

Abstract
The heart hypertrophies in response to developmental signals as well as increased workload. Although adult-onset hypertrophy can ultimately lead to disease, cardiac hypertrophy is not necessarily maladaptive and can even be beneficial. Progress has been made in our understanding of the structural and molecular characteristics of physiological cardiac hypertrophy, as well as of the endocrine effectors and associated signalling pathways that regulate it. Physiological hypertrophy is initiated by finite signals, which include growth hormones (such as thyroid hormone, insulin, insulin-like growth factor 1 and vascular endothelial growth factor) and mechanical forces that converge on a limited number of intracellular signalling pathways (such as PI3K, AKT, AMP-activated protein kinase and mTOR) to affect gene transcription, protein translation and metabolism. Harnessing adaptive signalling mediators to reinvigorate the diseased heart could have important medical ramifications.
The mammalian heart is a muscle, the fundamental function of which is to pump blood throughout the circulatory system to deliver oxygen and nutrients to organs and to transport carbon dioxide back to the lungs. In response to an increased workload, typically caused by pathological or physiological stimulation, the heart undergoes a growth process named hypertrophy, which decreases ventricular wall stress and maintains or even augments pump function (BOX 1). However, hypertrophy, from the Greek for ‘increased growth’, is a more complex phenomenon than this simple definition might suggest. Cardiomyocytes, which represent 85% of the heart mass, are the contracting cells of the heart, and they contain an arrayed series of basic contractile units called sarcomeres1–3 (BOX 1). Unlike other cell types that comprise the heart (that is, endothelial cells, fibroblasts, immune cells and progenitor cells), mammalian cardiomyocytes become terminally differentiated shortly after birth and mostly lose their ability to proliferate, although a low level of cardiomyocyte turnover occurs throughout life1,4,5. As a consequence, although cardiac mass can be increased by fibroblast proliferation, and possibly also by progenitor cell activity and some cardiomyocyte renewal, mass increase primarily occurs through the hypertrophy of individual cardiomyocytes (BOX 1). Cardiac hypertrophy commonly occurs in response to pathological conditions such as hypertension and myocardial infarction from coronary artery disease, which eventually give rise to ventricular remodelling and dilatation, fibrosis, and diminished cardiac output2,3. However, the myocardium can also undergo an adaptive form of cardiac hypertrophy called physiological hypertrophy, which is fundamentally different from pathological hypertrophy because the heart does not develop disease and can even benefit from it6. A heart undergoing physiological hypertrophy shows enhanced vascular perfusion and metabolism, and the growth process is initiated by molecular pathways specific to physiological hypertrophy. This Review discusses the unique structural, functional, metabolic and circulatory features of physiological hypertrophy, although we do not attempt to cover the even more elaborate literature related to pathological cardiac hypertrophy.
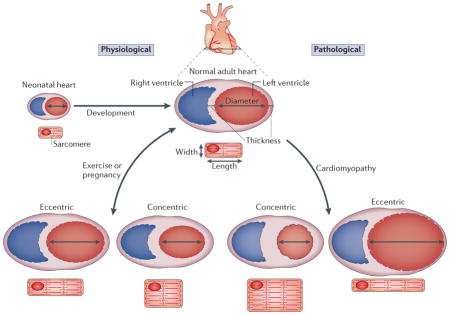
Box 1. Cardiac hypertrophy geometries.
The heart has the ability to increase its size and, depending on the stimulus, this results in physiological or pathological hypertrophy. Hypertrophy itself decreases ventricular wall stress by increasing the thickness of this wall. It follows Laplace’s law, which says wall stress (or tension) is an inverse function of wall thickness (tension = (pressure × radius)/(2 × wall thickness)). Cardiac hypertrophy can be classed as either eccentric or concentric growth, based on the geometries of the heart and individual cardiomyocytes (see the figure). Non-pathological eccentric hypertrophy is characterized by an increase in ventricular volume with a coordinated growth in wall and septal thicknesses, where individual cardiomyocytes grow in both length and width. However, eccentric hypertrophy under pathological conditions (myocardial infarction or dilated cardiomyopathy) can lead to wall dilation with preferential lengthening of cardiomyocytes. Physiological stimulation (such as pregnancy or endurance training) can also induce a less pronounced form of eccentric hypertrophy2,6. Concentric hypertrophy is characterized by a reduction in left ventricular chamber dimension and an increase in free wall and septal thicknesses, and individual cardiomyocytes typically increase in thickness more than in length (this results in a decreased length/width ratio)2,6. Concentric hypertrophy usually arises owing to pathological conditions such as chronic hypertension or valvular stenosis. Isometric exercise training, such as wrestling or weight-lifting, also induces a milder form of concentric cardiac hypertrophy that is not known to be pathological1. The heart can go from a normal state to a state of physiological hypertrophy and back, although pathological hypertrophy that produces heart failure may be less reversible.
Characteristics of physiological hypertrophy
Physiological cardiac hypertrophy drives the normal growth of the heart from birth to early adulthood (which is known as postnatal or maturational hypertrophy, hereafter referred to as postnatal hypertrophy), the growth of the maternal heart during pregnancy, and the growth of the heart in well-conditioned athletes as a result of extreme and/or repetitive exercise. Postnatal hypertrophy is induced by greater circulatory demands of the growing organism and by high circulatory levels of growth hormone and insulin-like growth factor 1 (IGF1)7. During postnatal hypertrophy, some of the cardiomyocytes become binucleated, expand their contractile apparatus, remodel their extracellular matrix as the heart chambers enlarge and wall and septal thicknesses increase, and improve excitation contraction coupling efficiency through more elaborate calcium cycling between the transverse-tubule system and sarcoplasmic reticulum8,9. In pregnancy-induced physiological hypertrophy, cardiac growth is the greatest in the third trimester, as it is associated with the largest expansion in circulatory volume and cardiac output requirements for the mother given the rapid growth of the fetus10. Finally, exercise-induced cardiac hypertrophy, which was initially empirically described in cross-country skiers in the late nineteenth century, was later defined more definitively by electrocardiographic studies in the mid-1950s in marathon runners11,12 (BOX 1).
Physiological hypertrophy is a mild form of growth, typically characterized by a 10–20% increase in heart weight normalized to body weight. Heart mass in professional athletes assessed by echocardiography routinely demonstrated a slightly, albeit significantly, increased value compared with sedentary age-matched control individuals13–15. In healthy women, pregnancy is associated with a transient 10–20% increase in total heart mass16. By contrast, in all mammals, postnatal hypertrophy typically results in a twofold or greater increase in left ventricular mass between birth and adulthood as a result of an increase in the average diameter of cardiomyocytes17,18.
Importantly, cardiac function is preserved during physiological cardiac hypertrophy. Measurements of systolic function, as assessed by echocardiography, are similar in both professional athletes and sedentary control individuals19. Similarly, diastolic function remains normal or is marginally enhanced in the athlete’s heart19. In addition, physiological cardiac hypertrophy induced by pregnancy or exercise is fully reversible. In healthy women the hypertrophy of the left ventricle normalizes 8 weeks after parturition10. Moreover, echocardiographic studies showed that posterior wall thickness and estimated left ventricular mass regressed within a few weeks of deconditioning in college- and Olympic-trained athletes20,21. This collectively suggests that physiological hypertrophy induced by exercise or pregnancy is likely to be harmless, and perhaps even beneficial, in healthy individuals, although pre-existing conditions such as inherited cardiomyopathies, arrhythmogenic channelopathies or angiogenic imbalance can promote disease and even premature death in athletes and mothers22,23.
There is also a net induction in angiogenesis during physiological hypertrophy. Exercise training enhances coronary blood flow capacity, coronary artery diameter and the capillary to cardiomyocyte ratio throughout the heart24–26. Finally, hypertrophy can affect the metabolic substrate profile of the myocardium. Early in cardiac development, glycolysis is the major source of energy for proliferating cardiomyocytes. However, after birth, mitochondrial capacity increases and fatty acid oxidation becomes the primary metabolic pathway for generating ATP in the heart27. Whereas pathological cardiac hypertrophy is initially associated with a switch from an oxidative to a glycolytic metabolic profile, exercise-induced hypertrophy relies on maintaining a proper balance of glycolytic and fatty acid oxidation, as both are augmented28,29.
An important molecular feature of physiological hypertrophy is the absence of induction of the molecular stress fetal gene programme that is classically associated with the development of pathological hypertrophy30. This pathology-induced stress programme includes induction of mRNA for atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), skeletal α-actin and β-myosin heavy chain (β-MHC; also known as myosin heavy chain 7)1. Genes encoding calcium-handling proteins also remain unchanged during physiological hypertrophy, whereas many such genes are altered in pathological hypertrophy or heart failure1. Moreover, physiological hypertrophy is not significantly associated with interstitial or replacement fibrosis as observed in pathological hypertrophy1. For example, levels of collagen I, which confers stiffness to the fibrotic matrix of the heart, remain unchanged in the left ventricles of rats subjected to endurance training31,32. Similarly, markers of myofibroblast activation that underlie pathological remodelling of the heart, such as smooth muscle α-actin, are not detected in exercised rat hearts32.
Physiological hypertrophy at the organ level is triggered at the cardiomyocyte level by a restricted number of hypertrophic stimuli, the signals from which converge on a limited number of intracellular signal transduction pathways that alter gene expression in the nucleus and increase the rates of protein and RNA synthesis to coordinate this unique type of growth response. The initiating stimuli for triggering physiological cardiac hypertrophy can be separated into stretch-sensitive mechanisms or biochemical signals that include neuro-endocrine factors and hormones, and will be discussed below.
Initiating signals: hormones and receptors
The biochemical signals that underlie physiological hypertrophy are well characterized and include ligands for a network of tyrosine kinase receptors and nuclear receptors. Whereas pathological cardiac hypertrophy is mediated by the neuro-endocrine hormones endothelin 1 and angiotensin II (BOX 2), physiological hypertrophy appears to be initiated by different ligands that include thyroid hormone, insulin, growth hormone and IGF1 (REF. 2) (FIG. 1). Cardiac concentrations of IGF1 but not endothelin 1 or angiotensin II were upregulated in professional athletes33. Vascular endothelial growth factors (VEGFs) also affect physiological cardiac hypertrophy. For example, postnatal heart growth was impaired in Vegfb-null mice34, and mice and rats overexpressing Vegfb in the heart developed cardiac hypertrophy characterized by enlarged cardiomyocytes with preserved cardiac function, at least early in life35,36.
Box 2. Pathological hypertrophy signalling pathways.
Pathological hypertrophy is induced by disorders such as systemic or pulmonary hypertension, myocardial infarction, coronary artery disease, genetic mutations in genes encoding sarcomeric proteins, diabetic and metabolic cardiomyopathy, viral and bacterial myocarditis, valvular insufficiency and congenital heart defects3. Pathological hypertrophy is associated with increased rates of myocyte death, fibrotic remodelling and decreased systolic and diastolic function that often progresses towards heart failure. Angiotensin II, endothelin 1 and catecholamines bind to seven-transmembrane receptors that are coupled to and activate heterotrimeric G proteins. In particular, Gq/11 signalling activates phospholipase C (PLC), which catalyses the synthesis of inositol 1,4,5-triphosphate (Ins(1,4,5) P3) and DAG3. Ins(1,4,5)P3 production induces intracellular Ca2+ release to then activate calcium/calmodulin-dependent protein kinase (CaMK) or calcineurin, which mediate cardiomyocyte growth2. Calcineurin is a calcium-dependent protein phosphatase that dephosphorylates nuclear factor of activated T cells (NFAT) transcription factors. NFAT forms complexes with the cofactors GATA4 or myocyte enhancer factor-2 (MEF2) to transactivate the transcription of hypertrophic target genes that are typically maladaptive, although some beneficial or cellular protective genes are also activated. For example, numerous genetic or pharmacologic gain- and loss-of-function approaches have demonstrated the importance of the calcium–calcineurin–NFAT pathway in regulating the pathological growth of the heart2 (FIG. 2), although calcineurin–NFAT signalling downstream of insulin-like growth factor 1 (IGF1) signalling could be adaptive62. Other examples of mediators of pathological cardiac hypertrophy are the p38 and c-Jun N-terminal kinase (JNK) branches of the MAPK cascade, which phosphorylate and activate the GATA4 transcription factor. Sustained p38 or JNK activation in the heart leads to cardiomyopathy and heart failure, whereas loss-of-function studies demonstrate their function as negative regulators of cardiac hypertrophy2. Catecholamines can also signal to adenylyl cyclase (AC) to induce activation of protein kinase A (PKA), which then phosphorylates an array of intracellular targets, leading to increased calcium release and enhanced contractility; the net effect of this appears to be cardiomyopathic, as it results in increased myocyte apoptosis and necrosis. α/β-AR, α- or β-adrenergic receptor; AT-R, angiotensin II receptor; Endo-R, endothelin 1 receptor; MAPKK, MAPK kinase; MAPKKK, MAPK kinase kinase.
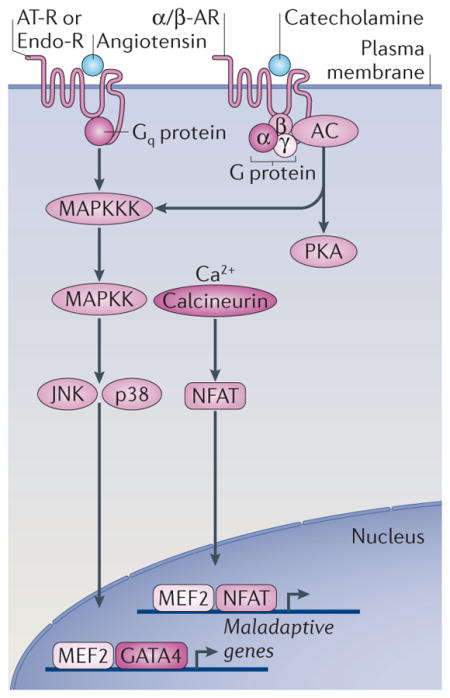
Figure 1. Physiological hypertrophy signalling pathways.

Physiological hypertrophy is an adaptive form of cardiac hypertrophy. The figure depicts central signalling pathways of physiological hypertrophy discussed in the text. Physiological hypertrophy is initiated by intermittent signals of triiodothyronine (T3), vascular endothelial growth factor B (VEGFB), insulin and insulin-like growth factor 1 (IGF1), as illustrated by the oscillating curve. The growth hormones activate membrane-localized tyrosine kinase receptors (VEGF receptor 1 (VEGFR1), IGF1 receptor (IGF1R) or insulin receptor (IR)) and nuclear receptors (thyroid hormone receptor (TR)), which trigger intracellular signalling pathways specific to physiological hypertrophy. These signalling pathways regulate the transcription of adaptive genes, protein synthesis, metabolism and energy production. The growth signals centre on common signalling branches controlled by ERK1/2, PI3K, AKT and mTOR complex 1 (mTORC1), whereas AMP-activated protein kinase (AMPK) governs metabolic adaptive reprogramming. 4EBP1, eukaryotic translation initiation factor 4E-binding protein 1; C/EBPβ, CCAAT/enhancer binding protein-β; CaMKKβ, calcium/calmodulin-dependent protein kinase kinase-β; CITED4, CBP/p300 interacting transactivator 4; eIF2Bε, eukaryotic translation initiation factor 2Bε; FOXO, forkhead box O; GSK3β, glycogen synthase kinase 3β; IRS1/2, insulin receptor substrate 1 or 2; LKB1, liver kinase B1; PDK1, phosphoinositide-dependent protein kinase 1; PGC1α, peroxisome proliferator-activated receptor-γ co-activator 1α; RHEB, RAS homologue enriched in brain; RXR, retinoic acid receptor; S6K, S6 kinase; SRF, serum response factor; TAK1, transforming growth factor β-activated kinase 1; TSC1/2, tuberous sclerosis complex 1 or 2.
Thyroid hormone as a transcriptional activator of post-natal growth
Although the influence of the thyroid hormone triiodothyronine (T3) on adult physiological hypertrophy remains controversial, its role in postnatal hypertrophy (developmental growth) is more accepted. Thyroid hormone levels increase dramatically in children after birth37. In mice, circulating T3 levels transiently increase after birth, reaching a maximum during the second postnatal week (a 2,000-fold increase compared to the level of T3 at birth) and returning to lower levels at the end of the third week of life38. T3 exerts a direct transcriptional effect on contractile and calcium handling proteins as the heart matures during postnatal development. In rodents, T3 regulates the postnatal switch from the transcription of Myh7 (which encodes β-MHC) to the transcription of genes encoding α-MHC39. T3 signals through two different thyroid hormone receptors (TRα and TRβ); by acting as a dimer with the retinoic acid receptor, these receptors function as transcription factors that can change gene expression40. In a similar way to α-MHC, T3 positively regulates expression of sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) while inhibiting β-MHC and phospholamban expression40–43. T3 also activates the transcription of the β1 adrenergic receptor, sodium and potassium channels, cardiac troponin I (CTNI), ANF, sodium/calcium exchanger (NCX), TRα1 and adenylyl cyclase (AC) types V and VI40. Interestingly, cytosolic TRα1 interacts with the regulatory p85 subunit of PI3K, which might be a potential mechanism underlying protein synthesis regulation in the heart41.
Insulin regulates protein synthesis and gene expression
In the heart, insulin signalling directly affects glucose transport, glycolysis, glucose oxidation, glycogen synthesis, fatty acid oxidation and protein synthesis44,45. Insulin binds to the tyrosine kinase insulin receptor (IR), the activation of which leads to the recruitment and phosphorylation of the adaptor proteins insulin receptor substrate 1 (IRS1) and IRS2, which activate the PI3K–AKT signalling pathway (see below). Mice null for Irs1 or Irs2 displayed a general postnatal growth retardation phenotype46,47. Heart-specific deletion of the Inrs (insulin receptor) gene confirmed the role of insulin in postnatal heart growth, as hearts from these mice were smaller and individual cardio-myocyte volumes were reduced48. Moreover, heart-specific Inrs-knockout mice showed exacerbated hypertrophy with pathological stimulation, which was associated with mitochondrial dysfunction, suggesting that in the absence of this physiological growth pathway hearts were more prone to pathological hypertrophy49–51. Altogether, these studies indicate that insulin signalling serves as a fundamental baseline regulator of physiological growth of the heart, and the heart is not only smaller in its absence but is also more prone to pathology.
IGF1 as an initiator of physiological growth signalling
The best-described and perhaps most crucial signalling pathway regulating physiological cardiac hypertrophy is that initiated by IGF1. This ligand is mostly synthesized and secreted by the liver in response to systemic growth hormone (GH) and then targeted to the tissues in which it mediates growth. However, IGF1 can also be produced by target tissues, where it directly acts in an autocrine and paracrine manner52. For example, cardiac IGF1 and serum GH and IGF1 levels were found to be transiently increased following exercise33,53. Systemic and local IGF1 signalling were also shown to be essential for postnatal growth in general54. IGF1 binds the IR and the IGF1 receptor (IGF1R), a transmembrane tyrosine kinase receptor that activates the PI3K–AKT–phosphoinositide-dependent protein kinase 1 (PDK1)–glycogen synthase kinase 3β (GSK3β) pathway (see below). Igf1-null mice or Igf1r-null mice display a growth retardation phenotype and perinatal lethality, confirming the centrality of this ligand in mediating developmental growth55.
Results obtained in genetically modified mice generally confirm the role of IGF1 in regulating physiological cardiac hypertrophy. Initial results obtained in mice with α-MHC-driven overexpression of IGF1 in the heart showed a physiological benefit, but this improvement was likely to have occurred through postnatally related cardiomyocyte hyperplasia56. Later, separate transgenic mice overexpressing IGF1 under the control of the skeletal α-actin promoter developed a physiological form of cardiac hypertrophy, although the prolongation of this signal well into adulthood resulted in pathological cardiac hypertrophy characterized by increased fibrosis and decreased cardiac function57. By comparison, hearts overexpressing IGF1R (driven by α-MHC) displayed cardiac hypertrophy characterized by an increase in cardiomyocyte volume, absence of histopathology and increased systolic function at 3 and 10 months of age58. However, Igfr1 deletion in adult cardiomyocytes using the α-MHC-Cre transgene, which induced recombination few days after birth, did not affect the baseline growth phenotype of the heart, although adult mice with this deletion were resistant to exercise-induced hypertrophy59,60. These results clearly indicate the centrality of IGF1 and its receptor in signalling the physiological growth of the heart.
Initiating signals: mechanosensors
Mechanotransduction is the fundamental process that permits the conversion of mechanical forces into biochemical signals. In cardiomyocytes, extracellular, intracellular or intercellular mechanical forces (changes in pressure, volume, stiffness, and so on) are sensed by specialized transducing proteins within the plasma membrane, by focal adhesion complexes or internally within the cardiac Z line that contains mechanosensing proteins (FIG. 2). These sensing systems then activate signalling pathways that initiate physiological cardiac hypertrophy (FIG. 1).
Figure 2. Stretch-mechanosensing in the initiation of physiological hypertrophy.

Mechanotransduction converts mechanical forces into biochemical signals. Activation of transient receptor potential canonical (TRPC) channels by stretch leads to calcium influx and the activation of hypertrophic signalling pathways. Changes in the extracellular matrix (ECM) are sensed by integrins that signal through RHO GTPases, integrin-linked kinase (ILK), focal adhesion kinase (FAK), protein kinase C (PKC) and the pro-hypertrophic PI3K and ERK1/2. Stretch is also sensed within the sarcomeres at the Z line by proteins such as muscle LIM protein (MLP), actinin-associated LIM protein (ALP), nebulette (NEB), cypher, telethonin (Tele), obscurin (Obs) and the giant protein titin that senses both strain and stretch.
Transient receptor potential (TRP) channels, which were originally identified in chicken skeletal myocytes61 and later identified in essentially all tissues and cell types, can function as stretch-sensitive signalling mediators that permeate calcium and other cations. This stretch-activated calcium current could then mediate growth signalling in the heart through downstream effectors such as calcineurin–nuclear factor of activated T cells (NFAT) signalling62. The TRP canonical (TRPC) subfamily is comprised of TRPC3, TRPC6 and TRPC7 (which are activated by diacylglycerol (DAG) that is generated by G protein-coupled receptor signalling) as well as TRPC1, TRPC4 and TRPC5 (which can be activated by DAG and depletion of intracellular calcium stores)61. TRPC1, TRPC4, TRPC5 and TRPC6 are also known to be activated by stretching, and might thus participate in stretch sensing and downstream signalling to induce hypertrophic heart growth. Indeed, TRPC1 and TRPC6 have been shown to be directly activated by stretching, and both have been implicated in regulating cardiac hypertrophy63–66.
Integrins can also mediate mechanotransduction through their attachment to the extracellular matrix. Integrins are plasma membrane-spanning heterodimers comprised of β and α subunits that bind extracellular matrix components such as fibronectin, collagen or laminin. The cytoplasmic tail of the β subunit can transmit information on the basis of the composition and stretching of the extracellular matrix through signalling proteins that reside within the focal adhesion complex inside the cell. These integrin-associated complexes are composed of non-receptor tyrosine kinases, such as focal adhesion kinase (FAK) or integrin-linked kinase (ILK), which recruit signalling proteins such as the RHO GTPases, PI3K and protein kinase C (PKC). Cardiac-specific ablation of Itgb1 (which encodes the β1 subunit) or a global deletion of Itgb3 (which encodes the β3 subunit) worsened hypertrophic disease induced by pressure overload67,68. However, although integrins certainly mediate stretch response signalling in fibroblasts and other cells, it remains unclear whether they function in an analogous manner in cardiomyocytes.
The cardiac Z line, which is the site of thin filament anchoring, has also been implicated in mechanotransduction and stretch sensing through a large array of structural proteins, such as muscle LIM protein (MLP; also known as CSRP3), telethonin, myopalladin, palladin, cypher (also known as LDB3), actinin-associated LIM protein (ALP; also known as PDLIM3), cardiac ankyrin repeat protein (CARP; also known as ANKRD1), ankyrin, nebulette and obscurin (FIG. 2). Deficiencies or mutations in some of these proteins lead to cardiomyopathy69,70. Titin is another interesting candidate for a role in mechanotransduction, as it spans from Z line to Z line (where it interacts with other sarcomeric proteins), provides direct structural support by regulating the distention of sarcomeres and binds well over 20 proteins with roles in stretch–spring sensing70 (FIG. 2). Moreover, mutations in titin in humans also cause dilated cardiomyopathy71. Thus, there are multiple systems in place that could regulate physiological hypertrophy through a direct load-sensing mechanism within the cardiomyocyte itself.
Mediators of physiological hypertrophy
Many of the growth-inducing hormones, such as T3, insulin or IGF1, and the stretch-sensitive signalling pathways, which are possibly initiated by integrins, TRPCs or sarcomeric Z-line proteins, converge on a finite number of intracellular signalling pathways. This enables them to directly transduce compensated or physiological growth signals into changes in gene expression, protein turnover, protein degradation and RNA processing to directly mediate the growth characteristics of the heart (FIG. 1). In addition, select microRNAs have been identified that are likely to directly regulate the physiological growth response of the myocardium. This area and the relevant references are discussed in BOX 3.
Box 3. MicroRNAs and physiological cardiac hypertrophy.
MicroRNAs (miRNAs) are highly conserved, small, non-coding RNAs of 18 25 nucleotides that generally downregulate gene or protein expression post-transcriptionally. With respect to adaptive growth regulation, the muscle-specific miRNAs miR-1 and miR-133 are downregulated in adult animal models of physiological cardiac hypertrophy116. Hypertensive rats undergoing exercise training show reduced muscle levels of miR-16 and miR-21 (which target vascular endothelial growth factor (VEGF) and BCL-2, respectively) but normal levels of miR-126 (which targets PI3K)117. However, other studies suggest that miRNAs might be important regulators of postnatal heart growth. For example, miR-195, a member of the miR-15 family, participates in the postnatal mitotic arrest of cardiomyocytes by downregulating cell cycle genes118. Transgenic mice overexpressing miR-195 under the control of the β-myosin heavy chain (β-MHC) promoter had hypoplastic hearts associated with downregulation of cell cycle genes that included the checkpoint kinase 1 (REF. 118). Similarly, the overexpression of miRNAs targeting thyroid hormone signalling, such as miR-208a, negatively regulates β-MHC expression through thyroid hormone receptor-associated protein 1 (THRAP1; also known as MED13)119,120. Finally, miR-27a silences the thyroid hormone β1 receptor, thereby negatively regulating β-MHC gene expression121. There are undoubtedly other miRNAs that will be uncovered as regulators of physiological hypertrophy.
PI3K–PTEN as a nodal signalling integrator
In response to insulin and IGF1 signal reception at the sarcolemma, signals converge on a common effector, PI3K. PI3Ks are heterodimeric lipid kinases localized at the plasma membrane, where they catalyse the formation of phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3). Once synthesized, PtdIns(3,4,5)P3 recruits cytosolic effectors through their pleckstrin homology (PH) domains. PtdIns(3,4,5)P3 is inactivated by lipid phosphatases such as PTEN, which acts as an endogenous PI3K inhibitor45. PI3Ks are subdivided into three classes based on sequence homologies of the catalytic domains and substrate specificity. Among them, class Ia PI3Ks (PI3Kα, PI3Kβ and PI3Kδ) are especially important for physiological hypertrophy because they are activated by the IR and IGF1R, and integrins. Class Ia PI3Ks are heterodimers composed of a regulatory subunit (p85β or p85α, or their truncated splice variants p50α or p55α, respectively) and a catalytic subunit (p110α, p110β or p110δ)45. Overexpression of a constitutively active p110α in the mouse heart under the control of the α-MHC promoter produced physiological growth of the heart72. Conversely, dominant negative p110α overexpression in the heart induced a non-pathological atrophy and repressed growth induced by IGF1R overexpression and exercise training58,72,73. These results were later confirmed by gene targeting. Muscle-specific deletion of PI3K regulatory subunit 1 (Pik3r1; which encodes p85α) in a Pik3r2 (which encodes p85β)-null background or cardiac-specific deletion of Pik3ca (which encodes p110α) confirmed that class Ia PI3Ks are essential for physiological growth of the heart74,75. Finally, cardiac-restricted Pten deletion promoted heart growth at the organ and cellular levels76. Thus, PI3K and PTEN are central signalling integrators of the physiological growth response.
AKT directly promotes protein translation
AKT is a serine/threonine protein kinase, the activity of which is primarily regulated by PtdIns(3,4,5)P3-mediated membrane recruitment and by PDK1 activity. PDK1 is a kinase that translocates to the membrane following PtdIns(3,4,5)P3 synthesis, where it activates AKT, atypical PKCs and the ribosomal p70 S6 kinase (S6K). PI3K-dependent PDK1 activation leads to direct AKT activation through phosphorylation of this kinase by PDK1 at Thr308 (REFS 45,77). Heart mass and individual cardiomyocyte volumes were significantly reduced in mice null for Pdk1 specifically in the heart, which suggests a role for this kinase during postnatal development78. There are three genes encoding AKT — Akt1, Akt2 and Akt3 — although Akt1 and Akt2 are the main cardiac isoforms45,79. Global loss of Akt2 in gene-targeted mice produced insulin resistance with no other discernible phenotype80. However, Akt1-null mice displayed general growth impairment and, remarkably, were refractory to physiological cardiac hypertrophy in response to swimming training81–83. Similarly, overexpression of a dominant negative AKT1 mutant (AKT1K179M) in the heart prevented hypertrophic growth84. Cardiac over-expression of a constitutively active AKT1 mutant (either AKT1T308D/S473D or AKT1E40) or a membrane-localized AKT1 mutant (myr-AKT1) initially promoted hypertrophy with physiological characteristics in young animals, although prolongation of such a signal was ultimately pathological84–86. Indeed, the magnitude of AKT1 activation and its cellular location are crucial determinants of its cellular effects. Conditional expression of the membrane-targeted myr-AKT1 in the mouse heart for 2 weeks induced a reversible physiological hypertrophy, whereas sustained AKT1 expression for 6 weeks caused heart failure87. By comparison, nuclear targeting of AKT1 did not affect heart growth but did induce hyper-plasia88,89. Mechanistically, AKT1 directly promotes protein translation, in part by inhibiting GSK3β activity, which negatively regulates eukaryotic translation initiation factor 2Bε (eIF2Bε)45,90,91 (FIG. 1). Consistent with these observations, overexpression of GSK3β in the adult heart blocked postnatal hypertrophy before ultimately inducing heart failure92. As a final mechanism, AKT was shown to inhibit the activity of forkhead box protein O3 (FOXO3), which results in reduced general protein turnover and catabolism mediated by the ubiquitin ligases atrogin 1 (also known as FBXO32) and muscle-specific RING finger protein 1 (MURF1; also known as TRIM63), and hence favours the net protein accumulation needed for hypertrophy93. Altogether these results indicate that short-term AKT activation promotes an adaptive cellular growth programme, whereas sustained AKT signalling leads to pathological hypertrophy and heart failure. Thus, the duration of a signal or its frequency is likely to be crucial in determining the overall beneficial versus detrimental effect on the heart.
mTOR increases mRNA translation in physiological hypertrophy
mTOR is part of two distinct serine/threonine kinase complexes, mTOR complex 1 (mTORC1) and mTORC2, the latter of which is not sensitive to rapamycin. Both complexes regulate cell growth and survival, and have also been implicated in controlling adaptive growth of the heart. mTORC1 contains mTOR, regulatory associated protein of mTOR (RAPTOR) and G protein β subunit-like (GβL; also known as LST8). mTORC1 is activated following AKT-mediated inhibition of tuberous sclerosis complex 1 (TSC1) and TSC2, the latter of which functions as a GTPase-activating protein for RAS homologue enriched in brain (RHEB), which then directly activates mTORC1 (REF. 45) (FIG. 1). mTORC1 can also be activated by amino acids and inhibited by AMP-activated protein kinase (AMPK). Once activated, mTORC1 promotes ribosomal protein production through direct regulation of S6Ks and by inhibiting eIF4E-binding protein 1 (4EBP1), which then allows unrestrained cap-dependent translation by eIF4E45. mTORC2 may also control adaptive growth through its ability to directly phosphorylate and activate AKT, which then secondarily leads to the activation of mTORC1.
mTOR signalling has been explored in the heart. For example, pharmacologic inhibition of mTORC1 with rapamycin reverses cardiac hypertrophy induced by AKT overexpression84,87. Similarly, deletion of Tsc1 in the heart induced early neonatal cardiac hypertrophy, leading to heart failure that could be delayed by rapamycin treatment94. RHEB overexpression in vitro induced a 4EBP1-dependent hypertrophy that was probably adaptive, given the lack of fetal gene re-expression95. However, overexpression of a dominant negative mTOR in the heart did not inhibit the hypertrophic response to swimming exercise in mice96. Moreover, cardiac-specific deletion of Mtor or Rptor resulted in heart failure without an initial phase of hypertrophy97,98. Just as perplexing, deletion or overexpression of S6K had no effect in various models of physiological hypertrophy in the mouse99. Taken together, these results underscore the complexity associated with mTOR signalling in the heart, such that pharmacologic inhibition with rapamycin can blunt physiological hypertrophy, but deletion of mTOR or other pathway regulators gives rise to cardiac dilatation and pathology. These findings suggest a crucial role for mTOR in basic heart homeostasis, as well as in adaptive hypertrophy.
C/EBPβ regulates physiological hypertrophy
CCAAT/enhancer binding protein-β (C/EBPβ) is a transcription factor that controls cellular proliferation in many cell types through changes in gene expression100. C/EBPβ was identified as a factor specifically downregulated by exercise training in a differential screen for physiological and pathological cardiac hypertrophy effectors101. Cardiomyocytes in which C/EBPβ was knocked down by siRNA or mice in which Cebpb was deleted heterozygously showed changes in gene expression profiles that mimic those seen in response to exercise. Cebpb-heterozygous mice displayed greater cardiomyocyte proliferation at baseline, as if they had been exercised, and were protected from pathological stress stimulation. Mechanistically, C/EBPβ functions downstream of AKT1 to inhibit CBP/p300-interacting transactivator 4 (CITED4)-induced proliferation. C/EBPβ is also proposed to compete with serum response factor (SRF) to regulate a transcriptome specific to exercise training101. This study shows that C/EBPβ is another important regulator of an adaptive or physiological hypertrophy response: downregulation of C/EBPβ permits the expression of beneficial or protective genes and allows the expression of genes that enhance cardiomyocyte proliferation and hence cell number in the heart. Thus, the inhibition of C/EBPβ activity could be used to possibly revitalize the damaged or cardiomyopathic heart.
ERK1/2 in physiological hypertrophy
ERK1 and ERK2 (ERK1/2; also known as MAPK3 and MAPK1, respectively) are activated in the heart or cultured cardiomyocytes by a wide array of stress stimuli, some of which, such as growth factors and stretching, are consistent with physiological growth. Indeed, overexpression of an activated MEK1 mutant protein in the heart, driven by the α-MHC promoter, induced constitutive ERK1/2 signalling; this produced a stable form of concentric cardiac hypertrophy without fibrosis and with a significant enhancement in cardiac function that was associated with protection from ischaemia–reperfusion injury to the heart102,103. Consistent with these observations, inhibition of ERK1/2 by overexpression of their dedicated phosphatase, dual specificity phosphatase 6 (DUSP6), resulted in more cardiac dilation on stress stimulation104. Similar results were observed in mice with combined disruption of Mapk3 and Mapk1 in the heart: such mice showed cardiac failure with stress stimulation, or even with ageing105. These lines of evidence suggest that ERK1/2 signalling provides a crucial component to the adaptive hypertrophic response that is protective, and it certainly protects cardiomyocytes from death following ischaemic injury106.
It thus appears that physiological hypertrophy is triggered by a restricted number of intracellular signalling pathways centred around the nodal mediators PI3K–PTEN, AKT and ERK1/2, which regulate the transcription of a specific set of genes (for example, C/EBPβ controls an exercise-specific transcriptome) and increase protein translation through mTOR.
AMPK in metabolic reprogramming
The coordination of cardiac growth with metabolism is essential for an adaptive response. AMPK is described as the nodal point energy sensor in all cells that coordinates increases in metabolic output with nutrient utilization (FIG. 1). AMPK is a heterotrimeric protein kinase composed of three subunits: the catalytic α subunit, the β subunit that links the α and γ subunits and binds glycogen, and the γ subunit that binds AMP, ADP or ATP in a mutually exclusive manner107. AMP binding to AMPK leads to its activation through conformational changes that allow phosphorylation of the α subunit by the upstream kinases liver kinase B1 (LKB1; also known as STK11), calcium/calmodulin-dependent protein kinase kinase-β (CaMKKβ; also known as CaMKK2) and transforming growth factor β-activated kinase 1 (TAK1; also known as MAP3K7). Falling energy levels lead to AMPK activation by AMP that favours ATP production by stimulating fatty acid oxidation, glucose uptake and glycolysis, as well as by reducing ATP-dependent processes such as transcription and protein synthesis107. AMPK is therefore crucial to cardiac homeostasis, and most studies suggest that long-term inhibition of AMPK exacerbates pathological hypertrophy, leading to heart failure, whereas intermittent AMPK activation could be cardioprotective. For example, mouse hearts null for Adipoq (an adipose tissue-secreted hormone), in which AMPK activity was decreased, were sensitized to pressure overload-induced hypertrophy108. Mice null for Prkaa2, which encodes AMPKα2, developed an exacerbated pathological response to hypertrophic stimuli109,110. Interestingly, deletion of Stk11 (which encodes LKB1) in the heart led to reduced ventricular cardiomyocyte size in young animals, suggesting a positive role of AMPK in permitting proper postnatal cardiac growth111,112. However, loss of AMPK activity in older animals induced hypertrophy that was normalized by a constitutively active AMPK mutant or with rapamycin treatment112. With respect to AMPK activation, treatment with the AMPK activator metformin improved cardiac function following ischaemia–reperfusion injury in mice through a mechanism involving activation of endothelial nitric oxide synthase (eNOS) and the mitochondrial transcription factor peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α)113. Collectively, these studies underscore the importance of metabolic reprogramming during the hypertrophic response to ensure proper adaptation during physiological heart growth. Thus, enhancing AMPK activity with specific pharmacologic agents could be an attractive therapeutic avenue to explore for treating pathological conditions of the heart.
Conclusions
The concept that cardiac hypertrophy is deleterious largely pertains to insults that are known to result in loss of cardiovascular homeostasis or proper gene function. However, in the absence of a pre-existing condition, the heart can clearly grow to a limited extent in response to physiological stimuli without an increase in morbidity or mortality. Moreover, the heart can increase in size by two- to threefold solely as a result of cellular hypertrophy during postnatal development. Physiological hypertrophy could therefore be considered as a way to prevent cardiac dysfunction and failure. Indeed, exercise conditioning by itself reversed or delayed the onset of disease and extended lifespan in mouse models of desmin-related cardiomyopathy or hypertrophic cardiomyopathy114,115. Studies in animal models largely support the selective hypothesis that failure of the heart to mount a compensated hypertrophy response through select nodal regulators of adaptive hypertrophic signalling ultimately leads to pathology and potentially heart failure. Moreover, one might even postulate that loss of adaptive hypertrophic ability and the underlying homeostatic hypertrophic pathways that support cardiomyocyte cross-sectional area growth is a molecular event that directly promotes dilation and possibly heart failure. For example, deletion of Mapk1 and Mapk3 from the mouse heart produced a phenotype in which cardiomyocytes were unable to grow in width (cross-sectional area) and instead defaulted to a molecular programme of myocyte lengthening that led to heart failure105. With respect to medical relevance, the collective work in the field suggests that the activation of adaptive or protective factors, even if they induce cardiac hypertrophy, could be a useful strategy in patients with heart failure. For example, selective induction of AMPK activity should be protective and help to restore energy balance to the cardiomyocyte to support adaptive hypertrophy. Several pharmaceutical companies have programmes devoted to the development and application of selective AMPK activators with just such a goal (cellular protection and supportive adaptive growth). Moreover, intermittent or controlled activation of AKT, PI3K, ERK1/2 or mTOR could be protective in supporting cardiac growth and cardiomyocyte viability to prevent a further decline in functional performance. However, a temporal component to such regulation might also be crucial. Indeed, physiological hypertrophy induced by exercise, and even by pregnancy, induces transient increases in cardiac load that are more phasic compared with sustained overload signals associated with pathological signalling (BOX 2). For example, it may be desirable to activate PI3K, AKT, ERK1/2 or AMPK for only a few hours each day to induce adaptive signals associated with exercise, which might selectively augment myocardial growth and reinvigorate metabolic functioning, while at the same time antagonizing ongoing cell death. Thus, the challenge moving forward will be to properly harness the molecular effectors that program beneficial or physiological cardiac growth in a controlled manner in an attempt to reinvigorate the failing heart.
Acknowledgments
This work was supported by grants from the US National Institutes of Health (J.D.M., J.H.v.B. and M.M.), and the Howard Hughes Medical Institute (J.D.M).
Glossary
- Myocardium
From the Greek mys (muscle) and kardia (heart). It is the thick middle muscular layer of the heart that contracts
- Valvular stenosis
Also called heart valve disease. Valvular stenosis occurs in response to stiffening, thickening, fusion or blockage of one or more valves of the heart. The heart comprises four valves: the mitral, aortic, tricuspid and pulmonic valves
- Transverse-tubule system
Also called the T-tubule system. A T-tubule is a deep invagination of the sarcolemma (cardiomyocyte plasma membrane) enriched in excitation–contraction coupling molecules. T-tubule system refers to the network of T-tubules within an adult cardiomyocyte
- Systolic function
The performance of the left ventricle during systole, which is the contraction of the heart. The best index of left ventricle systolic function is ejection fraction, which is calculated as the difference between end-diastolic and end-systolic left ventricle volume, divided by the end-diastolic left ventricle volume
- Diastolic function
The performance of the left ventricle during diastole, which is the relaxation of the heart and the filling of the ventricle
- Arrhythmogenic channelopathies
Genetic or acquired cardiac ion channel diseases. Ion channels (sodium, potassium and calcium channels) control the electrical activity of the heart. Abnormal electrical activity can lead to cardiac arrhythmias (irregular cardiac rhythm) and sudden death
- Myocarditis
Inflammation of the heart caused by a viral or bacterial infection or an autoimmune disease. Myocarditis sometimes induces eccentric hypertrophy and heart failure
- Stretch–spring sensing
Translation of changes in the cardiomyocyte extracellular environment (stretch) and the sarcomeres elasticity (spring) into biochemical hypertrophic signals
- Sarcolemma
Specialized plasma membrane of a myocyte
Footnotes
Competing interests statement
The authors declare no competing financial interests.
FURTHER INFORMATION
Jeffery D. Molkentin’s homepage: http://www.cincinnatichildrens.org/molkentinlab
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191–227. doi: 10.1016/j.pharmthera.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nature Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 3.Van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. doi: 10.1172/JCI62839. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergmann O, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kajstura J, et al. Cardiomyogenesis in the aging and failing human heart. Circulation. 2012;126:1869–1881. doi: 10.1161/CIRCULATIONAHA.112.118380. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Dorn GW. The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–970. doi: 10.1161/HYPERTENSIONAHA.106.079426. [DOI] [PubMed] [Google Scholar]
- 7.Porrello ER, Widdop RE, Delbridge LM. Early origins of cardiac hypertrophy: does cardiomyocyte attrition programme for pathological ‘catch-up’ growth of the heart? Clin Exp Pharmacol Physiol. 2008;35:1358–1364. doi: 10.1111/j.1440-1681.2008.05036.x. [DOI] [PubMed] [Google Scholar]
- 8.Hopkins SF, Jr, McCutcheon EP, Wekstein DR. Postnatal changes in rat ventricular function. Circ Res. 1973;32:685–691. doi: 10.1161/01.res.32.6.685. [DOI] [PubMed] [Google Scholar]
- 9.Clubb FJ, Jr, Bishop SP. Formation of binucleated myocardial cells in the neonatal rat. An index for growth hypertrophy. Lab Invest. 1984;50:571–577. [PubMed] [Google Scholar]
- 10.Eghbali M, Wang Y, Toro L, Stefani E. Heart hypertrophy during pregnancy: a better functioning heart? Trends Cardiovasc Med. 2006;16:285–291. doi: 10.1016/j.tcm.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 11.Winsor T, Beckner G. Hypertrophy of the heart; electrocardiographic distinction between physiologic and pathologic enlargement. Calif Med. 1955;82:151–158. [PMC free article] [PubMed] [Google Scholar]
- 12.Henschen S. Skilanglauf and skiwettlauf: ein medizinische sportstudie. Mitt Med Klin Uppsala. 1899;2:15–18. [Google Scholar]
- 13.Nishimura T, Yamada Y, Kawai C. Echocardiographic evaluation of long-term effects of exercise on left ventricular hypertrophy and function in professional bicyclists. Circulation. 1980;61:832–840. doi: 10.1161/01.cir.61.4.832. [DOI] [PubMed] [Google Scholar]
- 14.Sugishita Y, Koseki S, Matsuda M, Yamaguchi T, Ito I. Myocardial mechanics of athletic hearts in comparison with diseased hearts. Am Heart J. 1983;105:273–280. doi: 10.1016/0002-8703(83)90527-6. [DOI] [PubMed] [Google Scholar]
- 15.Dickhuth HH, Reindell H, Lehmann M, Keul J. Capacity for regression of the athletic heart. Z Kardiol. 1985;74(Suppl 7):135–143. [PubMed] [Google Scholar]
- 16.Schannwell CM, et al. Left ventricular hypertrophy and diastolic dysfunction in healthy pregnant women. Cardiology. 2002;97:73–78. doi: 10.1159/000057675. [DOI] [PubMed] [Google Scholar]
- 17.Janz KF, Dawson JD, Mahoney LT. Predicting heart growth during puberty: The Muscatine Study. Pediatrics. 2000;105:e63. doi: 10.1542/peds.105.5.e63. [DOI] [PubMed] [Google Scholar]
- 18.Hew KW, Keller KA. Postnatal anatomical and functional development of the heart: a species comparison. Birth Defects Res B Dev Reprod Toxicol. 2003;68:309–320. doi: 10.1002/bdrb.10034. [DOI] [PubMed] [Google Scholar]
- 19.Pluim BM, Zwinderman AH, van der Laarse A, van der Wall EE. The athlete’s heart. A meta-analysis of cardiac structure and function. Circulation. 2000;101:336–344. doi: 10.1161/01.cir.101.3.336. [DOI] [PubMed] [Google Scholar]
- 20.Ehsani AA, Hagberg JM, Hickson RC. Rapid changes in left ventricular dimensions and mass in response to physical conditioning and deconditioning. Am J Cardiol. 1978;42:52–56. doi: 10.1016/0002-9149(78)90984-0. [DOI] [PubMed] [Google Scholar]
- 21.Maron BJ, Pelliccia A, Spataro A, Granata M. Reduction in left ventricular wall thickness after deconditioning in highly trained Olympic athletes. Br Heart J. 1993;69:125–128. doi: 10.1136/hrt.69.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarquella-Brugada G, et al. Genetics of sudden cardiac death in children and young athletes. Cardiol Young. 2012;24:1–15. doi: 10.1017/S1047951112001138. [DOI] [PubMed] [Google Scholar]
- 23.Patten IS, et al. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature. 2012;485:333–338. doi: 10.1038/nature11040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heiss HW, et al. Studies on the regulation of myocardial blood flow in man. I.: Training effects on blood flow and metabolism of the healthy heart at rest and during standardized heavy exercise. Bas Res Cardiol. 1976;71:658–675. doi: 10.1007/BF01906411. [DOI] [PubMed] [Google Scholar]
- 25.Pelliccia A, et al. Coronary arteries in physiological hypertrophy: echocardiographic evidence of increased proximal size in elite athletes. Int J Sports Med. 1990;11:120–126. doi: 10.1055/s-2007-1024775. [DOI] [PubMed] [Google Scholar]
- 26.Laughlin MH, Bowles DK, Duncker DJ. The coronary circulation in exercise training. Am J Physiol Heart Circ Physiol. 2012;302:H10–H23. doi: 10.1152/ajpheart.00574.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. 2010;56:130–140. doi: 10.1097/FJC.0b013e3181e74a14. [DOI] [PubMed] [Google Scholar]
- 28.Gertz EW, Wisneski JA, Stanley WC, Neese RA. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J Clin Invest. 1988;82:2017–2025. doi: 10.1172/JCI113822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abel ED, Doenst T. Mitochondrial adaptations to physiological versus pathological cardiac hypertrophy. Cardiovasc Res. 2011;90:234–242. doi: 10.1093/cvr/cvr015. In-depth review of the mitochondrial adaptations to physiological or pathological cardiac hypertrophic signals. Describes how the distinct cardiac metabolic profiles associated with physiological and pathological hypertrophy are initiated by specific signalling pathways: PI3K, AMPK and PGC1α. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilkins BJ, et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 31.Burgess ML, et al. Exercise- and hypertension-induced collagen changes are related to left ventricular function in rat hearts. Am J Physiol. 1996;270:H151–H159. doi: 10.1152/ajpheart.1996.270.1.H151. [DOI] [PubMed] [Google Scholar]
- 32.Jin H, et al. Effects of exercise training on cardiac function, gene expression, and apoptosis in rats. Am J Physiol Heart Circ Physiol. 2000;279:H2994–H3002. doi: 10.1152/ajpheart.2000.279.6.H2994. [DOI] [PubMed] [Google Scholar]
- 33.Neri Serneri GG, et al. Increased cardiac sympathetic activity and insulin-like growth factor-I formation are associated with physiological hypertrophy in athletes. Circ Res. 2001;89:977–982. doi: 10.1161/hh2301.100982. [DOI] [PubMed] [Google Scholar]
- 34.Bellomo D, et al. Mice lacking the vascular endothelial growth factor-B gene (Vegfb) have smaller hearts, dysfunctional coronary vasculature, and impaired recovery from cardiac ischemia. Circ Res. 2000;86:e29–e35. doi: 10.1161/01.res.86.2.e29. [DOI] [PubMed] [Google Scholar]
- 35.Karpanen T, et al. Overexpression of vascular endothelial growth factor-B in mouse heart alters cardiac lipid metabolism and induces myocardial hypertrophy. Circ Res. 2008;103:1018–1026. doi: 10.1161/CIRCRESAHA.108.178459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bry M, et al. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. 2010;122:1725–1733. doi: 10.1161/CIRCULATIONAHA.110.957332. [DOI] [PubMed] [Google Scholar]
- 37.Stubbe P, Gatz J, Heidemann P, Muhlen A, Hesch R. Thyroxine-binding globulin, triiodothyronine, thyroxine and thyrotropin in newborn infants and children. Horm Metab Res. 1978;10:58–61. doi: 10.1055/s-0028-1093482. [DOI] [PubMed] [Google Scholar]
- 38.Hadj-Sahraoui N, Seugnet I, Ghorbel MT, Demeneix B. Hypothyroidism prolongs mitotic activity in the post-natal mouse brain. Neurosci Lett. 2000;280:79–82. doi: 10.1016/s0304-3940(00)00768-0. [DOI] [PubMed] [Google Scholar]
- 39.Morkin E. Regulation of myosin heavy chain genes in the heart. Circulation. 1993;87:1451–1460. doi: 10.1161/01.cir.87.5.1451. [DOI] [PubMed] [Google Scholar]
- 40.Arsanjani R, McCarren M, Bahl JJ, Goldman S. Translational potential of thyroid hormone and its analogs. J Mol Cell Cardiol. 2011;51:506–511. doi: 10.1016/j.yjmcc.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 41.Kenessey A, Ojamaa K. Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6K pathways. J Biol Chem. 2006;281:20666–20672. doi: 10.1074/jbc.M512671200. [DOI] [PubMed] [Google Scholar]
- 42.Kenessey A, Sullivan EA, Ojamaa K. Nuclear localization of protein kinase C-α induces thyroid hormone receptor-α1 expression in the cardiomyocyte. Am J Physiol Heart Circ Physiol. 2006;290:H381–H389. doi: 10.1152/ajpheart.00576.2005. [DOI] [PubMed] [Google Scholar]
- 43.Belakavadi M, Saunders J, Weisleder N, Raghava PS, Fondell JD. Repression of cardiac phospholamban gene expression is mediated by thyroid hormone receptor-α1 and involves targeted covalent histone modifications. Endocrinology. 2010;151:2946–2956. doi: 10.1210/en.2009-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brownsey RW, Boone AN, Allard MF. Actions of insulin on the mammalian heart: metabolism, pathology and biochemical mechanisms. Cardiovasc Res. 1997;34:3–24. doi: 10.1016/s0008-6363(97)00051-5. [DOI] [PubMed] [Google Scholar]
- 45.Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–3365. doi: 10.1101/gad.1492806. Comprehensive review on the role of PI3K–AKT signalling pathways in regulating physiological cardiac hypertrophy, cardiac contractile function and coronary angiogenesis. [DOI] [PubMed] [Google Scholar]
- 46.Araki E, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372:186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 47.Burks DJ, et al. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature. 2000;407:377–382. doi: 10.1038/35030105. [DOI] [PubMed] [Google Scholar]
- 48.Belke DD, et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest. 2002;109:629–639. doi: 10.1172/JCI13946. Shows that insulin signalling controls postnatal cardiac growth. Cardiomyocyte-specific IR knockout was shown to result in cardiomyocytes with a reduced volume, and postnatal contractile and metabolic switches were altered. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu P, et al. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol. 2003;285:H1261–H1269. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 50.Sena S, et al. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J Mol Cell Cardiol. 2009;46:910–918. doi: 10.1016/j.yjmcc.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boudina S, et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009;119:1272–1283. doi: 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Efstratiadis A. Genetics of mouse growth. Int J Dev Biol. 1998;42:955–976. [PubMed] [Google Scholar]
- 53.Sutton J, Lazarus LA. Growth hormone in exercise: comparison of physiological and pharmacological stimuli. J Appl Physiol. 1976;41:523–527. doi: 10.1152/jappl.1976.41.4.523. [DOI] [PubMed] [Google Scholar]
- 54.Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- 55.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. References 54 and 55 show that IGF1 signalling is essential for postnatal growth. [PubMed] [Google Scholar]
- 56.Reiss K, et al. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc Natl Acad Sci USA. 1996;93:8630–8635. doi: 10.1073/pnas.93.16.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delaughter MC, Taffet GE, Fiorotto ML, Entman ML, Schwartz RJ. Local insulin-like growth factor I expression induces physiologic, then pathologic, cardiac hypertrophy in transgenic mice. FASEB J. 1999;13:1923–1929. doi: 10.1096/fasebj.13.14.1923. [DOI] [PubMed] [Google Scholar]
- 58.McMullen JR, et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110α) pathway. J Biol Chem. 2004;279:4782–4793. doi: 10.1074/jbc.M310405200. [DOI] [PubMed] [Google Scholar]
- 59.Kim J, et al. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol. 2008;22:2531–2543. doi: 10.1210/me.2008-0265. In this study, IGF1R targeted deletion in adult cardiomyocytes did not result in any baseline hypertrophic phenotype in young mice. IGF1R-targeted hearts were resistant to exercise induced hypertrophy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moellendorf S, et al. IGF-IR signaling attenuates the age-related decline of diastolic cardiac function. Am J Physiol Endocrinol Metab. 2012;303:e213–e222. doi: 10.1152/ajpendo.00538.2011. [DOI] [PubMed] [Google Scholar]
- 61.Patel A, et al. Canonical TRP channels and mechanotransduction: from physiology to disease states. Pflugers Arch. 2010;460:571–581. doi: 10.1007/s00424-010-0847-8. [DOI] [PubMed] [Google Scholar]
- 62.Musarò A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999;400:581–585. doi: 10.1038/23060. [DOI] [PubMed] [Google Scholar]
- 63.Maroto R, et al. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nature Cell Biol. 2005;7:179–185. doi: 10.1038/ncb1218. [DOI] [PubMed] [Google Scholar]
- 64.Spassova MA, Hewavitharana T, Xu W, Soboloff J, Gill DL. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci USA. 2006;103:16586–16591. doi: 10.1073/pnas.0606894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seth M, et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res. 2009;105:1023–1030. doi: 10.1161/CIRCRESAHA.109.206581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu X, Eder P, Chang B, Molkentin JD. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci USA. 2010;107:7000–7005. doi: 10.1073/pnas.1001825107. This study, along with reference 65, showed that TRPC channels are necessary mediators of cardiac hypertrophy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shai SY, et al. Cardiac myocyte-specific excision of the β1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res. 2002;90:458–464. doi: 10.1161/hh0402.105790. [DOI] [PubMed] [Google Scholar]
- 68.Johnston RK, et al. β3 integrin-mediated ubiquitination activates survival signaling during myocardial hypertrophy. FASEB J. 2009;23:2759–2771. doi: 10.1096/fj.08-127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cox L, Umans L, Cornelis F, Huylebroeck D, Zwijsen A. A broken heart: a stretch too far: an overview of mouse models with mutations in stretch-sensor components. Int J Cardiol. 2008;131:33–44. doi: 10.1016/j.ijcard.2008.06.049. [DOI] [PubMed] [Google Scholar]
- 70.Linke WA. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. 2008;77:637–648. doi: 10.1016/j.cardiores.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 71.Herman DS, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shioi T, et al. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 2000;19:2537–2548. doi: 10.1093/emboj/19.11.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McMullen JR, et al. Phosphoinositide 3-kinase(p110α) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci USA. 2003;100:12355–12360. doi: 10.1073/pnas.1934654100. This study, along with reference 72, showed that PI3Kα regulates the physiological growth of the heart in gain and loss of function studies. PI3Kα activity controls exercise-induced physiological cardiac hypertrophy but not pathological hypertrophy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luo J, et al. Class IA phosphoinositide 3-kinase regulates heart size and physiological cardiac hypertrophy. Mol Cell Biol. 2005;25:9491–9502. doi: 10.1128/MCB.25.21.9491-9502.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu Z, et al. Loss of cardiac phosphoinositide 3-kinase p110α results in contractile dysfunction. Circulation. 2009;120:318–325. doi: 10.1161/CIRCULATIONAHA.109.873380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crackower MA, et al. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–749. doi: 10.1016/s0092-8674(02)00969-8. This study showed that PTEN deletion in cardiomyocytes promotes heart growth at the organ and cellular level. Overexpression of dominant negative p110α downstream of PTEN normalizes the phenotype. [DOI] [PubMed] [Google Scholar]
- 77.McManus EJ, et al. The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J. 2004;23:2071–2082. doi: 10.1038/sj.emboj.7600218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mora A, et al. Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. EMBO J. 2003;22:4666–4676. doi: 10.1093/emboj/cdg469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oudit GY, et al. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J Mol Cell Cardiol. 2004;37:449–471. doi: 10.1016/j.yjmcc.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 80.Cho H, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 81.Chen WS, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBα is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 83.DeBosch B, et al. Akt1 is required for physiological cardiac growth. Circulation. 2006;113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 84.Shioi T, et al. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matsui T, et al. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem. 2002;277:22896–22901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- 86.Condorelli G, et al. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci USA. 2002;99:12333–12338. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shiojima I, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. Showed that conditional expression of AKT1 in the heart for 2 weeks induces a reversible physiological hypertrophy while sustained AKT1 expression for 6 weeks causes heart failure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shiraishi I, et al. Nuclear targeting of Akt enhances kinase activity and survival of cardiomyocytes. Circ Res. 2004;94:884–891. doi: 10.1161/01.RES.0000124394.01180.BE. [DOI] [PubMed] [Google Scholar]
- 89.Rota M, et al. Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ Res. 2005;97:1332–1341. doi: 10.1161/01.RES.0000196568.11624.ae. [DOI] [PubMed] [Google Scholar]
- 90.Haq S, et al. Glycogen synthase kinase-3β is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol. 2000;151:117–130. doi: 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Antos CL, et al. Activated glycogen synthase-3β suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2002;99:907–912. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Michael A, et al. Glycogen synthase kinase-3β regulates growth, calcium homeostasis, and diastolic function in the heart. J Biol Chem. 2004;279:21383–21393. doi: 10.1074/jbc.M401413200. [DOI] [PubMed] [Google Scholar]
- 93.Skurk C, et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J Biol Chem. 2005;280:20814–20823. doi: 10.1074/jbc.M500528200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Malhowski AJ, et al. Smooth muscle protein-22-mediated deletion of Tsc1 results in cardiac hypertrophy that is mTORC1-mediated and reversed by rapamycin. Hum Mol Genet. 2011;20:1290–1305. doi: 10.1093/hmg/ddq570. Showed that Tsc1 deletion results in lethal developmental and postnatal cardiac hypertrophy that is reversed by rapamycin treatment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y, et al. Rheb activates protein synthesis and growth in adult rat ventricular cardiomyocytes. J Mol Cell Cardiol. 2008;45:812–820. doi: 10.1016/j.yjmcc.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 96.Shen WH, et al. Cardiac restricted overexpression of kinase-dead mammalian target of rapamycin (mTOR) mutant impairs the mTOR-mediated signaling and cardiac function. J Biol Chem. 2008;283:13842–13849. doi: 10.1074/jbc.M801510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang D, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120:2805–2816. doi: 10.1172/JCI43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shende P, et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123:1073–1082. doi: 10.1161/CIRCULATIONAHA.110.977066. This study, along with reference 97, shows that cardiac-specific mTOR or RAPTOR ablation results in heart failure without an initial phase of hypertrophy. [DOI] [PubMed] [Google Scholar]
- 99.McMullen JR, et al. Deletion of ribosomal S6 kinases does not attenuate pathological, physiological, or insulin-like growth factor 1 receptor-phosphoinositide 3-kinase-induced cardiac hypertrophy. Mol Cell Biol. 2004;24:6231–6240. doi: 10.1128/MCB.24.14.6231-6240.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tsukada J, Yoshida Y, Kominato Y, Auron PE. The CCAAT/enhancer (C/EBP) family of basic-leucine zipper (bZIP) transcription factors is a multifaceted highly-regulated system for gene regulation. Cytokine. 2011;54:6–19. doi: 10.1016/j.cyto.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 101.Bostrom P, et al. C/EBPβ controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036. Shows that C/EBPβ is specifically downregulated by exercise training. C/EBPβ downregulation increases cardiomyocyte proliferation and leads to the transcription of genes specific to exercise. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bueno OF, et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350. doi: 10.1093/emboj/19.23.6341. Demonstrates that activation of MEK1–ERK1/2 signalling in the mouse heart induces a non-pathological form of compensated cardiac hypertrophy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lips DJ, et al. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation. 2004;109:1938–1941. doi: 10.1161/01.CIR.0000127126.73759.23. [DOI] [PubMed] [Google Scholar]
- 104.Purcell NH, et al. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc Natl Acad Sci USA. 2007;104:14074–14079. doi: 10.1073/pnas.0610906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kehat I, et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ Res. 2011;108:176–183. doi: 10.1161/CIRCRESAHA.110.231514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kehat I, Molkentin JD. Extracellular signal-regulated kinase 1/2 (ERK1/2) signaling in cardiac hypertrophy. Ann NY Acad Sci. 2010;1188:96–102. doi: 10.1111/j.1749-6632.2009.05088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Horman S, Beauloye C, Vanoverschelde JL, Bertrand L. AMP-activated protein kinase in the control of cardiac metabolism and remodeling. Curr Heart Fail Rep. 2012;9:164–173. doi: 10.1007/s11897-012-0102-z. [DOI] [PubMed] [Google Scholar]
- 108.Shibata R, et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nature Med. 2004;10:1384–1389. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zarrinpashneh E, et al. AMPKα2 counteracts the development of cardiac hypertrophy induced by isoproterenol. Biochem Biophys Res Commun. 2008;376:677–681. doi: 10.1016/j.bbrc.2008.09.057. [DOI] [PubMed] [Google Scholar]
- 110.Zhang P, et al. AMP activated protein kinase-α2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008;52:918–924. doi: 10.1161/HYPERTENSIONAHA.108.114702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sakamoto K, et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKα2 but not AMPKα1. Am J Physiol Endocrinol Metab. 2006;290:e780–e788. doi: 10.1152/ajpendo.00443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ikeda Y, et al. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J Biol Chem. 2009;284:35839–35849. doi: 10.1074/jbc.M109.057273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gundewar S, et al. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res. 2009;104:403–411. doi: 10.1161/CIRCRESAHA.108.190918. Shows that metformin exerts its cardioprotective effects through AMPK activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Maloyan A, et al. Exercise reverses preamyloid oligomer and prolongs survival in αB-crystallin-based desmin-related cardiomyopathy. Proc Natl Acad Sci USA. 2007;104:5995–6000. doi: 10.1073/pnas.0609202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Konhilas JP, et al. Exercise can prevent and reverse the severity of hypertrophic cardiomyopathy. Circ Res. 2006;98:540–548. doi: 10.1161/01.RES.0000205766.97556.00. [DOI] [PubMed] [Google Scholar]
- 116.Care A, et al. MicroRNA-133 controls cardiac hypertrophy. Nature Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 117.Fernandes T, et al. Exercise training prevents the microvascular rarefaction in hypertension balancing angiogenic and apoptotic factors: role of microRNAs-16, -21, and -126. Hypertension. 2012;59:513–520. doi: 10.1161/HYPERTENSIONAHA.111.185801. [DOI] [PubMed] [Google Scholar]
- 118.Porrello ER, et al. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res. 2011;109:670–679. doi: 10.1161/CIRCRESAHA.111.248880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.van Rooij E, et al. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 120.Callis TE, et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. 2009;119:2772–2786. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nishi H, et al. MicroRNA-27a regulates β cardiac myosin heavy chain gene expression by targeting thyroid hormone receptor β1 in neonatal rat ventricular myocytes. Mol Cell Biol. 2011;31:744–755. doi: 10.1128/MCB.00581-10. [DOI] [PMC free article] [PubMed] [Google Scholar]