

Abstract
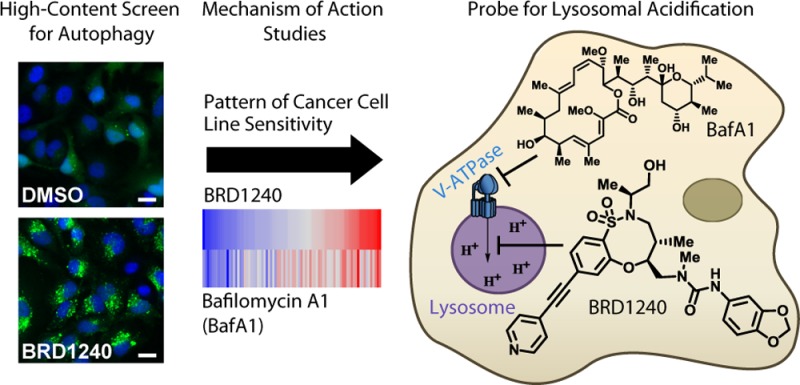
Lysosomes perform a critical cellular function as a site of degradation for diverse cargoes including proteins, organelles, and pathogens delivered through distinct pathways, and defects in lysosomal function have been implicated in a number of diseases. Recent studies have elucidated roles for the lysosome in the regulation of protein synthesis, metabolism, membrane integrity, and other processes involved in homeostasis. Complex small-molecule natural products have greatly contributed to the investigation of lysosomal function in cellular physiology. Here we report the discovery of a novel, small-molecule modulator of lysosomal acidification derived from diversity-oriented synthesis through high-content screening.
Introduction
Studies of human genetics and physiology have implicated autophagy in several inflammatory, neurodegenerative, infectious, and autoimmune diseases, revealing the importance of cellular homeostasis in human disease and motivating the discovery of small-molecule probes to investigate the different stages of this complex pathway.1−3 Lysosomes are the site of degradation and recycling in eukaryotic cells for macromolecules, organelles, and pathogens engulfed through autophagy, endocytosis, and phagocytosis. Degradation within the lysosome is facilitated by lysosomal hydrolases, including proteases, peptidases, phosphatases, nucleases, glycosidases, and lipases, and products are released by diffusion or carrier-mediated transporters for reuse by cells.4 In some cell types, lysosomes can also deliver cargo to pathogen recognition receptors (PRRs), or preserve degraded cargo for antigen presentation.5 The acidic pH in the lysosomal lumen (pH = 4.5–5.0), which is required for optimal hydrolase activity, is generated and maintained by the vacuolar-type H+-ATPase (V-ATPase), a protein complex on lysosomal membranes that hydrolyzes ATP to drive protons into lysosomes.6 Mutations in various subunits of the V-ATPase complex have been linked to osteopetrosis,7,8 x-linked myopathy,9 distal renal tubular acidosis, sensorineural deafness,10,11 and pulmonary tuberculosis,12 and the complex has been studied as a potential dependency of certain cancer cells.6,13,14
Beyond its degradative function, recent studies have identified the lysosome as a critical component of various signaling pathways. For example, amino acids in the lysosomal lumen promote the recruitment of the mechanistic target of rapamycin (mTOR) to the lysosomal membrane and the activation of mTOR signaling in a V-ATPase- and ragulator-dependent manner,15,16 leading to enhancement of cell growth and protein synthesis and inhibition of autophagy.17 mTOR also regulates TFEB, a transcription factor that promotes expression of genes required for the biogenesis of lysosomes and activation of the endolysosomal system and autophagic catabolism.18,19 Lysosomes and lysosomal proteins, such as Niemann-Pick disease C1 (NPC1) and NPC2, additionally maintain cholesterol homeostasis by controlling cholesterol efflux from the lysosomal lumen.20,21 The lysosome is also involved in exocytosis to promote intercellular signaling and plasma membrane repair through fusion with the plasma membrane to restore membrane integrity.22
The study of lysosomes has been greatly enabled by the discovery of small-molecule probes that perturb lysosomal function through distinct mechanisms, including direct inhibition of lysosomal proteases, inhibition of the V-ATPase, extrusion and degradation of enzymes from the lysosomal membrane, or perturbation of lysosomal pH through protonation and accumulation in lysosomes.23,24 Many of these modulators are derived from natural sources, including the protease inhibitors leupeptin, pepstatin A, and E64d, as well as several classes of V-ATPase modulators, including the plecomacrolides, bafilomycin A1 and concanamycin A; the macrolides, archazolid A and palmerolide A; and the benzolactone enamides, apicularen A and salicylihalamide A.25,26 Additional small molecules that perturb the lysosome may serve as useful tools to study its role in cellular physiology and human disease biology.
Diversity-oriented synthesis (DOS) aims to synthesize candidate probes and therapeutics having novel mechanisms of action not easily found in other sources of synthetic compounds. The short and modular synthetic pathways that result from the build/couple/pair (BCP) strategy, which mimics the strategy used in nature to synthesize natural products, ensure ease of chemical optimization of starting points found using screening. This chemistry has yielded compounds enriched for sp3-hybridized skeletal atoms and often results in all possible stereoisomers to maximize diversity of scaffold shape.27−30
Here we report the discovery of a novel small-molecule inhibitor of lysosomal acidification (BRD1240) through high-content screening of a DOS-derived compound collection. We identified BRD1240 on the basis of its ability to increase numbers of autophagosomes, as measured by GFP-LC3 punctae accumulation. Among screening hits, BRD1240 displayed a particularly striking dependence of activity on stereochemistry, suggesting a potentially selective interaction with a protein target. Subsequent experiments revealed that BRD1240 blocks the maturation of autophagosomes to autolysosomes, likely due to its ability to interfere with lysosomal acidification.
To study the mechanism of action of BRD1240, we measured the sensitivity of 83 cancer cell lines to BRD1240 and compared the resulting sensitivity profile to those of 480 other small molecules spanning a range of protein targets; the profile of BRD1240 correlated most strongly with that of bafilomycin A1, a potent, specific inhibitor of the V-ATPase. Biochemical assays confirmed that BRD1240 can suppress V-ATPase function, though with kinetics different than that of bafilomycin A1, suggesting it may operate through a different molecular mode of action. BRD1240 may serve as a probe to study how lysosomal acidification is regulated by the multisubunit molecular machine, V-ATPase, and how it affects cellular physiology.
Results and Discussion
BRD1240 Increases Autophagosome Number by Inhibiting Autophagosomal Turnover
We performed a high-throughput screen (HTS) of 59 541 stereochemically and skeletally diverse compounds derived from DOS for compounds that modulate autophagosome number. The primary HTS was conducted in HeLa cells stably expressing a GFP-LC3 fusion protein (Figure 1). LC3 normally displays a diffuse cytosolic pattern (LC3-I), but upon induction of autophagy, LC3 is cleaved and conjugated to phosphatidylethanolamine (LC3-II), and accumulates on autophagosome membranes. As such, the number of autophagosomes can be estimated by counting the number of GFP-LC3 “punctae/cell” by microscopy.31 Hits were selected on the basis of their ability to elicit statistically significant changes in the number of punctae/cell compared with the observed number in DMSO-treated wells. Significance was assessed by computing the area under the curve of the test distribution in compound-treated wells beyond the critical value corresponding to 95% confidence of the null distribution estimated from the observed number of “punctae/cell” in DMSO-treated wells.
Figure 1.
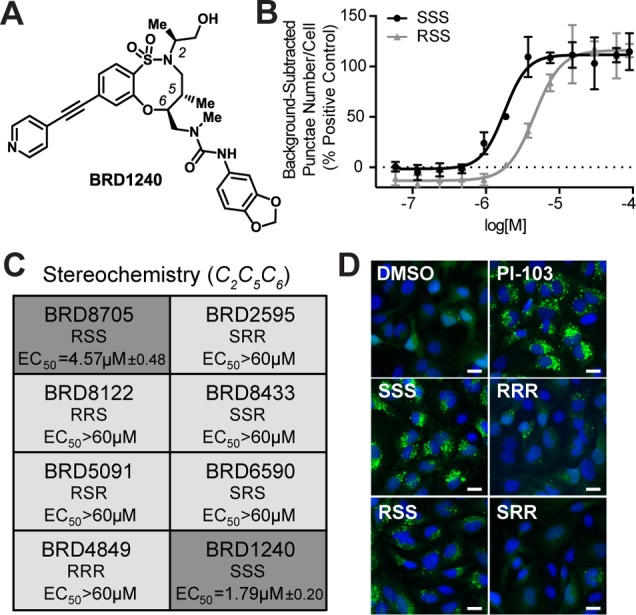
BRD1240 increases the GFP-LC3 punctae number, and this activity is dependent on stereochemistry. (A) Chemical structure of BRD1240. (B) Dose–response curves of BRD1240 (SSS) and BRD8705 (RSS) in the GFP-LC3 punctae formation assay. The positive control for increased punctae number is PI-103, a dual PI3K/mTOR inhibitor. Values are presented as the average ± SD of three independent experiments, each run in duplicate. (C) Dose–response data (EC50) for all stereoisomers in the GFP-LC3 punctae formation assay. Values are presented as average ± SEM. (D) Representative images from the GFP-LC3 punctae formation assay following treatment with DMSO; PI-103 (5 μM); BRD1240 and its enantiomer, BRD4849 (RRR); and BRD8705 (RSS) and its enantiomer, BRD2595 (SRR) (all 20 μM). Blue (Hoechst 33342), green (eGFP). Scalar bars represent 10 μm.
Among confirmed hits, BRD1240 was particularly potent and displayed a striking structure–activity relationship (SAR) with respect to the stereochemistry within the ring (Figure 1). Only two of the eight stereoisomers were active, the SSS and RSS diastereomers, with the sole difference being the configuration of the extra-annular methyl group; all other stereoisomers were inactive (Figure 1C,D and Supporting Information Figure S1). The dependence of BRD1240 activity on its stereochemistry suggests a small-molecule–protein interaction that is highly dependent on the three-dimensional structure of the molecule. Notably, the enantiomer of BRD1240 (BRD4849), which has the same physicochemical properties as BRD1240, does not cause an increase in GFP-LC3 punctae number, supporting a hypothesis that BRD1240 does not simply perturb lysosomal function by accumulating nonspecifically within the organelle as has been observed for other small molecules.23
The number of GFP-LC3 punctae in the cell can be increased by either activation of autophagy or inhibition of autophagy at a late step in the process, such as by disrupting lysosomal function. To test BRD1240 for these activities, we performed an assay for autophagosome formation and maturation (flux) that relies on visualizing an LC3 protein tagged with both mCherry and eGFP. The assay exploits the differential sensitivities of mCherry and eGFP to the acidic lysosomal environment to infer autophagic flux. The eGFP signal is attenuated in autolysosomes, while the mCherry signal remains stable;32 as a result, autophagosomes (eGFP+/mCherry+: yellow puncta) and autolysosomes (eGFP–/mCherry+: red puncta) can be distinguished and counted by high-throughput imaging. This assay was optimized using a known activator of autophagy, PI-103 (dual inhibitor of mTOR and PI3K), and a known inhibitor of lysosomal acidification, bafilomycin A1 (V-ATPase inhibitor). As expected, PI-103 robustly increased the number of autolysosomes (red puncta), while bafilomycin A1 (BafA1) increased the number of autophagosomes (yellow puncta) by blocking autophagic flux (Figure 2A). Similar to BafA1, BRD1240 and its active diastereomer (BRD8705) robustly inhibited autolysosome formation, causing a significant accumulation of autophagosomes (Figure 2A). These results suggest that BRD1240 blocks autophagosomal turnover. In further support of this hypothesis, we found that treatment with BRD1240, when cotreated with the lysosomal protease inhibitors, E64d and pepstatin A (pepA), caused no additional increase in LC3-II/LC3-I ratio by Western blot (Figure 2B).
Figure 2.

BRD1240 blocks the later stages of autophagy. (A) Representative images from the mCherry-eGFP-LC3 assay following treatment with DMSO, PI-103 (5 μM), BafA1 (100 nM), BRD1240 (10 μM), BRD8705 (10 μM), and BRD4849 (10 μM). Blue (Hoechst 33342), red (mCherry), green (eGFP). Scalar bars represent 10 μm. (B) Western blot for LC3-I to LC3-II shift in HeLa cells treated with BafA1, BRD1240, and BRD4849 ± 10 μg/mL E64d/pepA. (C) Normalized average fluorescence intensity of punctae per cell for BafA1 (100 nM), BRD1240 (SSS) (10 μM), and all seven stereoisomers (10 μM) in HeLa cells in the LysoTracker displacement assay. (D) Normalized average punctae number per cell for BafA1 (200 nM), chloroquine (CQ) (50 μM), E64d/pepA (10 μg/mL), BRD1240 (20 μM), and all seven stereoisomers (20 μM) in HeLa cells in the DQ-BSA assay. In parts C and D data are presented as the average ± SD of three independent experiments, each run in duplicate.
BRD1240 Modulates Lysosomal Function
To determine whether BRD1240 disrupts autophagic flux by perturbing lysosomal function, we first tested its effects on cellular staining with a pH-sensitive cationic fluorescent dye (LysoTracker Red) which accumulates in acidic cellular compartments.33 The fluorescent signal was greatly diminished after BRD1240 and BRD8705 treatment, suggesting that the compounds may inhibit lysosomal acidification (Figure 2C, Supporting Information Figure S2B). In contrast, none of the inactive stereoisomers was capable of diminishing the fluorescent LysoTracker signal. We then tested whether BRD1240 affected the ability of lysosomal proteases, which require an acidic environment for optimal activity, to process a fluorogenic substrate (DQ-BSA).34 As expected, treatment with compounds that inhibit lysosomal proteases directly (E64d/pepA) or indirectly by increasing lysosomal pH (BafA1 and chloroquine) significantly decreased both DQ-BSA punctae number and intensity (Figure 2D, Supporting Information Figure S2C). We found that treatment with BRD1240 and BRD8705 for 6 h also robustly decreased DQ-BSA punctae number and intensity, whereas the inactive stereoisomers had no activity in the assay. These data implicate the lysosome as a potential site of action of BRD1240.
BRD1240 Has Notable Structure–Activity Relationships
To understand better the structural elements that, in addition to the stereochemistry of the ring, are required for the activity of BRD1240, we tested analogues of BRD1240 in both the GFP-LC3 and LysoTracker assays (Table 1 and Supporting Information Table S1). Movement of the nitrogen of the pyridine from the 4-position to the 3-position, or replacement of the pyridine with a phenyl ring, resulted in a complete loss of activity, confirming the importance of the nitrogen heteroatom at this position. It is possible that this nitrogen participates in a critical hydrogen bonding interaction with a protein target of BRD1240. Changes to the urea moiety were slightly more tolerated. Complete removal of the piperonyl urea ablated activity as did replacement of the urea with various amides; however, exchange of the piperonyl urea moiety with a meta-methoxy urea moiety yielded an active analogue. These observations suggest that an electron-rich phenyl urea is optimal for activity, but subtle changes at this position may be acceptable. More drastic changes to either position resulted in completely inactive analogues. Importantly, the SAR trends observed in the GFP-LC3 assay are paralleled in the LysoTracker assay, strengthening the hypothesis that BRD1240’s effects on autophagosome number are linked to its ability to perturb lysosomal function.
Table 1. BRD1240 Analogues Reveal Structure–Activity Relationships in GFP-LC3a and LysoTracker Assaysb.
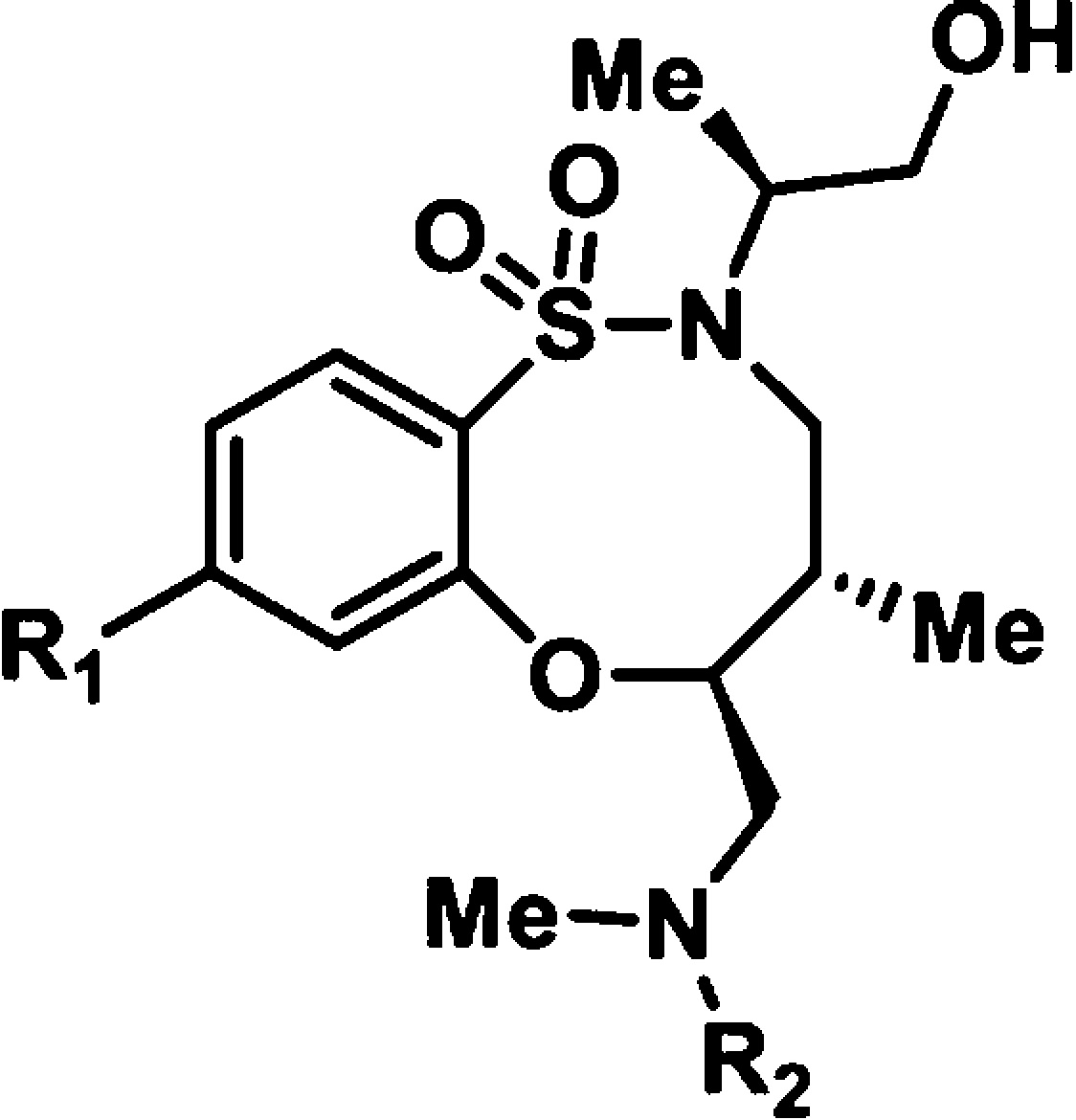
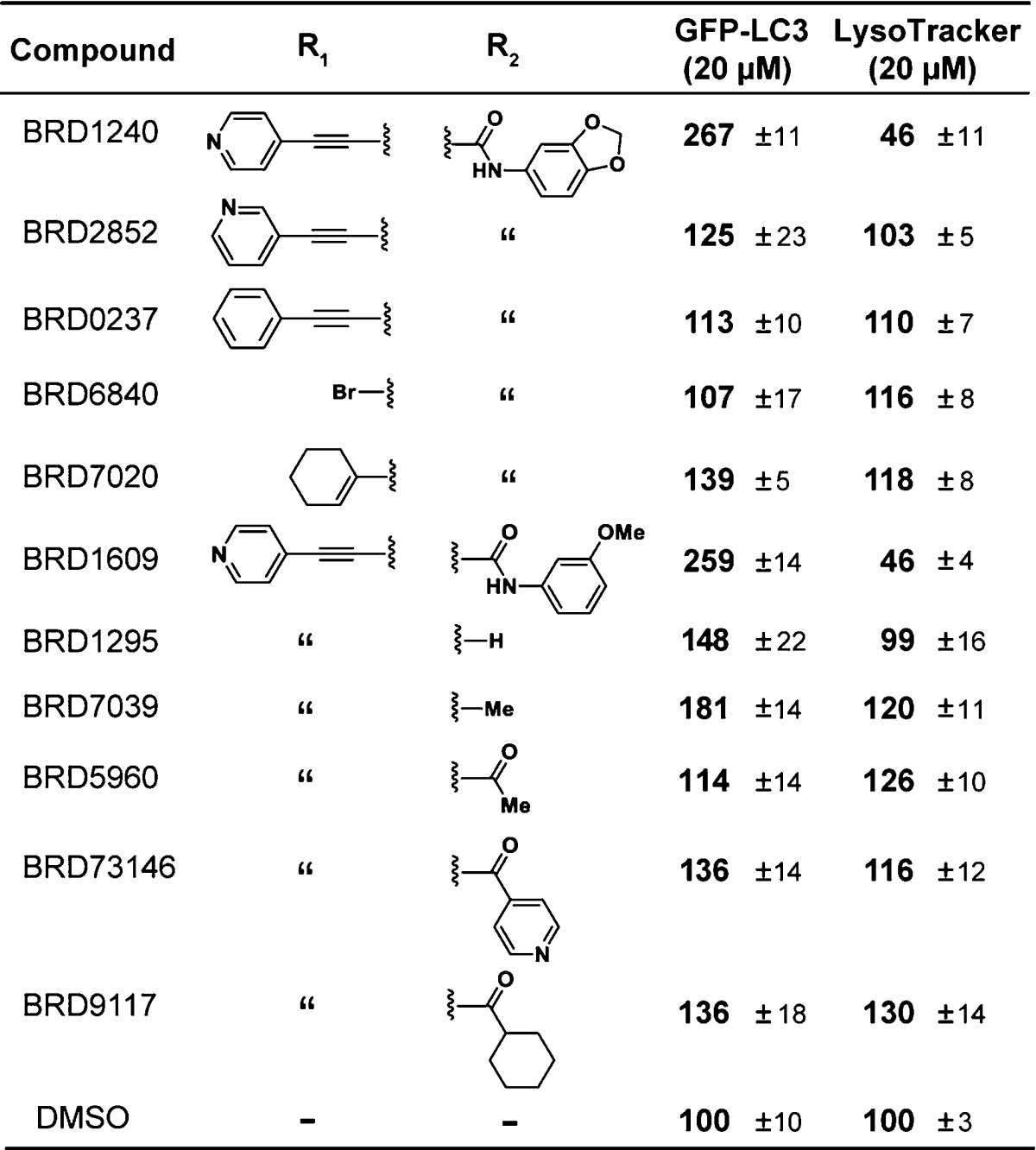
GFP-LC3 punctae formation data are presented as the average of three independent experiments run in duplicate and are reported as relative average % intensity ± SEM at 20 μM.
LysoTracker displacement data are presented as the average of two independent experiments run in duplicate and are reported as relative average % intensity ± SEM at 20 μM.
BRD1240 Kills a Similar Subset of Cancer Cells as the V-ATPase Inhibitor, Bafilomycin A1
Profiling the sensitivity of cancer cell lines to small molecules has emerged as a powerful tool for gaining insights into their mechanisms of action.35 Compounds with similar mechanisms of action have been shown to elicit a similar pattern of responses across panels of cancer cell lines.35−37 We measured the sensitivity of 83 cancer cell lines to BRD1240 and its inactive enantiomer, BRD4849, and compared it to the patterns of sensitivity measured for 479 other small molecules spanning a diverse range of known mechanisms of action gathered as part of the NCI’s Cancer Target Discovery and Development Network.38 We found that the patterns of sensitivity elicited by BRD1240 and BafA1 were highly similar, while such correlation was not observed between the inactive stereoisomer BRD4849 and BafA1 (Figure 3B,C). Furthermore, among the 481 compounds tested in these 83 cancer cell lines in the project, the correlation between BRD1240 and BafA1 is the strongest (Figure 3D). These data suggest that BRD1240 and BafA1 may share a common mechanism of action, and that BRD1240 may inhibit lysosomal acidification by directly suppressing the activity of V-ATPase.
Figure 3.

Comparison of 481 compounds in 83 cancer cell lines reveals a significant correlation between BRD1240, but not inactive stereoisomer BRD4849, and bafilomycin A1, a potent V-ATPase inhibitor. (A) Chemical structures of BRD1240 and BafA1. (B, C) Scatter plots presenting the area under the curve (AUC) for each cell line in response to compound treatments. r values indicate the Spearman correlation coefficient calculated on the basis of each data plot. (D) Box and whisker plot showing the Spearman correlation coefficient between sensitivities of BRD1240 or BafA1 and the other 480 compounds tested in 83 cancer cell lines.
BRD1240 Suppresses V-ATPase Function in Biochemical Assays Using Membrane Fractions
To determine whether BRD1240 perturbs lysosomal acidification by modulating V-ATPase activity, we tested its activity in a previously described in vitro V-ATPase activity assay using cellular membrane fractions (Figure 4A).16 Both BRD1240 and BafA1 suppressed lysosomal acidification in this assay. Interestingly, the activity of BRD1240 was dependent on pretreating the system with the compound for 1 h, while BafA1 performed identically regardless of whether a pretreatment step was included (Figure 4B,C). Our results suggest that BRD1240 perturbs lysosomal acidification by suppressing V-ATPase function, but potentially by a mode of action different than that of BafA1. Additional work will be required to identify the specific protein target of BRD1240, and whether this target is among the multiple subunits of the V-ATPase complex or its accessory regulatory factors.
Figure 4.

BafA1 and BRD1240 deacidify lysosomes in an in vitro V-ATPase activity assay. (A) Scheme of the assay protocol. (B) Performances of DMSO, BafA1 (100 nM), and BRD1240 (10 μM) in the in vitro V-ATPase assay over 30 min after ATP addition and after introduction of 10 μg/mL nigericin when no pretreatment was applied. Higher relative fluorescence values are indicative of higher pH. (C) Performances of these compounds in the assay when 1 h pretreatment was applied. In parts B and C, representative results from three independent experiments are shown. Values are presented as the average ± SD of two technical replicates.
Conclusion
Small-molecule probes have been critical to understanding the dynamic and multifunctional role of the lysosome in cellular physiology. We have identified BRD1240 as a novel small-molecule inhibitor of lysosomal acidification potentially operating via suppression of V-ATPase function. BRD1240 was derived from diversity-oriented synthesis, and its activity displays striking dependence on the stereochemistry of the ring. It promotes cell death in numerous cancer cell lines with a sensitivity profile that correlates significantly to that of the naturally occurring V-ATPase inhibitor, bafilomycin A1. These results highlight the power of DOS and HTS to yield novel, potent, and selective probes to study cellular pathways of biological and medical interest.
Acknowledgments
The authors would like to thank the Compound Management Team (Broad Institute) for providing compound plates and synthetic intermediates; Dr. Stephen Johnston (Broad Institute) for analytical support; and Dr. Michelle Stewart, Dr. Kara Lassen, Dr. Vlado Dančík, and Dr. Paul Clemons (Broad Institute) for valuable discussions. This research was supported by the Helmsley Charitable Trust (500203, S.L.S. and R.J.X.), the NIAID-sponsored Center for Excellence in Translational Research (U19AI109725, S.L.S., A.F.S., R.J.X.), and the National Institutes of Health (DK097485, R.J.X., and U01CA176152, S.L.S.). S.L.S. is an Investigator of the Howard Hughes Medical Institute.
Supporting Information Available
Experimental procedures, characterization data, and supplemental figures and tables. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
○ Alnylam Pharmaceuticals, Cambridge, Massachusetts 02142, United States.
Author Present Address
◆ Roche, Nonnenwald 2, DE-82377 Penzberg, Germany.
Author Present Address
¶ Koch Institute for Integrative Cancer Research, Cambridge, Massachusetts 02142, United States.
Author Contributions
L.N.A. and S.-Y.K. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Levine B.; Mizushima N.; Virgin H. W. Nature 2011, 469, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V.; Saitoh T.; Akira S. Nat. Rev. Immunol. 2013, 13, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochfeld W. E.; Lee S.; Rubinsztein D. C. Acta Pharmacol. Sin. 2013, 34, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen E.-L.; Tanaka Y.; Saftig P. Trends Cell Biol. 2003, 13, 137. [DOI] [PubMed] [Google Scholar]
- Watts C. Biochim. Biophys. Acta 2012, 1824, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshansky V.; Rubinstein J. L.; Gruber G. Biochim. Biophys. Acta 2014, 1837, 857. [DOI] [PubMed] [Google Scholar]
- Bhargava A.; Voronov I.; Wang Y.; Glogauer M.; Kartner N.; Manolson M. F. J. Biol. Chem. 2012, 287, 26829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochotny N.; Flenniken A. M.; Owen C.; Voronov I.; Zirngibl R. A.; Osborne L. R.; Henderson J. E.; Adamson S. L.; Rossant J.; Manolson M. F.; Aubin J. E. J. Bone Miner. Res. 2011, 26, 1484. [DOI] [PubMed] [Google Scholar]
- Ramachandran N.; Munteanu I.; Wang P.; Ruggieri A.; Rilstone J. J.; Israelian N.; Naranian T.; Paroutis P.; Guo R.; Ren Z. P.; Nishino I.; Chabrol B.; Pellissier J. F.; Minetti C.; Udd B.; Fardeau M.; Tailor C. S.; Mahuran D. J.; Kissel J. T.; Kalimo H.; Levy N.; Manolson M. F.; Ackerley C. A.; Minassian B. A. Acta Neuropathol. 2013, 125, 439. [DOI] [PubMed] [Google Scholar]
- Batlle D.; Haque S. K. Nephrol., Dial., Transplant. 2012, 27, 3691. [DOI] [PubMed] [Google Scholar]
- Fuster D. G.; Zhang J.; Xie X. S.; Moe O. W. Kidney Int. 2008, 73, 1151. [DOI] [PubMed] [Google Scholar]
- Capparelli R.; Palumbo D.; Iannaccone M.; Iannelli D. Genes Immun. 2009, 10, 641. [DOI] [PubMed] [Google Scholar]
- Cotter K.; Capecci J.; Sennoune S.; Huss M.; Maier M.; Martinez-Zaguilan R.; Forgac M. J. Biol. Chem. 2015, 290, 3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Gupta M.; Wu X.; Yoon C.; Giobbie-Hurder A.; Hodi F. S. Cancer Immunol. Res. 2015, 3, 59. [DOI] [PubMed] [Google Scholar]
- Sancak Y.; Bar-Peled L.; Zoncu R.; Markhard A. L.; Nada S.; Sabatini D. M. Cell 2010, 141, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R.; Bar-Peled L.; Efeyan A.; Wang S.; Sancak Y.; Sabatini D. M. Science 2011, 334, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M.; Sabatini D. M. Cell 2012, 149, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C.; Di Malta C.; Polito V. A.; Garcia Arencibia M.; Vetrini F.; Erdin S.; Erdin S. U.; Huynh T.; Medina D.; Colella P.; Sardiello M.; Rubinsztein D. C.; Ballabio A. Science 2011, 332, 1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C.; Zoncu R.; Medina D. L.; Vetrini F.; Erdin S.; Erdin S.; Huynh T.; Ferron M.; Karsenty G.; Vellard M. C.; Facchinetti V.; Sabatini D. M.; Ballabio A. EMBO J. 2012, 31, 1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz J. C.; Sugii S.; Yu C.; Chang T. Y. J. Biol. Chem. 2000, 275, 4013. [DOI] [PubMed] [Google Scholar]
- Infante R. E.; Wang M. L.; Radhakrishnan A.; Kwon H. J.; Brown M. S.; Goldstein J. L. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 15287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P.; Klumperman J. Nat. Rev. Mol. Cell Biol. 2009, 10, 623. [DOI] [PubMed] [Google Scholar]
- Yang Y. P.; Hu L. F.; Zheng H. F.; Mao C. J.; Hu W. D.; Xiong K. P.; Wang F.; Liu C. F. Acta Pharmacol. Sin. 2013, 34, 625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhuber J.; Muehlbacher M.; Trapp S.; Pechmann S.; Friedl A.; Reichel M.; Muhle C.; Terfloth L.; Groemer T. W.; Spitzer G. M.; Liedl K. R.; Gulbins E.; Tripal P. PLoS One 2011, 6, e23852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss M.; Wieczorek H. J. Exp. Biol. 2009, 212, 341. [DOI] [PubMed] [Google Scholar]
- Diyabalanage T.; Amsler C. D.; McClintock J. B.; Baker B. J. J. Am. Chem. Soc. 2006, 128, 5630. [DOI] [PubMed] [Google Scholar]
- Marcaurelle L. A.; Comer E.; Dandapani S.; Duvall J. R.; Gerard B.; Kesavan S.; Lee M. D.; Liu H.; Lowe J. T.; Marie J.-C.; Mulrooney C. A.; Pandya B. A.; Rowley A.; Ryba T. D.; Suh B.-C.; Wei J.; Young D. W.; Akella L. B.; Ross N. T.; Zhang Y.-L.; Fass D. M.; Reis S. A.; Zhao W.-N.; Haggarty S. J.; Palmer M.; Foley M. A. J. Am. Chem. Soc. 2010, 132, 16962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard B.; Duvall J. R.; Lowe J. T.; Murillo T.; Wei J.; Akella L. B.; Marcaurelle L. A. ACS Comb. Sci. 2011, 13, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald M. E.; Mulrooney C. A.; Duvall J. R.; Wei J.; Suh B. C.; Akella L. B.; Vrcic A.; Marcaurelle L. A. ACS Comb. Sci. 2012, 14, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen T. E.; Schreiber S. L. Angew. Chem., Int. Ed. 2008, 47, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N.; Yamamoto A.; Matsui M.; Yoshimori T.; Ohsumi Y. Mol. Biol. Cell 2004, 15, 1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S.; Noda T.; Yoshimori T. Autophagy 2007, 3, 452. [DOI] [PubMed] [Google Scholar]
- Lemieux B.; Percival M. D.; Falgueyret J. P. Anal. Biochem. 2004, 327, 247. [DOI] [PubMed] [Google Scholar]
- Vázquez C. L.; Colombo M. I. Methods Enzymol. 2009, 452, 85. [DOI] [PubMed] [Google Scholar]
- Shoemaker R. H. Nat. Rev. Cancer 2006, 6, 813. [DOI] [PubMed] [Google Scholar]
- Boyd M. R.; Farina C.; Belfiore P.; Gagliardi S.; Kim J. W.; Hayakawa Y.; Beutler J. A.; McKee T. C.; Bowman B. J.; Bowman E. J. J. Pharmacol. Exp. Ther. 2001, 297, 114. [PubMed] [Google Scholar]
- Adams D. J.; Ito D.; Rees M. G.; Seashore-Ludlow B.; Puyang X.; Ramos A. H.; Cheah J. H.; Clemons P. A.; Warmuth M.; Zhu P.; Shamji A. F.; Schreiber S. L. ACS Chem. Biol. 2014, 9, 2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A.; Bodycombe N. E.; Cheah J. H.; Price E. V.; Liu K.; Schaefer G. I.; Ebright R. Y.; Stewart M. L.; Ito D.; Wang S.; Bracha A. L.; Liefeld T.; Wawer M.; Gilbert J. C.; Wilson A. J.; Stransky N.; Kryukov G. V.; Dancik V.; Barretina J.; Garraway L. A.; Hon C. S.; Munoz B.; Bittker J. A.; Stockwell B. R.; Khabele D.; Stern A. M.; Clemons P. A.; Shamji A. F.; Schreiber S. L. Cell 2013, 154, 1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.