

Abstract
Objective
To examine the role of interferon regulatory factor 1 (IRF-1) in tumor necrosis factor α (TNFα)–induced interleukin-18 binding protein a (IL-18BPa) expression in rheumatoid arthritis synovial fibroblasts (RASFs).
Methods
TNFα-induced IRF-1 expression was assessed by real-time quantitative polymerase chain reaction and Western blotting. The effect of TNFα on IRF-1 was assessed using nuclear and cytoplasmic extracts, Western blots, and immunofluorescence. Chemical inhibitors of NF-κB or MAP kinases were used to analyze the signaling pathways of TNFα-induced IRF-1 expression and IRF-1 nuclear translocation. Control and IRF-1 small interfering RNA (siRNA) were used to analyze the effect of IRF-1 down-regulation on TNFα-induced IL-18BP expression. IL-18BPa expression was assessed by enzyme-linked immunosorbent assay, and IL-18 was assessed at the transcription and bioactivity levels using KG-1 cells.
Results
TNFα induced RASF IRF-1 expression at the messenger RNA and protein levels, with a maximal effect at 2 hours (P < 0.05; n ≥ 3). Furthermore, TNFα induced nuclear translocation of IRF-1, with maximal translocation at 2 hours (~6 fold-induction) (P < 0.05; n = 4). Blocking of the NF-κB or JNK-2 pathways reduced TNFα-induced IRF-1 nuclear translocation by 35% and 50%, respectively (P < 0.05; n ≥ 4). Using siRNA to knock down IRF-1, we observed reduced IL-18BPa expression. Additionally, IL-18 bioactivity was higher when siRNA was used to knock down IRF-1 expression.
Conclusion
These results show that IRF-1 is a key regulator of IL-18BPa expression and IL-18 bioactivity in RASFs. Regulation of IRF-1 will be a new therapeutic target in RA.
Rheumatoid arthritis (RA) is the most common chronic articular inflammatory disease involving joint destruction. The inflamed RA synovial tissue is characterized by a massive infiltration of lymphocytes and macrophages and proliferation of synovial fibroblasts (SFs). Interleukin-18 (IL-18) is considered to be a key cytokine in the pathogenesis of RA (1). Initially described as an interferon-γ (IFNγ)–inducing factor (2), IL-18 is a member of the IL-1 family (3), which is important as a regulator of innate and acquired immune responses. IL-18 promotes the release of Th1 cytokines by T cells as well as Th1 cell differentiation (4), influences the cytotoxic activity of natural killer cells through up-regulation of Fas ligand (2,4–9), and promotes angiogenesis (10–13). It is synthesized as a precursor, proIL-18, which is cleaved by the IL-1β–converting enzyme (caspase 1), resulting in the active 18.3-kd mature protein (14,15). IL-18 expression has been detected in antigen-presenting cells such as activated macrophages, Kupffer cells, dendritic cells, Langerhans cells, and epidermal cells, as well as in articular chondrocytes, osteoblasts, and activated SFs (1,2,16–20).
Compared with other proinflammatory cytokines, IL-18 is regulated in a unique way. Constitutive levels of IL-18 messenger RNA (mRNA) and protein are present in unstimulated human and murine cells and in the tissues of untreated normal mice (21,22). In unstimulated cells or tissues, IL-18 is primarily present in the precursor form, which requires conversion by caspase 1 to the mature and bioactive molecule (14,15,18,21). The other way to regulate IL-18 bioactivity is via its natural inhibitor, IL-18 binding protein (IL-18BP). In humans, 4 isoforms of IL-18BP (isoforms a, b, c, and d) have been described and are produced as a result of alternative splicing. Only isoforms a and c are able to neutralize IL-18 bioactivity. IL-18BPa is the major constitutively secreted splicing variant, with the highest binding affinity for IL-18, and thereby acts as a soluble decoy receptor (23). IL-18 bioactivity is ~17-fold lower than its total protein level, suggesting that inhibition of IL-18 bioactivity can be attained by increasing the level of IL-18BPa (16).
As we recently suggested, inhibition of IL-18 bioactivity can be attained by increasing the level of IL-1BPa (20). Interferon regulatory factor 1 (IRF-1) is a transcription factor that was initially described to be strongly induced by IFNγ and to bind to IFN-stimulated response elements within promoters, thereby activating transcription (24). It was recently shown to be a key regulator of IL-18BP expression (25). The IL-18BP promoter contains an IRF-1 binding site (25). Indeed, the expression of IL-1BP was reduced or undetectable in IRF-1–knockout mice (25–27). In the same mice, IL-18 expression was unaffected (27), suggesting an increase in IL-18 bioactivity. Recently, we demonstrated that tumor necrosis factor α (TNFα) was able to induce both IL-18 and IL-18BP expression by RASFs, and that blocking of the ERK signaling pathway reduced TNFα-induced IL-18 bioactivity by inhibiting IL-18 expression (20). Here, we examined the role of IRF-1 in TNFα-induced IL-18BPa expression.
MATERIALS AND METHODS
Cytokines and culture of human RASFs
TNFα was purchased from R&D Systems. Fibroblasts were isolated, according to an institutional review board–approved protocol, from synovium obtained from RA patients who had undergone total joint replacement or synovectomy and were processed as described previously (20,28,29). RASFs were grown in RPMI 1640 with 10% fetal bovine serum supplementation. All of the experiments were performed in serum-free media.
RNA extraction and real-time quantitative polymerase chain reaction (qPCR)
RNA was isolated from RASFs using RNeasy Mini kits in conjunction with QIAshredders, according to the manufacturer’s protocol (Qiagen), as described previously (20,28). Following isolation, RNA was quantified and checked for purity by spectrophotometry (NanoDrop Technologies). Complementary DNA (cDNA) was then prepared using a Reverse-iT MAX 1st Strand Synthesis Kit (ABgene), as described previously (20,28). Quantitative PCR was performed using Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen) according to the manufacturer’s protocol, with the following specific primer sequences: for IRF-1, forward 5′-AGCCAACATGCCCATCACTCGG-3′, reverse 5′-TGCTACGGTGCACAGGGAATGG-3′ (30); for IL-18, forward 5′-CTTGAATCTAAATTATCAGTC-3′, reverse 5′-GAAGATTCAAATTGCATCTTAT-3′ (20); and for β-actin, forward 5′-GTCAGGCAGCTCGTAGCTCT-3′, reverse 5′-CCATGTACGTTGCTATCCA-3′ (20,28).
Diluted cDNA was briefly mixed with Platinum SYBR Green qPCR SuperMix-UDG and forward and reverse primers specific for each gene (0.2 μM final concentration) and was incubated under the following conditions: 50°C for 2 minutes, 95°C for 2 minutes, and 40 cycles of 95°C for 30 seconds, 55°C for 30 seconds, and 68°C for 30 seconds using an Eppendorf Mastercycler ep realpex thermal cycler. All samples were run in duplicate and analyzed using Eppendorf software. For quantification, the relative abundance of each gene was normalized to β-actin.
Cell lysis and Western blotting
To study the effect of TNFα on IRF-1 production, RASFs were incubated with TNFα (20 ng/ml) in serum-free RPMI 1640, and assessed at several time points. Cells were lysed in cell lysis buffer containing 1 mM phenylmethylsulfonyl fluoride (PMSF) and protease inhibitors (Thermo Scientific), as previously described (29). Protein was measured using a BCA Protein Assay kit (Pierce). Equal amounts of protein (15 μg) were loaded and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes (Bio-Rad). Nitrocellulose membranes were blocked with 5% nonfat milk in Tris buffered saline–Tween 20 (TBST) for 60 minutes at room temperature. Blots were incubated overnight at 4°C with optimally diluted specific primary antibody in TBST containing 5% nonfat milk. A mouse anti-human IRF-1 monoclonal antibody (Santa Cruz Biotechnology) was used as the primary antibody. Blots were washed 3 times and incubated in horseradish peroxidase–conjugated antibody (1:1,000 dilution) for 1 hour at room temperature. Protein bands were detected using enhanced chemiluminescence (Pierce) in accordance with the manufacturer’s instructions. After stripping, blots were probed again using a rabbit anti–β-actin polyclonal antibody. Blots were scanned and analyzed for band intensities using UN-SCAN-IT version 5.1 software (Silk Scientific).
Nuclear and cytoplasmic extracts
RASFs (2 × 106/well) were grown to confluence in 6-cm dishes and treated with TNFα (20 ng/ml) with or without preincubation with inhibitors. Cytoplasmic and nuclear fractions were prepared as previously described (31). Upon termination of the reaction, cells were washed twice with ice-cold phosphate buffered saline (PBS) (pH 7.4), scraped, collected in Eppendorf tubes, and centrifuged at 1,500g for 5 minutes at 4°C. The pellet obtained was suspended in 300 μl of buffer A (10 mM HEPES buffered saline [pH 7.9], 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol [DTT], and 0.5 mM PMSF), gently mixed, and placed on ice for 15 minutes. Twenty-five micro-liters of cold 10% Nonidet P40 was added to individual samples, and the samples were vortexed and centrifuged at 14,000g for 1 minute. The supernatant (cytoplasmic fraction) was collected, and the nuclear pellet obtained was suspended in 60 μl of buffer C (20 mM HEPES buffered saline [pH 7.9], 0.4M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 1 mM PMSF) and rocked for 45 minutes at 4°C. Samples were centrifuged for 15 minutes at 14,000g at 4°C. The supernatant (nuclear fraction) was collected and stored at −80°C. Nuclear and cytoplasmic cell lysates (15 μg) were used for the detection of IRF-1 by Western blotting. After stripping, blots were probed successively with a rabbit anti–histone H3 polyclonal antibody (Abcam) and anti–β-actin antibodies to check the purity of both fractions.
Immunofluorescence staining
RASFs were plated at 10,000 cells/well in 8-well Lab-Tek chamber slides. After overnight serum starvation, cells were left untreated or were stimulated with TNFα (20 ng/ml) for 2 hours. Cells were washed with PBS, fixed with 4% formalin for 30 minutes at room temperature, washed again with PBS, and fixed with ice-cold methanol for 5 minutes. Blocking was performed by adding 5% serum in PBS for 1 hour at room temperature. IRF-1 primary antibody (5 μg/ml) in blocking buffer was added for 1 hour at room temperature. Following washing, Alexa Fluor–conjugated goat anti-rabbit antibody (10 μg/ml) was added for 1 hour at room temperature. Cells were washed with PBS, and DAPI nuclear stain (1:2,000 dilution; Invitrogen) was added for 5 minutes. Slides were dehydrated, mounted, and coverslipped. Images were obtained using an Olympus FluoView 500 confocal fluorescence microscope. A 60× objective was used, and 0.5-μm optical sections were obtained.
Chemical inhibitors
To study the signaling mechanism of IRF-1 expression and nuclear translocation by TNFα, RASFs were incubated with MAPK inhibitors (ERK-1/2 [PD98059], p38 [SB202190], and JNK-2 [SP600125]) or with an NF-κB inhibitor (pyrrolidine dithiocarbamate [PDTC]) for 1 hour, followed by stimulation with TNFα (20 ng/ml). The concentration of the inhibitors (10 μM for ERK-1/2, p38, and JNK-2; 200 μM for PDTC) was based on our previous studies (20,28). At these concentrations, RASF viability was not affected (32). All inhibitors were purchased from Calbiochem.
Immunoprecipitation
For immunoprecipitation, nuclear fractions containing equal amounts of protein were incubated with rabbit anti-human phosphorylated NF-κB p65, rabbit anti-human phosphorylated c-Jun, or rabbit IgG control overnight at 4°C, using the Pierce Classic Immunoprecipitation kit according to the protocol described by the manufacturer. Equal amounts were then loaded and run on Western blots to detect IRF-1. Next, the blots were stripped and reprobed with the antibodies used for immunoprecipitation.
Transfection studies
To analyze the effect of IRF-1 knockdown, we used a transfection approach with small interfering RNA (siRNA). RASFs were maintained in RPMI 1640 with 10% fetal bovine serum and plated in 6-well plates to grow to ~60% confluence. Then, they were transfected with either 100 nM control siRNA (Invitrogen) or IRF-1 siRNA (Dharmacon) using TransIT-TKO reagent (Mirus). After 48 hours, the cells were serum starved in RPMI 1640 overnight and then stimulated with TNFα (20 ng/ml) for 2 hours or 48 hours. Conditioned media, cell lysates, and mRNA were processed as described above.
IL-18 bioactivity
The biologic activity of IL-18 was measured by using human myelomonocytic KG-1 cells, as previously described (20). KG-1 cells (3 × 106 cells/ml; 100 μl), with or without mouse monoclonal anti–IL-18 antibody at 1 μg/ml (R&D Systems), were dispensed into the wells of 96-well Falcon microtiter plates (Becton Dickinson). Next, 100 μl of samples or recombinant human IL-18 standards was added to each well. The plates were incubated, and culture supernatants were harvested 24 hours later. The IFNγ concentration in this media was determined by enzyme-linked immunosorbent assay (Invitrogen). IL-18 bioactivity was determined based on the difference in IFNγ levels between cultures with and those without mouse anti–IL-18 monoclonal antibody.
Statistical analysis
Student’s t-tests were performed to calculate statistical differences between the means of the different variables. Two-tailed P values less than 0.05 were considered significant.
RESULTS
Time-dependent TNFα-induced IRF-1 in RASFs
To examine the effect of TNFα (20 ng/ml) on IRF-1 production, we investigated its effects at the mRNA and protein levels. TNFα stimulation of RASFs was performed for various amounts of time, up to 48 hours. IRF-1 was induced by TNFα at the mRNA level, with a maximal effect at 2 hours (P < 0.05; n ≥ 3) (Figure 1A). Similar results were observed at the protein level (P < 0.05; n ≥ 6) (Figure 1B). The results suggested that TNFα induced IRF-1, with a significant effect observed after 2 hours of stimulation at the transcription and protein levels.
Figure 1.

Up-regulation of interferon regulatory factor 1 (IRF-1) in rheumatoid arthritis synovial fibroblasts (RASFs) exposed to tumor necrosis factor α (TNFα). RASFs (2 × 105/well; 2 ml/well) were stimulated with TNFα in serum-free media for 0–48 hours. IRF-1 expression was assessed at the mRNA level (A) and at the protein level (B). Bars show the mean ± SEM. * = P < 0.05. NS = nonstimulated. n = number of donors and independent experiments.
MAPK and NF-κB signaling pathway–independence of TNFα-induced IRF-1 expression by RASFs
To identify the signaling events that are critical for TNFα-induced IRF-1 expression, RASFs were incubated with chemical signaling inhibitors for 1 hour, followed by stimulation with TNFα (20 ng/ml) for 2 hours. The results of this study showed that none of the inhibitors (NF-κB [PDTC], ERK-1/2 [PD98059], p38 [SB202190], or JNK-2 [SP600125]) significantly inhibited TNFα-induced IRF-1 expression at either the transcription level (Figure 2A) or the protein level (Figure 2B). These results indicated that TNFα induction of IRF-1 in RASFs is not regulated by the MAPK pathway or the NF-κB pathway.
Figure 2.

No effect of signaling inhibition on TNFα-induced IRF-1 levels in RASFs. RASFs were preincubated with different inhibitors for 1 hour, followed by stimulation with TNFα for 2 hours. IRF-1 expression was assessed at the mRNA level (A) and at the protein level (B). Bars show the mean ± SEM. PDTC = pyrrolidine dithiocarbamate (see Figure 1 for other definitions).
Time-dependent TNFα-induced IRF-1 nuclear translocation in RASFs
Because we hypothesized that IRF-1 is the key regulator of TNFα-induced IL-18BP in RASFs, we investigated IRF-1 nuclear translocation induced by TNFα (20 ng/ml) at 2, 4, and 8 hours. IRF-1 in the nuclear and cytoplasmic lysates was detected by Western blotting after TNFα stimulation. IRF-1 nuclear translocation was induced by TNFα at 2 hours, 4 hours, and 8 hours (6.7-, 5.3-, and 5.4-fold compared with unstimulated controls) (P < 0.05; n = 4) (Figure 3A). After successive stripping, the purity of the nuclear and cytoplasmic fractions was confirmed by probing for anti–histone H3 and anti–β-actin, respectively. We also confirmed that an equal amount of protein was loaded in each fraction. We then confirmed the effect of TNFα on IRF-1 nuclear translocation using dual confocal immunofluorescence microscopy with anti–IRF-1 antibody and DAPI staining. Without stimulation, IRF-1 was predominantly detected in the cytoplasm of RASFs (Figure 3B). After 2 hours of TNFα stimulation, IRF-1 was observed predominantly in the nucleus (Figure 3B), confirming the results observed using nuclear extracts and Western blots.
Figure 3.
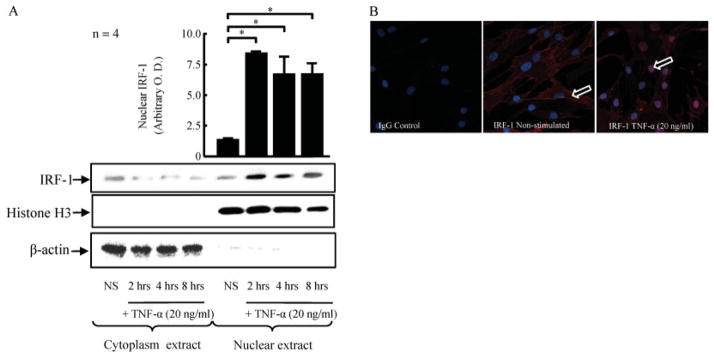
Nuclear translocation of IRF-1 in RASFs exposed to TNFα. A, RASFs (2 × 106/well) were stimulated with TNFα for 2, 4, or 8 hours in serum-free media. The cytoplasmic and nuclear fractions were prepared as described in Materials and Methods. Blots were probed successively for IRF-1, histone H3, and β-actin. Bars show the mean ± SEM. * = P < 0.05. B, Localization of IgG control and IRF-1 on RASFs with or without stimulation with TNFα (20 ng/ml for 2 hours) was examined by immunofluorescence and confocal microscopy (60× objective and 0.5-μm optical sections). Arrows indicate IRF-1, which was predominantly expressed in the cytoplasm without TNFα stimulation (middle) and was observed in the nucleus after 2 hours of TNFα stimulation (right). The images are representative of 4 independent experiments. OD = optical density (see Figure 1 for other definitions).
Roles of the JNK-2 and NF-κB, but not the ERK-1/2 or p38, signaling pathways in TNFα-induced IRF-1 nuclear translocation
Because we previously observed that NF-κB and JNK-2 are critical signaling pathways for TNFα-induced expression of IL-18BP (20), we investigated the effect of blocking these signaling pathways on TNFα-induced IRF-1 nuclear translocation. RASFs were incubated for 1 hour with chemical inhibitors of signaling, followed by stimulation with TNFα (20 ng/ml) for 2 hours. The inhibitors of the NF-κB (PDTC) and JNK-2 (SP600125) signaling pathways significantly (P < 0.05) repressed TNFα-induced IRF-1 nuclear translocation in RASFs (by 48% and 57%, respectively, versus no inhibition) (Figure 4A). In contrast, the inhibitors of ERK-1/2 (PD98059) and p38 (SB202190) did not significantly inhibit TNFα-induced IRF-1 nuclear translocation. These results indicated that induction of IRF-1 in RASFs by TNFα occurs via the NF-κB and JNK-2 signaling pathways, but not via the ERK-1/2 pathway or the p38 signaling pathway.
Figure 4.
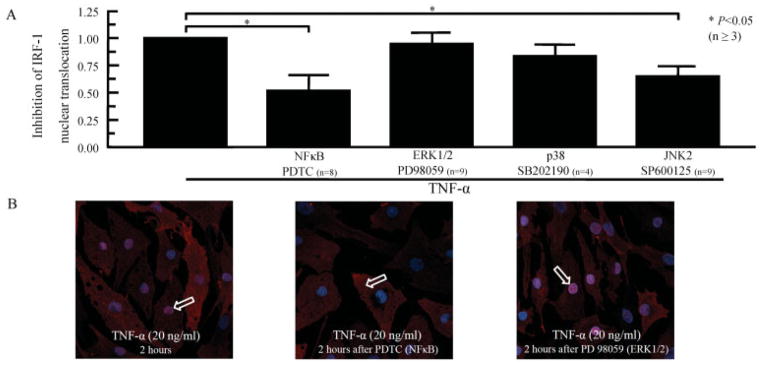
TNFα-induced IRF-1 nuclear translocation. A, RASFs were preincubated for 2 hours with pyrrolidine dithiocarbamate (PDTC; 200 μM), PD98059 (10 μM), SB202190 (10 μM), or SP600125 (10 μM) and then stimulated for 2 hours with TNFα (20 ng/ml). IRF-1 nuclear translocation was determined by Western blotting, using the nuclear extract from RASFs. Bars show the mean ± SEM. B, RASFs were preincubated for 2 hours with PDTC (200 μM) or PD98059 (10 μM) and then stimulated with TNFα (20 ng/ml) for 2 hours. Localization of IRF-1 on RASFs with or without TNFα stimulation was examined using immunofluorescence and confocal microscopy. After 2 hours of stimulation with TNFα, IRF-1 (arrows) was expressed in the nucleus (left). After preincubation with PDTC, IRF-1 expression was more pronounced in the cytoplasm (middle), whereas preincubation with PD98059 did not affect the location of IRF-1 in the nucleus. The images are representative of 4 independent experiments. See Figure 1 for other definitions.
We then confirmed the key role of the NF-κB signaling pathway by using confocal immunofluorescence microscopy. With blocking of the NF-κB signaling pathway, TNFα-induced expression of IRF-1 remained predominantly in the cytoplasm (Figure 4B). Furthermore, with blocking of the ERK-1/2 signaling pathway, TNFα-induced IRF-1 was preferentially expressed in the nucleus (Figure 4B), with expression similar to that observed following TNFα stimulation alone (Figure 4B). This confirmed that the NF-κB signaling pathway is required for TNFα-induced IRF-1 nuclear translocation.
Role of IRF-1 in forming a complex with NF-κB and c-Jun in the nucleus
To investigate whether IRF-1 forms a complex in the nucleus, we performed immunoprecipitation with antibodies against phosphorylated NF-κB p65 or phosphorylated c-Jun, with or without stimulation with TNFα (20 ng/ml) for 2 hours. After immunoprecipitation for phosphorylated NF-κB p65, we observed a low level of IRF-1 without stimulation, with an increase of at least 20-fold in the presence of TNFα (Figure 5). After immunoprecipitation for phosphorylated c-Jun, we observed a low level of IRF-1 without stimulation, with an increase of at least 6-fold in the presence of TNFα (Figure 5). After stripping was performed, we confirmed the specificity of immunoprecipitation by reprobing the blots with the antibodies used for immunoprecipitation. These results suggested that IRF-1 binds to phosphorylated NF-κB p65 and phosphorylated c-Jun in the nucleus.
Figure 5.

IRF-1 binds to phosphorylated NF-κB p65 and phosphorylated c-Jun in the nucleus after TNFα stimulation. Equalized (0.75 mg) protein from the RASF nuclear fraction, with or without TNFα stimulation (20 ng/ml for 2 hours), was immunoprecipitated with anti–phosphorylated NF-κB p65 or anti–phosphorylated c-Jun antibodies, and Western blotting was performed using an anti–IRF-1 antibody. After stripping, the blots were probed for phosphorylated NF-κB p65 or phosphorylated c-Jun. The results shown are representative of 2–4 assays. Bars show the mean ± SEM. * = P < 0.05. OD = optical density (see Figure 1 for other definitions).
Role of blocking IRF-1 in reducing IL-18BPa expression and increasing TNFα-induced IL-18 bioactivity in RASFs
To investigate the direct effect of IRF-1 on IL-18BPa production, we transfected RASFs with control or IRF-1 siRNA for 48 hours, followed by TNFα stimulation. We first checked the knockdown efficacy by assessing IRF-1 at the protein level after 2 hours of TNFα induction. Compared with control siRNA, IRF-1 siRNA was reduced by ~80% at the protein level (P < 0.05; n = 3) (Figure 6A). We then assessed IL-18BPa expression at the protein level after 48 hours of TNFα stimulation, in the presence or absence of IRF-1 knockdown. We observed that blocking of IRF-1 reduced IL-18BPa by 70% at the mRNA and protein levels (P < 0.05; n = 6) (Figure 6B). When TNFα-induced IL-18 expression was assessed at the mRNA level, we observed that blocking of IRF-1 had no effect on its expression (Figure 6C). We then assessed IL-18 bioactivity in the conditioned medium using KG-1 cells. In cells transfected with IRF-1 siRNA, TNFα induced almost 8-fold higher levels of IL-18 bioactivity compared with control siRNA (P < 0.05; n = 6) (Figure 6C). These results indicated a crucial role of IRF-1 in regulating TNFα-induced IL-18 bioactivity.
Figure 6.
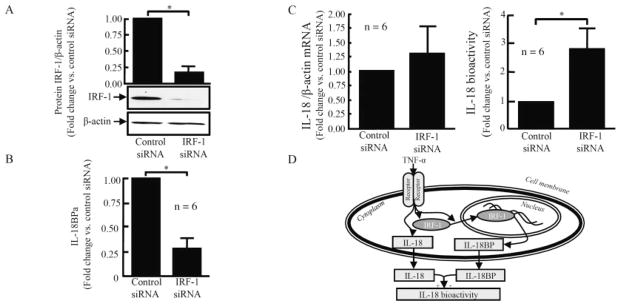
Blocking of IRF-1 regulates TNFα-induced interleukin-18 (IL-18) bioactivity by reducing IL-18 binding protein a (IL-18BPa). RASFs were transfected with control or IRF-1 small interfering RNA (siRNA) for 48 hours, using TransIT-TKO reagent. The transfected RASFs were then stimulated with TNFα (20 ng/ml). A, After 2 hours of TNFα stimulation, the transfection efficiency was checked at the protein level. Results are representative of 3 assays. B, After 48 hours of TNFα stimulation, the amount of IL-18BPa was assessed at the protein level. C, IL-18 was assessed at the mRNA level by real-time quantitative polymerase chain reaction, and its bioactivity was assessed using KG-1 cells. Bars show the mean ± SEM. * = P < 0.05. D, Schematic representation of the mechanism of TNFα induction of IL-18 and IL-18BPa through IRF-1. TNFα induces IL-18BPa through IRF-1 nuclear translocation and regulates IL-18 bioactivity. See Figure 1 for other definitions.
Summary of the mechanism of TNFα induction of IL-18 and IL-18BPa through IRF-1 nuclear translocation
The results from this study suggested that TNFα induces IL-18BP through IRF-1 nuclear translocation. Knocking down IRF-1 reduced this induction and changed the balance between IL-18 and IL-18BPa by increasing IL-18 bioactivity in RASFs (Figure 6D).
DISCUSSION
Recently, we showed that TNFα induced IL-18, IL-18BPa, and IL-18 bioactivity in RASFs (20). In addition, blocking of the ERK-1/2 signaling pathway decreased IL-18 bioactivity by blocking TNFα-induced IL-18 expression, without an effect on IL-18BPa expression (20). Checking critical pathways for TNFα-induced IL-18BPa expression showed that the NF-κB and JNK-2 signaling pathways were relevant without affecting IL-18 expression (20), and blocking of IL-18BPa expression would allow an increase in IL-18 bioactivity. IFNγ has been described to induce IRF-1 expression and bind to the IL-18BP promoter (25). Because IFNγ levels are low in the RA joint (33), and based on these previous observations, we hypothesized that IRF-1 is a key regulator of TNFα-induced IL-18 bioactivity in RASFs by regulating IL-18BPa.
In 1989, IRF-1 was described to be induced by type I IFN and to induce IFN-inducible genes (24). IRF-1 was then shown to mediate IL-18BP expression induced by IFNγ (25,27). Therefore, according to our hypothesis, we first demonstrated that TNFα induced IRF-1 at the transcription and protein levels. Only a few other cytokines besides IFNγ have been reported to induce IRF-1 expression. IL-12 and IL-18, alone or in combination, were reported to induce IRF-1 in mouse splenocytes (26). Recently, TNFα was described to induce IL-32 through IRF-1 (34). TNFα was also recently described to induce IRF-1 expression in human umbilical vein endothelial cells (HUVECs) (35), whereas IL-1β or TNFα had no effect on IRF-1 expression in human keratinocytes (36). Furthermore, lipopolysaccharide was also reported to induce IRF-1 expression in murine macrophages, using a microarray approach (37). The time course of TNFα-induced IRF-1 expression is in accordance with the time course previously described in murine L929 cells (24), murine macrophages (37), human keratinocytes (36), or HUVECs (35). The discrepancy between the time course of mRNA and protein expression could be explained, in part, by posttranscription and translation regulation, as well as the short IRF-1 half-life of 30 minutes (38), which suggests no accumulation of IRF-1 at the protein level.
The second step was to explore how TNFα also induced IRF-1 nuclear translocation. We observed that IRF-1 nuclear translocation was maximal after 2 hours of TNFα stimulation. Based on our hypothesis, we investigated the effect of blocking of the NF-κB or MAPK signaling pathways on TNFα-induced IRF-1 nuclear translocation. After blocking the NF-κB or JNK-2 signaling pathway, we observed a reduction of IRF-1 nuclear translocation, suggesting that both the NF-κB and JNK-2 signaling pathways are required for nuclear translocation, whereas the p38 and ERK-1/2 signaling pathways are not required. Because we previously showed that the NF-κB and/or JNK-2 signaling pathways are critical for TNFα-induced IL-18BPa expression (20), our data provided indirect evidence that IRF-1 is associated with IL-18BP production.
We then demonstrated at the mRNA and protein levels that TNFα-induced IRF-1 expression was not affected by blocking the NF-κB or JNK-2 signaling pathway. Thus, our results suggested that the NF-κB and/or JNK-2 signaling pathways are required for TNFα-induced IRF-1 nuclear translocation without an effect on its expression. Minimal data are available on signaling pathways regulating IRF-1 expression and/or its nuclear translocation. Furthermore, in human keratinocytes, blocking of the p38 signaling pathway was described to reduce IFNγ-induced IRF-1 by half after 2 hours of stimulation (36). Blocking of the JAK/STAT-3 signaling pathway reduced TNFα-induced IRF-1 expression in HUVECs (35).
IRF-1 and other members of the IRF family have been reported to form dimers among themselves and form complexes with other transcription factors, including members of the NF-κB signaling pathway (39). By using immunoprecipitation, we established that IRF-1 was associated with NF-κB and c-Jun in the nucleus, confirming the presence of a complex combining IRF-1 with NF-κB or c-Jun. This is consistent with a previous report that NF-κB can physically interact with IRF-1 (40) and can influence its binding to the basal transcription complex. Previously, the function of IRF-1 has been shown to be associated with NF-κB p65 (41). Our data confirmed that IRF-1 interacts with NF-κB for nuclear translocation in RASFs after TNFα stimulation. However, the nuclear complex between IRF-1 and c-Jun has not been described previously.
The next step was to use a functional assay to confirm the role of IRF-1 in TNFα-induced IL-18BP. Our hypothesis was based on the fact that IL-18BP was initially described as a key regulator of the Th1 immune response. IL-18, as an important mediator of Th1 cell differentiation, induces IFNγ production.
The biologic activities of cytokines are frequently controlled by endogenous inhibitory proteins. IL-18 is under the control of IL-18BP, and the result is IL-18 bioactivity. Thus, a negative feedback loop was described, in which IL-18BP is the key mediator regulating IL-18 bioactivity (42–45). However, to avoid premature termination of IL-18 activity, induction of IL-18BP must occur after a delay. Usually, IFNγ signals quickly through the JAK/STAT signaling pathway (25). However, for this feedback loop and IL-18BP expression, IFNγ required de novo synthesis of the transcription factor IRF-1, which activates the IL-18BP promoter (25). Because IRF-1 can bind to the IL-18BP promoter and activate its transcription (40,41), we explored the effect of IRF-1 knockdown on TNFα-induced IL-18BP expression in RASFs. We employed an approach using siRNA to knock down IRF-1 expression. After confirming the reduction in IRF-1 at the protein level in the presence of IRF-1 siRNA, we observed a reduction in TNFα-induced IL-18BPa at the protein level.
The second component of IL-18 bioactivity is IL-18 itself. Because IL-18 levels are similar in the presence or absence of the knockdown of IRF-1, IL-18 bioactivity was increased in the case of IRF-1 knockdown. A discrepancy between antigenic protein and functional bioactivity for TNFα has been reported previously (46). This study is the first to show that IRF-1 is a mediator of TNFα-induced IL-18BPa. This concept was previously described in various animal models in vivo. Indeed, IRF-1–knockout mice have reduced or undetectable expression of IL-18BP (25–27). In the same mice, IL-18 expression was unaffected (27), suggesting an increase in IL-18 bioactivity. However, the IL-18 bioactivity in vivo was not examined in those studies.
In this study, we showed that IRF-1 is a key regulator of IL-18BPa expression in RASFs and can thus regulate IL-18 bioactivity. Hence, strategies to augment IRF-1 activity may be beneficial for the treatment of RA.
Acknowledgments
Dr. Marotte’s work was supported by the Bettencourt-Schueller Foundation and the Philippe Foundation. Dr. Rabquer’s work was supported by the NIH (grant HL-094017). Dr. Koch’s work was supported by the NIH (grants AI-40987 and AR-48267), the Frederick G. L. Huetwell and William D. Robinson, MD, Professorship in Rheumatology, and the Office of Research and Development, Medical Research Service, Department of Veterans Affairs.
We thank the National Disease Research Interchange for providing RA synovial tissue and Dr. K. W. Janczak for providing the KG-1 cell line. The confocal microscopy work was performed in the Microscopy & Image Analysis Laboratory (MIL) at the University of Michigan, Department of Cell and Developmental Biology, with the assistance of Shelley Almburg. The MIL is a multiuser imaging facility supported by the National Institutes of Health–National Cancer Institute, the O’Brien Renal Center, the University of Michigan Medical School, an Endowment for the Basic Sciences, Department of Cell and Developmental Biology, and the University of Michigan.
Footnotes
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Koch had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Marotte, Koch.
Acquisition of data. Marotte, Tsou, Rabquer, Pinney, Fedorova, Lalwani.
Analysis and interpretation of data. Marotte.
References
- 1.Gracie JA, Forsey RJ, Chan WL, Gilmour A, Leung BP, Greer MR, et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest. 1999;104:1393–401. doi: 10.1172/JCI7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 3.Bazan JF, Timans JC, Kastelein RA. A newly defined interleukin-1? Nature. 1996;379:591. doi: 10.1038/379591a0. [DOI] [PubMed] [Google Scholar]
- 4.Ushio S, Namba M, Okura T, Hattori K, Nukada Y, Akita K, et al. Cloning of the cDNA for human IFN-γ-inducing factor, expression in Escherichia coli, and studies on the biologic activities of the protein. J Immunol. 1996;156:4274–9. [PubMed] [Google Scholar]
- 5.Okamura H, Nagata K, Komatsu T, Tanimoto T, Nukata Y, Tanabe F, et al. A novel costimulatory factor for γ interferon induction found in the livers of mice causes endotoxic shock. Infect Immun. 1995;63:3966–72. doi: 10.1128/iai.63.10.3966-3972.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohno K, Kataoka J, Ohtsuki T, Suemoto Y, Okamoto I, Usui M, et al. IFN-γ-inducing factor (IGIF) is a costimulatory factor on the activation of Th1 but not Th2 cells and exerts its effect independently of IL-12. J Immunol. 1997;158:1541–50. [PubMed] [Google Scholar]
- 7.Micallef MJ, Ohtsuki T, Kohno K, Tanabe F, Ushio S, Namba M, et al. Interferon-γ-inducing factor enhances T helper 1 cytokine production by stimulated human T cells: synergism with interleukin-12 for interferon-γ production. Eur J Immunol. 1996;26:1647–51. doi: 10.1002/eji.1830260736. [DOI] [PubMed] [Google Scholar]
- 8.Tsutsui H, Nakanishi K, Matsui K, Higashino K, Okamura H, Miyazawa Y, et al. IFN-γ-inducing factor up-regulates Fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J Immunol. 1996;157:3967–73. [PubMed] [Google Scholar]
- 9.Tsutsui H, Matsui K, Kawada N, Hyodo Y, Hayashi N, Okamura H, et al. IL-18 accounts for both TNF-α- and Fas ligand-mediated hepatotoxic pathways in endotoxin-induced liver injury in mice. J Immunol. 1997;159:3961–7. [PubMed] [Google Scholar]
- 10.Morel JC, Park CC, Woods JM, Koch AE. A novel role for interleukin-18 in adhesion molecule induction through NF κB and phosphatidylinositol (PI) 3-kinase-dependent signal transduction pathways. J Biol Chem. 2001;276:37069–75. doi: 10.1074/jbc.M103574200. [DOI] [PubMed] [Google Scholar]
- 11.Amin MA, Mansfield PJ, Pakozdi A, Campbell PL, Ahmed S, Martinez RJ, et al. Interleukin-18 induces angiogenic factors in rheumatoid arthritis synovial tissue fibroblasts via distinct signaling pathways. Arthritis Rheum. 2007;56:1787–97. doi: 10.1002/art.22705. [DOI] [PubMed] [Google Scholar]
- 12.Park CC, Morel JC, Amin MA, Connors MA, Harlow LA, Koch AE. Evidence of IL-18 as a novel angiogenic mediator. J Immunol. 2001;167:1644–53. doi: 10.4049/jimmunol.167.3.1644. [DOI] [PubMed] [Google Scholar]
- 13.Morel JC, Park CC, Kumar P, Koch AE. Interleukin-18 induces rheumatoid arthritis synovial fibroblast CXC chemokine production through NFκB activation. Lab Invest. 2001;81:1371–83. doi: 10.1038/labinvest.3780351. [DOI] [PubMed] [Google Scholar]
- 14.Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of interferon-γ inducing factor mediated by interleukin-1β converting enzyme. Science. 1997;275:206–9. doi: 10.1126/science.275.5297.206. [DOI] [PubMed] [Google Scholar]
- 15.Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-γ-inducing factor and regulates LPS-induced IFN-γ production. Nature. 1997;386:619–23. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 16.Yamamura M, Kawashima M, Taniai M, Yamauchi H, Tanimoto T, Kurimoto M, et al. Interferon-γ–inducing activity of interleukin-18 in the joint with rheumatoid arthritis. Arthritis Rheum. 2001;44:275–85. doi: 10.1002/1529-0131(200102)44:2<275::AID-ANR44>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 17.Brossart P, Grunebach F, Stuhler G, Reichardt VL, Mohle R, Kanz L, et al. Generation of functional human dendritic cells from adherent peripheral blood monocytes by CD40 ligation in the absence of granulocyte-macrophage colony-stimulating factor. Blood. 1998;92:4238–47. [PubMed] [Google Scholar]
- 18.Olee T, Hashimoto S, Quach J, Lotz M. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J Immunol. 1999;162:1096–100. [PubMed] [Google Scholar]
- 19.Udagawa N, Horwood NJ, Elliott J, Mackay A, Owens J, Okamura H, et al. Interleukin-18 (interferon-γ-inducing factor) is produced by osteoblasts and acts via granulocyte/macrophage colony-stimulating factor and not via interferon-γ to inhibit osteoclast formation. J Exp Med. 1997;185:1005–12. doi: 10.1084/jem.185.6.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marotte H, Ahmed S, Ruth JH, Koch AE. Blocking ERK-1/2 reduces tumor necrosis factor α–induced interleukin-18 bioactivity in rheumatoid arthritis synovial fibroblasts by induction of interleukin-18 binding protein A. Arthritis Rheum. 2010;62:722–31. doi: 10.1002/art.27269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fantuzzi G, Reed DA, Dinarello CA. IL-12-induced IFN-γ is dependent on caspase-1 processing of the IL-18 precursor. J Clin Invest. 1999;104:761–7. doi: 10.1172/JCI7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1β are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci U S A. 1999;96:2256–61. doi: 10.1073/pnas.96.5.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999;10:127–36. doi: 10.1016/s1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- 24.Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, et al. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell. 1989;58:729–39. doi: 10.1016/0092-8674(89)90107-4. [DOI] [PubMed] [Google Scholar]
- 25.Hurgin V, Novick D, Rubinstein M. The promoter of IL-18 binding protein: activation by an IFN-γ-induced complex of IFN regulatory factor 1 and CCAAT/enhancer binding protein β. Proc Natl Acad Sci U S A. 2002;99:16957–62. doi: 10.1073/pnas.262663399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fantuzzi G, Reed D, Qi M, Scully S, Dinarello CA, Senaldi G. Role of interferon regulatory factor-1 in the regulation of IL-18 production and activity. Eur J Immunol. 2001;31:369–75. doi: 10.1002/1521-4141(200102)31:2<369::aid-immu369>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 27.Siegmund B, Sennello JA, Lehr HA, Senaldi G, Dinarello CA, Fantuzzi G. Frontline: interferon regulatory factor-1 as a protective gene in intestinal inflammation: role of TCR γδ T cells and interleukin-18-binding protein. Eur J Immunol. 2004;34:2356–64. doi: 10.1002/eji.200425124. [DOI] [PubMed] [Google Scholar]
- 28.Ahmed S, Marotte H, Kwan K, Ruth JH, Campbell PL, Rabquer BJ, et al. Epigallocatechin-3-gallate inhibits IL-6 synthesis and suppresses transsignaling by enhancing soluble gp130 production. Proc Natl Acad Sci U S A. 2008;105:14692–7. doi: 10.1073/pnas.0802675105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmed S, Silverman MD, Marotte H, Kwan K, Matuszczak N, Koch AE. Down-regulation of myeloid cell leukemia 1 by epigallocatechin-3-gallate sensitizes rheumatoid arthritis synovial fibroblasts to tumor necrosis factor α–induced apoptosis. Arthritis Rheum. 2009;60:1282–93. doi: 10.1002/art.24488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang MC, Lin TL, Lee TL, Huang HT, Lin CL, Liao CF. IRF-1-mediated CAS expression enhances interferon-γ-induced apoptosis of HT-29 colon adenocarcinoma cells. Mol Cell Biol Res Commun. 2001;4:353–8. doi: 10.1006/mcbr.2001.0303. [DOI] [PubMed] [Google Scholar]
- 31.Ahmed S, Pakozdi A, Koch AE. Regulation of interleukin-1β–induced chemokine production and matrix metalloproteinase 2 activation by epigallocatechin-3-gallate in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2006;54:2393–401. doi: 10.1002/art.22023. [DOI] [PubMed] [Google Scholar]
- 32.Morel J, Audo R, Hahne M, Combe B. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces rheumatoid arthritis synovial fibroblast proliferation through mitogen-activated protein kinases and phosphatidylinositol 3-kinase/Akt. J Biol Chem. 2005;280:15709–18. doi: 10.1074/jbc.M414469200. [DOI] [PubMed] [Google Scholar]
- 33.Bas S, Kvien TK, Buchs N, Fulpius T, Gabay C. Lower level of synovial fluid interferon-γ in HLA-B27-positive than in HLA-B27-negative patients with Chlamydia trachomatis reactive arthritis. Rheumatology (Oxford) 2003;42:461–7. doi: 10.1093/rheumatology/keg163. [DOI] [PubMed] [Google Scholar]
- 34.Alsaleh G, Sparsa L, Chatelus E, Ehlinger M, Gottenberg JE, Wachsmann D, et al. Innate immunity triggers IL-32 expression by fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther. 2010;12:R135. doi: 10.1186/ar3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nizamutdinova IT, Kim YM, Chung JI, Shin SC, Jeong YK, Seo HG, et al. Anthocyanins from black soybean seed coats preferentially inhibit TNF-α-mediated induction of VCAM-1 over ICAM-1 through the regulation of GATAs and IRF-1. J Agric Food Chem. 2009;57:7324–30. doi: 10.1021/jf900856z. [DOI] [PubMed] [Google Scholar]
- 36.Kanda N, Shimizu T, Tada Y, Watanabe S. IL-18 enhances IFN-γ-induced production of CXCL9, CXCL10, and CXCL11 in human keratinocytes. Eur J Immunol. 2007;37:338–50. doi: 10.1002/eji.200636420. [DOI] [PubMed] [Google Scholar]
- 37.Scott MG, Rosenberger CM, Gold MR, Finlay BB, Hancock RE. An α-helical cationic antimicrobial peptide selectively modulates macrophage responses to lipopolysaccharide and directly alters macrophage gene expression. J Immunol. 2000;165:3358–65. doi: 10.4049/jimmunol.165.6.3358. [DOI] [PubMed] [Google Scholar]
- 38.Oshima S, Nakamura T, Namiki S, Okada E, Tsuchiya K, Okamoto R, et al. Interferon regulatory factor 1 (IRF-1) and IRF-2 distinctively up-regulate gene expression and production of interleukin-7 in human intestinal epithelial cells. Mol Cell Biol. 2004;24:6298–310. doi: 10.1128/MCB.24.14.6298-6310.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kroger A, Koster M, Schroeder K, Hauser H, Mueller PP. Activities of IRF-1. J Interferon Cytokine Res. 2002;22:5–14. doi: 10.1089/107999002753452610. [DOI] [PubMed] [Google Scholar]
- 40.Drew PD, Franzoso G, Becker KG, Bours V, Carlson LM, Siebenlist U, et al. NF κB and interferon regulatory factor 1 physically interact and synergistically induce major histocompatibility class I gene expression. J Interferon Cytokine Res. 1995;15:1037–45. doi: 10.1089/jir.1995.15.1037. [DOI] [PubMed] [Google Scholar]
- 41.Faggioli L, Merola M, Hiscott J, Furia A, Monese R, Tovey M, et al. Molecular mechanisms regulating induction of interleukin-6 gene transcription by interferon-γ. Eur J Immunol. 1997;27:3022–30. doi: 10.1002/eji.1830271140. [DOI] [PubMed] [Google Scholar]
- 42.Muhl H, Kampfer H, Bosmann M, Frank S, Radeke H, Pfeilschifter J. Interferon-γ mediates gene expression of IL-18 binding protein in nonleukocytic cells. Biochem Biophys Res Commun. 2000;267:960–3. doi: 10.1006/bbrc.1999.2064. [DOI] [PubMed] [Google Scholar]
- 43.Stoll S, Muller G, Kurimoto M, Saloga J, Tanimoto T, Yamauchi H, et al. Production of IL-18 (IFN-γ-inducing factor) messenger RNA and functional protein by murine keratinocytes. J Immunol. 1997;159:298–302. [PubMed] [Google Scholar]
- 44.Kampfer H, Kalina U, Muhl H, Pfeilschifter J, Frank S. Counter-regulation of interleukin-18 mRNA and protein expression during cutaneous wound repair in mice. J Invest Dermatol. 1999;113:369–74. doi: 10.1046/j.1523-1747.1999.00704.x. [DOI] [PubMed] [Google Scholar]
- 45.Pages F, Berger A, Henglein B, Piqueras B, Danel C, Zinzindohoue F, et al. Modulation of interleukin-18 expression in human colon carcinoma: consequences for tumor immune surveillance. Int J Cancer. 1999;84:326–30. doi: 10.1002/(sici)1097-0215(19990621)84:3<326::aid-ijc22>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 46.Marotte H, Maslinski W, Miossec P. Circulating tumour necrosis factor-α bioactivity in rheumatoid arthritis patients treated with infliximab: link to clinical response. Arthritis Res Ther. 2005;7:R149–55. doi: 10.1186/ar1465. [DOI] [PMC free article] [PubMed] [Google Scholar]