleosome potential in the promoter region may be related to the fact that expression of this region is inducible and tissue-specific.
Figure 3.

Representation of the results of an analysis in graphical mode using the example of the mouse GM–CSF gene with the option ‘Standardization by dispersion’ switched (A) off and (B) on. Arrows indicate positions of the nucleosome centers.
Operation of the program RECON can also be illustrated by the example of the Homo sapiens alpha-fetoprotein gene (AC M16110), in whose fourth intron [7728, 9213] two nucleosome formation sites [positions (8126, 8271) and (8335, 8480)] were discovered experimentally (16). Nucleosome positioning is connected with the presence of Alu repeats in this intron. Both experimental (16) and theoretical (9) arguments exist that Alu repeats favor nucleosome positioning. The profiles of nucleosome potential calculated for the fourth intron of the gene in question and the adjacent exons are shown in Figure 4. It is evident that both nucleosome formation sites display increased values compared with the adjacent regions. Note also the regions in the vicinity of the borders with the adjacent exons, which also show increased values of nucleosome formation; this may be interpreted as formation of nucleosome sites in the vicinity of splicing sites (9,17).
Figure 4.

Nucleosome formation potential ϕ(X) profiles for the Homo sapiens alpha-fetoprotein gene. The ovals denote nucleosome formation site regions (16). The intron and exon locations are shown below.
Further improvement of the program for calculating nucleosome potential requires development of new methods for detection of nucleosome code and involvement of new experimental data on nucleosome formation sites, which we are now collecting (12). Then, using these data, we plan to develop the next version of RECON as well as other software for the detection of nucleosome formation sites.
Acknowledgments
ACKNOWLEDGEMENTS
The author is grateful to Dr S. V. Lavryushev for interface design, and to Drs D. P. Furman and A.V. Katokhin for useful discussions. The work was supported by the Russian Foundation for Basic Research (grants nos. 03-04-48555, 03-04-48469, 03-04-48829, 02-07-90355 and 03-07-90181) and NATO (grant no. LST.CLG.979816); grant MCB RAS (No. 10.4); SB RAS (integration project No. 119); Russian Ministry of Industry, Science, and Technologies (grant Nos 43.073.1.1.1501).
REFERENCES
- 1.Luger K., Mader,A.W., Richmond,R.K., Sargent,D.F. and Richmond,T.J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 2.Kornberg R.D. and Lorch,Y. (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell, 98, 285–294. [DOI] [PubMed] [Google Scholar]
- 3.Richmond T.J. and Widom,J. (2000) ‘Nucleosome and chromatin Structure’, In Workman,J.L. and Elgin,S.C. (eds), Chromatin Structure and Gene Expression, 2nd edn. Oxford University Press, Oxford, UK, pp. 1–23. [Google Scholar]
- 4.Trifonov E.N. (1997) Genetic level of DNA sequences is determined by superposition of many codes. Mol. Biol. (Mosk), 31, 759–767. [PubMed] [Google Scholar]
- 5.Kiyama R. and Trifonov,E.N. (2002) What positions nucleosomes?—A model. FEBS Lett., 523, 7–11. [DOI] [PubMed] [Google Scholar]
- 6.Levitsky V.G., Podkolodnaya,O.A., Kolchanov,N.A. and Podkolodny,N.L. (2001) Nucleosome formation potential of eukaryotic DNA: tools for calculation and promoters analysis. Bioinformatics, 17, 998–1010. [DOI] [PubMed] [Google Scholar]
- 7.Widlund H.R., Cao,H., Simonsson,S., Magnusson,E., Simonsson,T., Nielsen,P.E., Kahn,J.D., Crothers,D.M. and Kubista,M. (1997) Identification and characterization of genomic nucleosome-positioning sequences. J. Mol. Biol., 267, 807–817. [DOI] [PubMed] [Google Scholar]
- 8.Cao H., Widlund,H.R., Simonsson,T. and Kubista,M. (1998) TGGA repeats impair nucleosome formation. J. Mol. Biol., 281, 253–260. [DOI] [PubMed] [Google Scholar]
- 9.Levitsky V.G., Podkolodnaya,O.A., Kolchanov,N.A. and Podkolodny,N.L. (2001) Nucleosome formation potential of exons, introns, and Alu repeats. Bioinformatics, 17, 1062–1064. [DOI] [PubMed] [Google Scholar]
- 10.Podkolodnaia O.A., Levitskii V.G. and Podkolodnyi,N.L. (2001) Locus control regions: description in the LCR-TRRDatabase. Mol. Biol. (Mosk), 35, 943–951. [PubMed] [Google Scholar]
- 11.Kutsenko A.S., Gizatullin,R.Z., Al-Amin,A.N., Wang,F., Kvasha,S.M., Podowski,R.M., Matushkin,Y.G., Gyanchandani,A., Muravenko,O.V., Levitsky,V.G., Kolchanov,N.A., Protopopov,A.I., Kashuba,V.I., Kisselev,L.L., Wasserman,W., Wahlestedt,C. and Zabarovsky,E.R. (2002) NotI flanking sequences: a tool for gene discovery and verification of the human genome. Nucleic Acids Res. 30, 3163–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levitsky V.G., Katokhin,A.V., Podkolodnaya,O.A. and Furman,D.P. (2004) Nucleosomal DNA organization: an integrated information system. In Kolchanov,N. and Hofestaedt,R. (eds), Bioinformatics of Genome Regulation and Structure. Kluwer Academic Publishers, Boston/Dordrecht/London, pp. 3–12. [Google Scholar]
- 13.Ioshikhes I. and Trifonov,E.N. (1993) Nucleosomal DNA sequence database. Nucleic Acids Res., 21, 4857–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karlin S. and Ladunga,I. (1994) Comparisons of eukaryotic genomic sequences. Proc. Natl Acad. Sci. USA, 91, 12832–12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holloway A.F., Rao,S., Chen,X. and Shannon,M.F. (2003) Changes in chromatin accessibility across the GM–CSF promoter upon cell activation are dependent on nuclear factor kappaB proteins. J. Exp. Med., 197, 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Englander E.W. and Howard,B.H. (1995) Nucleosome positioning by human Alu elements in chromatin. J. Biol. Chem., 270, 10091–10096. [DOI] [PubMed] [Google Scholar]
- 17.Denisov D.A., Shpigelman,E.S. and Trifonov,E.N. (1997) Protective nucleosome centering at splice sites as suggested by sequence-directed mapping of the nucleosomes. Gene, 205, 145–149. [DOI] [PubMed] [Google Scholar]