

Summary
LAGLIDADG homing endonucleases (also referred to as ‘meganucleases’) are compact DNA cleaving enzymes that specifically recognize long target sequences (approximately 20 base pairs), and thus serve as useful tools for therapeutic genome engineering. While stand-alone meganucleases are sufficiently active to introduce targeted genome modification, they can be fused to additional sequence-specific DNA binding domains in order to improve their performance in target cells. In this chapter, we describe an approach to retarget meganucleases to DNA targets of interest (such as sequences found in genes and cis regulatory regions), which is feasible in an academic laboratory environment. A combination of two selection systems, in vitro compartmentalization and two-plasmid cleavage assay in bacteria, allow for efficient engineering of meganucleases that specifically cleave a wide variety of DNA sequences.
Keywords: Meganuclease, Protein Engineering, Selection, Genome Engineering, nanodrops
1. Introduction
Over the past decade, several technologies have been developed to introduce site-specific mutagenesis in highly complex genomes of various eukaryotes. Both oligonucleotide-based methods (such as triplex-forming oligonucleotides coupled to DNA modifying enzyme domains) and protein-based approaches (using sequence-specific nucleases and recombinases) have been shown to promote targeted genome modifications such as gene disruption, integration and sequence modification (1). Engineered nucleases that have been most widely employed include zinc finger nucleases (ZFNs) (2,3), transcription activator-like effector nucleases (TALENs) (4-6), RNA-guided CRISPR/Cas9 endonucleases (CRISPR) (7,8), and LAGLIDADG homing endonucleases (also referred to as meganucleases) (9,10). These enzymes generate double strand breaks (DSBs) at their genomic target sites, and stimulate intrinsic DSB repair pathways. Template-independent repair, such as nonhomologous end joining, often gives rise to small deletion and insertion, while homology-driven repair can lead to duplication of a donor template sequence (resulting in a specific sequence alteration, insertion or deletion) (3,11,12). In addition, recent studies have demonstrated that simultaneous generation of DSBs at multiple genomic loci promotes more dynamic chromosomal mutations including deletion, inversion, and translocation (13-15).
Homing endonucleases (‘meganucleases’) are naturally-occurring enzymes that are found in mitochondrial and chloroplast genomes (as well as archaea), and are often encoded in concert with surrounding intron or intein sequences (16). When expressed in host cells, these enzymes generate strand breaks at alleles that lack the intervening sequence, leading to homology-driven repair that results in duplication of sequences carrying the meganuclease genes. As a result, intron- or intein-encoded homing endonucleases function as exceptionally efficient mobile elements that are inherited in a dominant, non-Mendelian manner.
Meganucleases that possess a single conserved LAGLIDADG motif per protein chain form homodimeric proteins that cleave palindromic and nearly palindromic DNA target sequences, while those that contain two such motifs per protein chain form larger, pseudo-symmetric monomers that can target completely asymmetric DNA sequences. To prevent cleavage at off-target sites that could otherwise compromise the viability of host cells, meganucleases specifically recognize long (18 to 24 basepair) target sequences, and thereby exhibit extremely high cleavage specificity. Because of their small size, stable folds and exceptional DNA cleavage specificities, several wild-type meganucleases (particularly I-SceI, a monomeric endonuclease that targets a 18 base-pair sequence), have been used to generate site-specific DSBs on integrated reporters in a variety of eukaryotic cells, and shown to do so without significant cytotoxicity (10,12,17). More recently, these endonucleases have been redesigned to modify genomic target sites in mammalian and plant cells (9,18-21).
Two recent studies have shown that by combining meganucleases with additional sequence-specific TALe DNA binding domains, highly active and specific genome engineering reagents can be created (22,23). However, this strategy still requires engineering of meganucleases to display a desired new specificity, corresponding to the genomic target site of interest. The complete redesign of meganuclease specificity toward such genomic sites has generally been accomplished in biotech settings that make use of industrial scale engineering pipelines (9,19). Here, we describe a detailed protocol to effectively obtain customized meganucleases that employs routine benchtop molecular biology protocols, that are accessible for academic laboratories (23). All steps required to initially redirect the specificity of meganucleases are conducted using a method termed in vitro compartmentalization (IVC). This approach, which was originally developed as described in (24-26), allows for expression and selection of engineered proteins in a numerous number of individual compartments generated in an oilsurfactant mixture (approximately 1010/ml). Subsequently, the most active population of redesigned meganucleases are filtered for optimal gene targeting activity using a two-plasmid cleavage assay in bacteria (27) to select variant endonucleases that are significantly active in cellulo.
The use of in vitro compartmentalization, as a tool for protein engineering, facilitates construction of highly complex protein libraries, compared to previously described methods, and thereby allows investigators to query a significantly higher number of protein sequence variants at each stage of specificity redesign. While the methodology described in this chapter is specific for the alteration of nuclease specificity, the general approach is appropriate for the modification of any extensive protein recognition surface, and relies only on the development of a phenotypic selection strategy that can be utilized in the context of compartmentalized aqueous droplets.
2. Materials
2.1. Library Construction
PCR thermal cycler
pET-21d(+) plasmid (EMD millipore) containing a meganuclease gene with codons optimized for bacterial expression (Note 1)
Phusion High-Fidelity DNA Polymerase (Thermo Scientific/New England Biolabs)
10 mM dNTP mix
25 nmol scale degenerate PCR primers desalting grade
PCR primers commonly used to construct a library: ‘Up1’ (5′-CGT CCG GCG TAG AGG ATC GAG ATC-3′), ‘Up2’ (5′-AGA TCT CGA TCC CGC GAA ATT-3′), UpRev (5′-CCA TGG TAT ATC TCC TTC TTA AAG TTA AAC-3′), ‘MidFwd’ (5′-CAC TGA GAT CCG GCT GC TAA C-(tandem 2 copies of a target site)-CCG CTG AGC AAT AAC TAG-3′), ‘MidRev’ (5′-GTT AGC AGC CGG ATC TCA GTG-3′), ‘Down1’ (5′-CAC TCG TGC ACC CAA CTG ATC TTC-3′), and ‘Down2’ (5′-TCA GGG TTA TTG TCT CAT GAG CG-3′).
5 μg/mL ethidium bromide
UltraPure Agarose
50 × TAE buffer (pH 8.2-8.4 at 25°C): 2.0 M Tris acetate, .05 M EDTA
6 × Loading dye: 30% glycerol, 6 mM EDTA, and 0.1 % orange G; filtered through a 0.2 μm PVDF membrane
≥98% Guanosine
1kb plus DNA ladder
QIAquick Gel Extraction Kit (Qiagen)
2.2. In vitro selection using IVC
4 × Annealing buffer: 40 mM Tris-HCl (pH7.5), 600 mM NaCl (autoclaved)
Mineral oil for molecular biology (light oil)
ABIL EM90 (Evonik)
98% Triton X-100 for molecular biology, DNase, RNase, Protease free
1 M potassium glutamate (pH 7.5): 5.6 grams of KOH is dissolved in 50 mL of water, and titrated with glutamate powder to adjust pH to 7.5. Water is added to make a final volume of 100 ml, and the solution is filtered through a 0.2 μm PVDF membrane.
1 M magnesium acetate (pH 7.5): 14.2 grams of magnesium acetate is dissolved in water, titrated with glacial acetic acid to adjust pH to 7.5, and filtered through a 0.2 μm PVDF membrane.
1 M dithiothreitol (DTT) filtered through a 0.2 μm PVDF membrane
10 mg/mL bovine serum albumin (BSA) Fraction V filtered through a 0.2 μm PVDF membrane
14-mL polypropylene round-bottom tube
Octagonal magnetic stir bar (Length: 13 mm; autoclaved for 15 minutes)
Magnetic stirrer
PURExpress (New England Biolabs)
40 units/μl Murine RNase inhibitor (New England Biolabs)
Phenol/Chloroform/Isoamyl alcohol (25:24:1, water-saturated, pH 7.9)
3M sodium acetate (pH5.2): sodium acetate is dissolved in water, titrated with glacial acetate to adjust pH to 5.2 and autoclaved for 15 minutes.
Isopropanol
70% ethanol
RNase Cocktail Enzyme Mix (Life Technology)
QIAquick PCR Purification Kit (Qiagen)
400,000 units/ml T4 DNA ligase
DNA adaptors (see Table 1)
PCR primers: ‘Up3’ (5′-CGA AAT TAA TAC GAC TCA CTA TAG G-3′), ‘Up4’ (5′-CCC TCT AGA AAT AAT TTT GTT TAA CTT-3′), primers specific to DNA adaptors (see Table 1).
Table 1.
DNA adaptors used to recover active meganuclease genes
| Adaptor | Sequence |
|---|---|
| A | 5′-GTTTGCTCAGGCTCTCCCCGTGGAGGTAATAATTGNNNN-3′ 3′-CAAACGAGTCCGAGAGGGGCACCTCCATTATTAAC-5′ |
| B | 5′-GATCTTACCGCTGTTGAGATCCAGTTCGATGTAACCNNNN-3′ 3′-CTAGAATGGCGACAACTCTAGGTCAAGCTACATTGG-5 |
| C | 5′-CAAACGTCTGAACATCAATGCGGCCAAATCTTCATTCCNNNN-3′ 3′-GTTTGCAGACTTGTAGTTACGCCGGTTTAGAAGTAAGG-5′ |
| D | 5′-AGCTAACGTTGGTCCAAACAGGATACCTGCGGTGANNNN-3′ 3′-TCGATTGCAACCAGGTTTGTCCTATGGACGCCACT-5′ |
| E | 5′-CCTAGACGGATAACGCGTACTCTTTCCTCCGATTGGNNNN-3′ 3′-GGATCTGCCTATTGCGCATGAGAAAGGAGGCTAACC-5′ |
| F | 5′-GCTCGAGACTCTCGCGAAAAGTAAGAAGGCTACATCNNNN-3′ 3′-CGAGCTCTGAGAGCGCTTTTCATTCTTCCGATGTAG-5′ |
| G | 5′-ACTGGTAGTCTCCGGCCATTTGTTCCTCAGCAAAGTNNNN-3′ 3′-TGACCATCAGAGGCCGGTAAACAAGGAGTCGTTTCA-5′ |
| H | 5′-GACTATATTCTCCAATCTCGGAGCAAAGGGGCTCGCNNNN-3′ 3′-CTGATATAAGAGGTTAGAGCCTCGTTTCCCCGAGCG-5′ |
Four-base, 3′ overhangs (bold) are complementary to those of target sites generated by variant endonucleases. Underlined are sequences of primers used for adaptor-specific PCR. Avoid ligation with adaptors used in the last two rounds of selection.
2.3. Two-plasmid cleavage assay in bacteria
pEndo and pCcdB: these plasmids were originally described in (27) (Fig. 1).
DNA Clean & Concentrator-5 (Zymo Research)
Restriction enzymes (AflIII, BglII, NheI, SacII, NcoI, and NotI; New England Biolabs)
Quick Ligation Kit (New England Biolabs)
NovaXGF’ competent cells (EMD Millipore)
DNA purification Miniprep Kit
Oligonucleotides (used for cloning, PCR and sequencing): ‘CcdB_seq1’ (5′-GTT ATC GGG GAA GAA GTG GC-3′), ‘CcdB_seq2’ (5′-CGG GTG ATG CTG CCA ACT TA-3′), ‘Endo_colonyPCR_fwd’ (5′-CAC GGC AGA AAA GTC CAC ATT G-3′), Endo_colonyPCR_rev (5′-TGA GGG AGC CAC GGT TGA TG-3′), ‘Endo_seq_fwd’ (5′-CGG CGT CAC ACT TTG CTA TG-3′), and ‘Endo_seq_rev’ (5′-GAG CCA CGG TTG ATG AGA GCT TTG-3′).
SBG medium: 15 g tryptone, 10 g yeast extracts, 2.5 g NaCl, and 2.5 g glucose are dissolved in water and titrated with NaOH to adjust pH to 7.4. Water is added to make a final volume of 500 ml, and the medium is autoclaved for 15 minutes.
1 mM Hepes (pH 7.0): Hepes is dissolved in water, titrated with NaOH to adjust pH to 7.0, and autoclaved for 15 minutes.
10 %(v/v) glycerol (autoclaved for 15 minutes)
Gel DNA Recovery Kit (Zymo Research)
1kb DNA ladder
2 × Gibson Assembly Master Mix (New England Biolabs)
DH5α and DH10B chemical competent cells
Sterilized water
Cell scraper
Electroporation cuvette (0.2-cm path)
2 × YT medium
20 % L-arabinose filtered through a 0.2 μm PVDF membrane
10 × M9 salts: 60 g/l Na2HPO4, 30 g/l KH2PO4, 5 g/l NaCl, and 10 g/l NH4Cl
1 M Mg2SO4 autoclaved for 30 minutes
1 M CaCl2 autoclaved for 30 minutes
1 % (w/v) thiamine filtered through a 0.2 μm PVDF membrane
1 M isopropyl β-D-1-thiogalactopyranoside (IPTG) filtered through a 0.2 μm PVDF membrane
Control plate: 100 mL of 3 %(w/v) agar and 100 mL of 2 × M9 salt supplemented with 2 %(v/v) glycerol and 1.6 %(w/v) tryptone are separately autoclaved for 30 minutes, and combined. After the agar medium is cooled down to 50 °C, 200 μl of 1 M Mg2SO4, 200 μl of 1 M CaCl2, 200 μl of 100 mg/mL carbenicillin, and 40 μl of thiamine are quickly added, and poured in 100-mm petri dishes.
Selective plate: the plates are made by further adding 200 μl of 20 % L-arabinose and 80 μl of IPTG to 200 mL of the control agar medium.
GoTaq DNA Polymerase (Promega)
GeneMorph II Random Mutagenesis Kit (Agilent Technologies)
ElectroMax DH10B T1 phage-resistant competent cells (Life Technologies)
SOC medium
150-mm petri dish
Glass beads autoclaved for 15 minutes
Plasmid Maxi Kit
Figure 1.
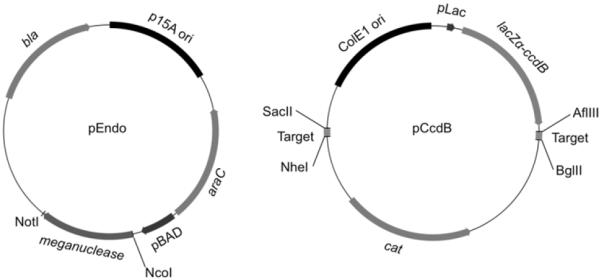
The pEndo and pCcdB plasmids used for the two-pasmid cleavage assay in bacteria (27). These plasmid maps were drawn using Flex Plasmid Draw (http://mavericktse.is-a-geek.com/PlasmidDrawv2/index.html).
3. Methods
The method described below is comprised of (1) a bioinformatic search for DNA sequences to which a meganuclease is to be retargeted (in this case, using the ‘LAHEDES’ web server; http://www.homingendonuclease.net) (28), (2) engineering of a customized meganuclease using In vitro compartmentalization (IVC), and (3) filtering and optimization of final pool of active enzymes using two-plasmid cleavage assay in bacteria. To create variant endonucleases that display cleavage activity against a target site of interest (e.g. clinically relevant genome site), site-directed saturation mutagenesis and several rounds of selections using IVC are iterated. An overview of this step is depicted in Fig. 2. A pool of meganucleases redesigned through in vitro selections is further screened in bacteria so as to isolate substantially active enzymes in cellulo.
Figure 2.

Schematics of (A) PCR-based construction of a library and (B) in vitro selection using IVC. (A) A parental meganuclease gene cloned into pET-21d(+) is used as a PCR template to introduce site-directed mutagenesis. All PCR products are purified by gel extraction prior to subsequent rounds of PCR. In separate reaction mixtures, the T7 promoter and an ORF encoding an endonuclease are PCR-amplified with multiple sets of primers, a portion of which contain wobble bases to introduce saturation mutagenesis (I). In parallel, a fragment containing target sites are generated using the Down1 primer and the MidFwd primer (that contains tandem two copies of a target site (black boxes)). All sequences generated are assembled by overlap extension PCR using the Up2 and Down2 primers (II). The resulting library containing variant endonuclease genes and tandem two copies of a target site (III) are compartmentalized together with in vitro protein synthesis reaction mixture in an oil-surfactant mixture (IV). After extraction from emulsion, fragments with cohesive ends generated by meganucleases are ligated to DNA adaptors containing their complementary ends (V). Sequences coupled to adaptors are amplified using the Up3 primer and an adaptor-specific primer (VI). Meganuclease genes are amplified again using the Up4 primer and the MidRev primer to eliminate an adaptor sequence (VII). In parallel, a fragment containing the T7 promoter and the ribosome binding site is generated using the Up1 primer and the UpRev primer. Two copies of a target site are added by another round of PCR (VIII). (B) IVC allows endonucleases (expressed in compartmentalized droplets) to access only target sites coupled to its own gene (black boxes), thus maintaining genotype-phenotype linkage during selection. After a library encoding meganucleases is extracted from emulsion, genes associated with cohesive ends that are generated by endonucleolytic cleavage are ligated to a DNA adaptor. PCR using a pair of primers, one of which is specific to an adaptor sequence, give rise to a DNA band containing meganuclease genes from a library subjected to in vitro selection (lane ‘+’), but not from the corresponding naive one (lane ‘−’).
3.1 Determining sequences to be targeted by engineered meganucleases
The previously developed ‘LAHEDES’ webserver is utilized to search for sequences that display high identities to the target sites for wild type meganucleases that have been crystallized in complexes with DNA (29). In addition to the homodimeric I-CreI meganuclease that has previously been well described for engineering and gene targeting, crystal structures of 8 monomeric meganucleases that recognize unique target sequences (I-SceI, I-OnuI, I-LtrI, I-LtrWI, I-PanMI, I-GzeMII, I-HjeMI, and ISmaMI) are now available. A ‘Central Four Search’, as described at that webserver, is used to explore candidate sites that an be targeted by engineered meganucleases (Fig. 3). This search option identifies sites with few nucleotide mismatches at the central 4 base-pair positions, where sequence-dependent DNA bending appears to significantly influence the cleavage activity of meganucleases (30). A target site for a redesigned meganuclease is chosen on the basis of its sequence identity to the original target sites for the wild type meganucleases and its location in a genomic target region. Using the approach described in this chapter, we have created variant endonucleases that target DNA sequences showing 50 % or higher identity to the original sites.
Figure 3.

Output of ‘Central Four Search’ in the LAHEDES web server. Sites are selected, based on both the local sequence identity (at the base-pair positions −2 to +2) and the overall homology to the original target sites for chosen meganucleases (wild type sequences). Nucleotides that differ from the wild type sequences are shown in lower cases.
3.2 Retargeting meganucleases to new sequences using IVC
3.2.1. Construction of a library encoding variant meganucleases using overlap extension PCR
The DNA interface of meganucleases is typically composed of a pair of four anti-parallel β-strands and their connecting loops, wherein clusters of 6 to 9 amino-acid residues dictase sequence specificity at 2 to 4 consecutive base-pair positions (10,29). Such a group of residues is termed ‘contact module’, and can be defined for every 3 consecutive base-pair positions except the central 4 positions, where the DNA target is bent as a result of meganuclease binding, far fewer protein-DNA contacts are observed and we have found reprogramming of specificity to be more difficult.
A meganuclease is first retargeted towards two sites containing only one cluster of base pairs that differ from the original target site, in either of the two half sites (termed as ‘round 1′ target sites) (Fig. 4). Up to 9 residues to be randomized are identified within a ‘contact module’ that covers these ‘round 1′ basepair substitutions (Note 2). To construct a library encoding variant meganucleases, DNA fragments containing a partial ORF of a meganuclease gene are PCR-amplfied using degenerate primers, which shuffle amino-acid residues within the ‘contact module’ against the altered DNA base pairs. A degenerate codon 5′-NNK-3′ is used to randomize amino-acid residues, but not included in approximately 10 nucleotides from the 3′ termini of the primers (Fig. 5A). In addition, degenerate primers are designed to generate PCR products that share approximately 20 base-pair sequences with their adjacent fragments at both ends. In the second round of PCR, all sequences containing a partial ORF of a meganuclease gene are assembled with a fragment containing 2 copies of a ‘round 1′ target site.
Figure 4.

Schematic of sequential protein engineering to reprogram meganuclease specificity. Meganucleases are first retargeted to ‘round 1′ targets, DNA sequences containing only one cluster of consecutive nucleotide substitutions from the original target (WT). Following rounds of in vitro selection, selected endonucleases are further mutated and screened to collect endonucleases that cleave next rounds of target sites, which contain additional base-pair mismatches. This iterative approach results in engineering of variant enzymes that target ‘half’ altered sites. Half domains of such proteins are shuffled to create meganucleases that recognize ‘fully’ altered targets.
Figure 5.
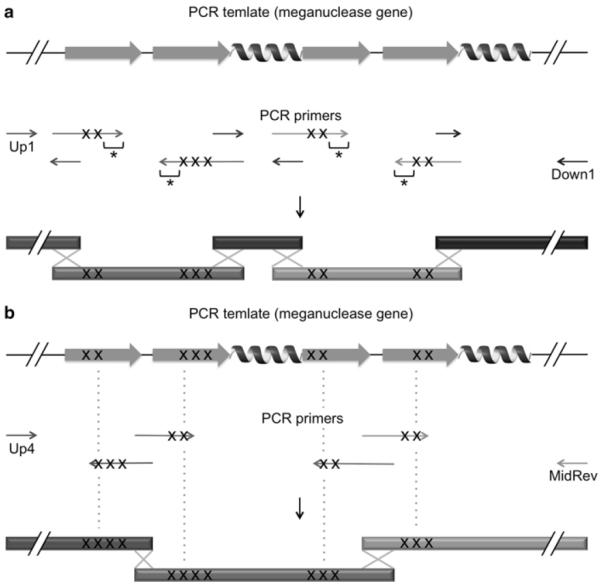
General guidelines for design of degenerate primers (A) No degenerate codon is incorporated in approximately 10 nucleotides from 3′ termini of primers (indicated by asterisks), and primers are designed such that resulting PCR fragments overlap at both ends with adjacent ones. Positions of degenerate codons are represented by ‘X’s. (B) In a case where degenerate primers including no wobble base in 9 nucleotides from 3′ termini cannot be design, the 3′ termini are extended beyond the closest residues that have been shuffled (indicated by dash lines), and these residues are randomized again. Such residues are also counted in 9 residues that are allowed to be randomized in one round of mutagenesis. Introduction of second-time saturation mutagenesis in 3′ terminal regions of degenerate primers ensures that at least a subset of degenerate primers stably form DNA duplexes with a PCR template, initiating PCR amplification.
Design degenerate primers as described above (also see Fig. 5A).
- To generate fragments containing a partial ORF of a meganuclease gene with targeted codons shuffled and a sequence containing 2 copies of a ‘round 1′ target site, set up the following PCR reaction mixture:
5 × Phusion HF Buffer 10 μl 10 mM dNTPs mix 1 μl Phusion High-Fidelity DNA polymerase 0.5 μl Water 32.5 μl 10 ng/μl pET-21d(+) plasmid (containing a meganuclease gene) 1 μl A pair of 10 pmol/μl PCR primers 2.5 μl
each Add 10 μl of 6 × the Loading dye in each PCR tube and separate 30 μl of each sample on an agarose-TAE gel containing 1 mM guanosine and 0.1 ng/mL ethidium bromide (Note 4).
Cut out the PCR products, and combine 2-3 gel slices in one tube (except a sequence containing a ‘round 1′ target site that can be reused to construct subsequent libraries) in order to save spin columns supplied with QIAquick Gel Extraction Kit.
Recover the DNA fragments using QIAquick Gel Extraction Kit, and elute them in 50 μl of supplied Buffer EB.
- To generate a library encoding variant meganucleases, set up the following PCR reaction mixture:
5 × Phusion HF Buffer 10 μl 10 mM dNTPs mix 1 μl Phusion High-Fidelity DNA Polymerase 0.5 μl Gel-extracted PCR fragments 1.5 μl each 10 pmol/μl the ‘Up2’ and ‘Down2’
primers2.5 μl each Water Added to make a final volume of 50 μl Add 10 μl of 6 × the Loading dye in a PCR product and separate 15 μl and 40 μl of the mixture in adjacent lanes on a 1% agarose-TAE gel containing 0.1 ng/mL ethidium bromide.
To prevent a PCR-generated library from UV-induced damage, divide two lanes where different volumes of the same PCR product are loaded, and mark the position where the library migrates only on a lane containing 15 μl of the sample under UV light. Match the two lanes again, and cut out DNA from the lane that is not exposed to UV (where 40 μl of a library is loaded).
Using QIAquick Gel Extraction Kit, recover and elute DNA in 30 μl of supplied Buffer EB (Note 5). Load the collected elution on the same spin column and centrifuge it again.
Separate 1-2 μl of the purified PCR fragment on a 1 % agarose-TAE gel along with 2.5 μl and 5 μl of 1kb plus DNA ladder, and quantify the library using a densitometry program such as ImageJ software (http://imagej.nih.gov/ij/).
3.2.2. First round of in vitro selection using IVC
Redesigned meganucleases are screened using IVC, where each of aqueous droplets that are compartmentalized in an oil phase contains an average of one DNA fragment encoding a variant endonuclease and an in vitro protein synthesis reaction mixture. A protein that is expressed in emulsion is retained in the same droplet throughout a time period for in vitro protein synthesis and selection, and thus has an opportunity to cleave only DNA target sites coupled to its own gene-coding sequence (Fig. 2B). After enzymatic reactions are terminated, extracted DNA fragments are subjected to a ligation reaction with a DNA adaptor containing cohesive ends that are compatible with a 3′, 4-base overhang generated by engineered meganucleases (Fig. 2). Genes linked to an adaptor are rescued by PCR using a pair of primers, one of which is specific to an adaptor (termed as adaptor-specific PCR). To eliminate the DNA adaptor from the adaptor-specific PCR product, another round of PCR is carried out and the resultant fragment is assembled with sequences containing the T7 promoter, the ribosome binding site, and 2 copies of ‘round 1′ target sites (Fig. 2A).
- To prepare a DNA adaptor, set up the following mixture:
4 × Annealing buffer 4 μl 1 mM complementary oligonucleotides (see Table 1) 1.6 μl each Water 8.8 μl Mix 40 μl of ABIL EM90 and 1 μl of Triton X-100 with 360 μl of light mineral oil in a 2-mL microcentrifuge tube (5 × oil-surfactant mixture), and vortex it for 1 minute (Note 6).
Mix 560 μl of light mineral oil with 140 μl of the 5 × oil-surfactant mixture in a 2-mL microcentrifuge tube, and vortex it for 1 minute.
Add 140 μl of Saturation buffer (100 mM potassium glutamate (pH 7.5), 10 mM magnesium acetate, 1 mM DTT, and 5 mg/mL BSA) in the same tube, and vortex it for one minute.
Incubate the tube at 37°C for 20 minutes, and centrifuge it at 16,000 × g for 15 min at 4°C.
Transfer 500 μl of the upper phase (white cloudy) into a 14-mL polypropylene round-bottom tube, and drop a stirrer bar in it.
- Set up the following in vitro protein synthesis reaction mixture:
PURExpress solution A 10 μl PURExpress solution B 7.5 μl Murine RNase inhibitor (40 units/μl) 0.5 μl Gel-extracted library (4 ng/μl) 2 μl 10 mg/mL BSA 3 μl Water 7 μl Stir the oil-surfactant mixture in ice water at 1,400 r.p.m, and slowly add 10 μl of the in vitro protein synthesis reaction mixture in the round-bottom tube (containing the oilsurfactant mixture) every 30 seconds. After all the aqueous mixture is added, continue stirring for additional 2.5 minutes.
Incubate the emulsified droplets at 30°C for 4 hours.
Heat the compartmentalized in vitro protein synthesis reaction mixture at 75°C for 15 minutes, and cool it down on ice for 1 minute.
Add 170 μl of supplied Buffer EB, vortex the emulsion for a few seconds and transfer it into a 1.5-mL microcentrifuge tube.
Centrifuge the tube at 16,000 × g for 20 min at 4 °C, and remove the upper (oil) phase.
Add 200 μl of Phenol/Chloroform/Isoamyl alcohol (pH 7.9) in the bottom (aqueous) phase, and mix them by vortexing.
Centrifuge the microcentrifuge tube again at 16,000 × g for 5 min at room temperature, and transfer the upper (aqueous) phase into a new 1.5-mL microcentrifuge tube.
Repeat Steps 13 and 14.
Add 200 μl of isopropanol and 20 μl of 3 M sodium acetate (pH 5.2) in the aqueous phase, and centrifuge the mixture at 16,000 × g for 15 min at 4°C.
Remove the supernatant, and add 500 μl of 70% ethanol to the pellet.
After centrifugation at 16,000 × g for 5 min at 4°C, remove the supernatant and dry the pellet at room temperature for 5-10 minutes.
Add 95 μl of supplied Buffer EB and 5 μl of RNase Cocktail Enzyme Mix in the tube, and incubate it at 37°C for 30 minutes.
Clean up the library using QIAquick PCR Purification Kit, and elute it in 40 μl of supplied Buffer EB (Note 5).
- Set up the following ligation reaction mixture:
10 × T4 DNA Ligase Reaction Buffer 1 μl T4 DNA ligase (400,000 units/ml) 0.5 μl 0.2 pmol/μl DNA adaptor (diluted in water) 1 μl Library extracted from emulsion
or
its corresponding naïve library (0.2 ng/μl)8 μl Incubate the reaction mixture at 16°C for 2 hours to overnight.
- Set up the following PCR premix for adaptor-specific PCR:
5 × Phusion HF Buffer 40 μl 10 mM dNTPs mix 4 μl Phusion High-Fidelity DNA Polymerase 2 μl 10 pmol/μl the Up3 primer and an adaptor-specific primer 2.5 μl each Water 33.5 μl Analyze 5 μl of adaptor-specific PCR products on a 1% agarose-TAE gel (Note 7).
If a DNA fragment corresponding to meganuclease genes ligated to a DNA adaptor is specifically amplified from a library subjected to selection, combine 10 μl each of the PCR products that are amplified in the three tubes, and separate them on a 1% agarose-TAE gel containing 1 mM guanosine and 0.1 ng/mL ethidium bromide.
Cut out a DNA band for variant endonuclease genes linked to a DNA adaptor, and purify the PCR product using QIAquick Gel Extraction Kit. Elute it in 50 μl of supplied Buffer EB.
- To generate a sequence containing meganuclease genes but not an adaptor sequence, and to prepare a fragment containing the T7 promoter and ribosome binding site, set up the following two PCR reaction mixtures:
5 × Phusion HF Buffer 10 μl 10 mM dNTPs mix 1 μl Phusion High-Fidelity DNA Polymerase 0.5 μl 10 pmol/μl the Up4 primer and the MidRev primer 2.5 μl each Gel-extracted DNA 3 μl Water 30.5 μl 5 × Phusion HF Buffer 10 μl 10 mM dNTPs mix 1 μl Phusion High-Fidelity DNA Polymerase 0.5 μl 10 pmol/μl the Up1 primer and the UpRev primer 2.5 μl each 10 ng/μl pET-21d(+) plasmid 1 μl Water 32.5 μl Separate the PCR products on a 1% agarose-TAE gel containing 1 mM guanosine and 0.1 ng/mL ethidium bromide, and individually purify them as described in Step 26.
- To construct a new library for the next round of selection, set up the following PCR reaction mixture:
5 × Phusion HF Buffer 10 μl 10 mM dNTPs mix 1 μl Phusion High-Fidelity DNA Polymerase 0.5 μl 10 pmol/μl the ‘Up2’ primer and the ‘Down2’
primer2.5 μl each Sequence containing 2 copies of target sites
(prepared in Subheading 3.2.1, Steps 2-5)1 μl Two PCR fragments purified in Step 28 1 μl each Water 30.5 μl Purify and quantify a PCR-amplified fragment, as described in Subheading 3.2.1, Steps 7-10.
3.2.3. Subsequent rounds of selection using IVC
A reconstructed library is screened under more stringent conditions (by reducing a time period for in vitro protein synthesis and DNA target cleavage and/or by elevating temperatures). Increasing rounds of selection (up to 6 rounds) result in enrichment of stable, active enzymes in collected populations: total 3 rounds are generally sufficient.
Prepare the oil surfactant mixture as described in Subheading 3.2.2, Steps 2-6.
- Set up the following in vitro protein synthesis reaction mixture:
PURExpress solution A 10 μl PURExpress solution B 7.5 μl Murine RNase inhibitor (40 units/μl) 0.5 μl Library prepared in Subheading 3.2.2, Step 30 (1 ng/μl) 2 μl 10 mg/mL BSA 3 μl Water 7 μl Compartmentalize the above mixture as described in Subheading 3.2.2, Step 8, and incubate the emulsified droplets at 42°C for 75 minutes (Note 7).
Extract the library from emulsion, and ligate it to a different DNA adaptor from that used in the previous round of selection, as described in Subheading 3.2.2, Steps 10-22.
Recover meganuclease genes by adaptor-specific PCR, and reconstruct a library, as shown in Subheading 3.2.2, Steps 23-30. Set up an adaptor-specific PCR reaction mixture in two tubes for a sample extracted from emulsion (instead of three in the first round of selection).
In the third and later rounds of selection, add 0.5 ng of a library encoding variant meganucleases in an in vitro protein synthesis reaction mixture (shown in Step 2), and carry out selection at 42°C (or 37°C) for 30 minutes as described above.
Extract a library from emulsion, and carry out ligation with a different DNA adaptor from that used in the last two rounds of selection, as shown in Subheading 3.2.2, Steps 10-22.
Set up the adaptor-specific PCR reaction mixture in a single tube for each library (see Subheading 3.2.2, Step 23), and purify an adaptor-specific PCR fragment (which is specifically amplified from a library subjected to selection), as described in Subheading 3.2.2, Steps 25 and 26.
3.2.4. Selection of meganucleases that cleave a target site containing more than one cluster of base-pair substitutions
Additional base-pair substitutions are introduced to a ‘round 1′ target site (‘round 2′ target site), and amino-acid residues of a ‘contact module’ corresponding to newly altered base-pairs are shuffled in ORFs of variant endonucleases enriched in previous rounds of selection. A library is generated through 2 rounds of PCR amplification as described above. However, since neighboring residues have been shuffled in an earlier round of mutagenesis, degenerate primers that include no wobble base in 9 nucleotides from 3′ termini often cannot be designed (Fig. 5B). If that is the case, 3′ termini of primers should be extended beyond one residue that have been randomized such that a subset of degenerate primers (containing a NNK codon near 3′ termini) form the correct Watson-Crick base pairs with a PCR template in 3′ terminal regions (which are required for DNA synthesis by a DNA polymerase). A residue to be shuffled again is also counted in up to 9 positions to be randomized in a single round of mutagenesis. The resulting library is screened through a few rounds of selection against a ‘round 2′ target site. This iterative approach is continued to obtain meganucleases that cleave a chimeric target site containing one half of the original target site for a parental enzyme and one half of a DNA sequence of interest (‘half’ altered target site) (Fig. 4). Half protein domains that are placed on half sites containing base-pair substitutions are shuffled to obtain variant meganucleases that target a ‘fully’ altered site.
Design degenerate primers as described above (also refer to Fig. 5B).
To generate sequences containing partial ORFs of variant endonucleases with particular residues randomized, carry out PCR as described in Subheading 3.2.1, Step 2 with the following modification: an adaptor-specific PCR product that is purified by gel extraction is used as a template (1-3 μl/reaction). A fragment containing 2 copies of a new target site and one containing the T7 promoter and the ribosome binding site are also PCR-amplified in parallel (refer to Subheading 3.2.1, Step 2 and Subheading 3.2.2, Step 27).
Construct a library as described in Subheading 3.2.1, Step 3-10.
Carry out selection as described in Subheadings 3.2.2. and 3.2.3.
Repeat Steps 1-4 to create variant endonucleases that cleave each of two ‘half’ altered target sites (see Fig. 4).
- Using adaptor-specific PCR products that are recovered in the last round of selections against ‘half’ altered target sites, PCR-amplify sequences encoding half domains of engineered enzymes (that are placed on half sites containing base-pair substitutions).
5 × Phusion HF Buffer 10 μl 10 mM dNTPs mix 1 μl Phusion High-Fidelity DNA Polymerase 0.5 μl 10 pmol/μl primers (see Note 9) 2.5 μl each Gel-extracted DNA 3 μl Water 30.5 μl Construct a library as described in Subheading 3.2.2, Step 28-30.
Carry out selection as described in Subheadings 3.2.2. and 3.2.3.
3.3. Selection using two-plasmid cleavage assay in bacteria
Two-plasmid cleavage assay that was originally developed by Doyon et al.(27) is carried out to isolate variant endonucleases that display substantial cleavage activity against a ‘fully’ altered target site in bacteria. In this assay, bacterial cells are transformed with two plasmids, pEndo and pCcdB: pEndo carries a redesigned meganuclease gene that is driven by the arabinose-inducible promoter, and pCcdB, which contains 2 copies of a ‘fully’ target site in two different positions, encodes a bacterial toxin (CcdB) that is expressed in the presence of IPTG (Fig. 1). A DSB generated by an expressed endonuclease at a ‘fully’ altered target site triggers degradation of the pCcdB plasmid by the endogenous RecBCD complex in transformed cells, resulting in colony formation on agar plates containing IPTG. Stringency can be modulated by varying both the temperature (30 or 37°C) and the time period in which an engineered meganuclease is expressed prior to induction of the ccdB gene expression (21,23).
3.3.1. Preparing competent cells harboring a pCcdB reporter plasmid containing ‘fully’ altered target sites for electroporation
- Synthesize two complementary oligonucleotides and anneal to generate double-stranded fragments containing 2 copies of a ‘fully’ altered target site with cohesive ends.
4 × Annealing buffer 4 μl 1 mM complementary oligonucleotides (see Note 10) 1.6 μl each Water 8.8 μl Digest 1 μg of the pCcdB plasmid with AflIII and BglII, and purify it using DNA Clean & Concentrator-5. Elute it in 10 μl of supplied Elution Buffer.
100-fold dilute the annealed DNA in water, and carry out a ligation in 10 μl of 1 × Quick Ligation Buffer containing 1 μl of the diluted, double-stranded oligonucleotide, 1 μl of approximately 20-30 ng/μl the linearized pCcdB plasmid and 0.5 μl of Qiuck T4 DNA ligase at room temperature for 15 minutes.
Transform NovaXGF’ competent cells with 1 μl of the reaction mixture, and spread cells on the LB plates containing 33 μg/mL chloramphenicol and 0.5% glucose (Note 11).
Purify pCcdB with 2 copies of a ‘fully’ altered target site (Note 12), and digest approximately 1 μg of the plasmid with NheI and SacII.
Clean up the linearized plasmid using DNA Clean & Concentrator-5, and carry out ligation and transformation as described in Step 3 in order to incorporate additional two copies of the same target site into the pCcdB plasmid.
Purify the pCcdB plasmid with 4 copies of an identical target site (Note 12), and verify sequences of the target sites and their flanking regions using the CcdB_seq1 primer and the CcdB_seq2 primer.
Transformed NovaXGF’ competent cells with 0.5 μl of a sequence-verified pCcdB plasmid, and spread cells on the LB plates containing 33 μg/mL chloramphenicol and 0.5 % glucose.
Inoculate a few colonies in 10 mL of the SBG medium supplemented with 33 μg/mL chloramphenicol, and grow cells at 37°C overnight.
Transfer all cells grown overnight into 500 mL of the SBG medium supplemented with 33 μg/mL chloramphenicol, and continue to culture cells at 37°C until O.D.600nm reaches 0.6-1.0.
Incubate the medium on ice for 15 minutes, and harvest cells at 2,700 × g for 15 minutes at 4E mL of 1 mM Hepes (pH 7.0).
Harvest cells at 2,700 × g for 15 minutes at 4°C, decant the supernatant, and gently suspend the pellet with 50 mL of 1 mM Hepes (pH 7.0).
Harvest cells at 2,700 × g for 15 minutes at 4°C, decant the supernatant, and gently suspend the pellet with 20 mL of 10 %(v/v) glycerol.
Harvest cells at 2,700 × g for 15 minutes at 4°C, decant the supernatant, and gently suspend the pellet with 1 mL of 10 %(v/v) glycerol.
Aliquot 50 μl of competent cells in each tube, which is frozen in liquid nitrogen, and stored at −80°C until use.
3.3.2. Selection of variant endonucleases that substantially cleave a ‘fully’ altered target site in bacteria
- PCR-amplify ORFs of meganuclease genes from a gel-extracted, adaptor-specific PCR product generated in the last round of selection against a ‘fully’ altered target site.
5 × Phusion HF Buffer 10 μl 10 mM dNTPs mix 1 μl Phusion High-Fidelity DNA Polymerase 0.5 μl 10 pmol/μl PCR primers (see Note 13) 2.5 μl each Adaptor-specific PCR product 3 μl Water 30.5 μl Digest approximately 1 μg of the pEndo plasmid with NcoI and NotI.
Separate the PCR-amplified ORFs and the linearized plasmid on a 1% agarose-TAE gel, and purify them using Gel DNA Recovery Kit. Elute them in 10 μl of Elution Buffer.
Separate 0.5 μl each of these two DNA fragments along with 5 μl of 1kb DNA ladder on an agarose gel, and quantify the gel-extracted DNA samples using a densitometry software.
- Set up the following reaction mixture to carry out Gibson Assembly:
2 × Gibson Assembly Master Mix 5 μl PCR fragment encoding variant endonucleases 30-40 ng Linearized pEndo plasmid 60-75 ng Water Added to make a final volume of 10 μl Incubate the reaction mixture at 50°C for an hour, and transform 50 μl of chemical competent cells (such as DH5α and DH10B) with 4 μl of the Gibson Assembly reaction mixture. Spread all cells on a two LB plates containing 50 μg/mL ampicillin.
Add 1-2 mL of sterilized water on each plate (where several hundred colonies are expected to be observed), scrape off colonies using a cell scraper, and collect cells in two 2-mL microcentrifuge tube.
Purify the pEndo library using two spin columns supplied with QIAprep Spin Miniprep Kit, and combine DNA eluted from the two columns.
Thaw an aliquot (50 μl) of NovaXGF’ competent cells that harbor pCcdB containing 4 copies of a ‘fully’ altered target site on ice, and quickly transform the cells (50 μl) with 30-50 ng of the pEndo library by electroporation (Note 14).
Immediately suspend the transformants with 1 mL of 2 × YT medium, and transfer all the cell suspension into a 50-mL conical tube.
Incubate cells at 37°C for 30 minutes, and add 10 mL of 2 × YT medium containing 100 μg/mL carbenicillin and 0.02 %(w/v) L-arabinose to induce expression of meganucleases encoded on pEndo.
Culture cells at 30°C for 4 hours, and harvest them at 1,100 × g for 5 minutes.
Resuspend cells in 1 mL of sterilized water, and spread 50 μl of the cell suspension on the 2-3 selective plates (Note 15). To calculate the transformation efficiency, dilute cells by 1,000-fold in sterilized water and spread 50 μl of the dilution on the control plate.
Incubate the plates at 30°C for approximately 36 hours.
Scrape off all colonies surviving on the selective plates, and purify plasmids using spin miniprep kit. If multiple spin columns are used, combine all eluted samples.
- To generate a PCR fragment encoding variant meganucleases, set up the following PCR reaction mixture:
5 × Phusion HF Buffer 10 μl 10 mM dNTPs mix 1 μl Phusion High-Fidelity DNA Polymerase 0.5 μl 10 pmol/μl PCR primers used in Step 1 2.5 μl each Purified plasmid 1 μl Water 32.5 μl To carry out selection under the same conditions, repeat Steps 3-14.
To reconstruct the pEndo library, follow Steps 15 and 16, then Steps 3-8.
Carry out electroporation as described in Step 9.
Suspend the transformants with 1 mL of 2 × YT medium supplemented with 0.02% (w/v) L-arabinose.
Transfer all the cell suspension into a 50-mL conical tube, and dilute it with 10 mL of the same medium.
Grow the cells at 37°C for one hour, and harvest cells from 1 mL of the culture at 1,100 × g for 5 minutes.
Resuspend cells in 0.5 mL of sterilized water, and spread 50 μl of the cell suspension on the selective plate (containing 0.4 mM IPTG and 0.02% (w/v) L-arabinose). To calculate the transformation efficiency, dilute cells by 200-fold in sterilized water and spread 50 μl of the dilution on the control plate.
Incubate the plates at 37°C for approximately 20 hours.
To carry out the second round of selection under the same conditions, follow Steps 18-24.
Randomly pick up colonies surviving on the selective plate, separately suspend them in 10 μl of water, and transfer 1 μl each of the cell suspension into a PCR tube.
Heat the PCR tubes at 95°C on a thermal cycler for 5 minutes with the lids open.
- Set up the following PCR premix (per 6 colonies):
5 × Colorless GoTaq Reaction Buffer 20 μl 10 mM dNTPs mix 2 μl GoTaq DNA polymerase (5 units/μl) 0.5 μl 10 pmol/μl the Endo_colonyPCR_fwd primer and
the Endo_colony PCR_rev primer2 μl each Water 73.5 μl Sequence colony PCR products using the Endo_seq_fwd primer and the Endo_seq_rev primer.
Test individual pEndo plasmids under the same conditions used in the final round of selection.
3.3.3. Supplementary protocol: random mutagenesis using error-prone PCR
If few colonies survive on the selective plates in Subheading 3.3.2, Step 17 (Note 16), a time period in which an engineered meganuclease is expressed prior to induction of the ccdB gene expression may be increased to 16 hours in order to reduce a level of stringency. Then, random mutagenesis may be introduced across the entire ORFs of selected variant genes to increase protein stability and/or overall activity.
Transform NovaXGF’ competent cells that harbor the pCcdB plasmid containing ‘fully’ altered target sites (50 μl) with 30-50 ng of the pEndo library (prepared in Subheading 3.3.2, Step 8) by electroporation.
Follow Subheading 3.3.2, Steps 10-11, and incubate cells at 30°C for 16 hours.
Make a serial dilution of the culture with sterilized water (1:102-1:105), and spread 50 μl of each dilution on the selective plate. To calculate the transformation efficiency, spread 50 μl of the culture that is diluted by 106-fold on the control plate.
Incubate the plates at 30°C for approximately 36 hours.
Recover meganuclease genes as described in Subheading 3.3.2, Steps 15 and 16, and reconstruct a pEndo library as described in and Subheading 3.3.2, Steps 3-8.
Carry out selection under the same conditions as in the previous round (refer to Steps1-4).
Purify plasmids from cells surviving on the selective plates as shown in Subheading 3.3.2, Steps 15.
- To generate a PCR fragment containing meganuclease genes, set up the following PCR reaction mixture:
5 × Phusion HF Buffer 10 μl 10 mM dNTPs mix 1 μl Phusion High-Fidelity DNA polymerase 0.5 μl 10 pmol/μl PCR primers used in Subheading 3.3.2, Step 28 2.5 μl each Purified plasmids 1 μl Water 32.5 μl Clean up the PCR product using DNA Clean & Concentrator-5, and elute DNA in 20 μl of supplied Elution Buffer.
- To carry out error-prone PCR using GeneMorph II Random Mutagenesis Kit, set up the following reaction mixtures with two different amounts of the PCR template purified above:
10 × Mutazyme II reaction mixture 5 μl 40 mM dNTP mix (supplied with the
kit)1 μl Mutazyme II DNA polymerase 1 μl 125 ng/μl PCR primers used in
Subheading 3.3.2, Step 11 μl each Purified PCR fragment 20 ng or 100 ng Water Added to make a final volume of 50 μl Digest the pEndo plasmid and purify the above PCR samples and the linearized plasmid as described in Subheading 3.3.2, Steps 3-4.
- To carry out Gibson Assembly, set up the following reaction mixtures:
2 × Gibson Assembly Master Mix 10 μl PCR fragment amplified from 20 ng or 100 ng of a PCR template 60-80 ng Linearized pEndo plasmid 120-150 ng Water Added to make a final
volume of 20 μl Incubate the reaction mixtures at 50°C for an hour, and clean up the two reaction mixtures individually, using DNA Clean & Concentrator-5. Elute DNA in 10 μl of supplied Elution Buffer.
Tramsform 50 μl of ElectroMax DH10B T1 phage-resistant competent cells with 3 μl each of the purified Gibson Assembly samples by electroporation, and separately culture the cells in 2 mL of SOC medium at 37°C for an hour.
To calculate the transformation efficiency, mix 2 μl of the culture with 100 μl of sterilized water and spread 50 μl of the dilution on a LB plate supplemented with 50 μg/mL ampicillin (100-mm dish). Then, spread the rest of the cells, using glass beads, on LB agar plates supplemented with 50 μg/mL ampicillin (160-200 μl/150-mm petri dish), and incubate all the plates at 37°C overnight.
To calculate the total number of transformants, count colonies on a LB plate where 50 μl of 50-fold dilution of competent cells are spread, and multiply the number by 2,000. Hundreds of thousands of transformants are generally obtained for each of two error-prone PCR samples. Scrape off all colonies that are formed on 150-mm plates, and purify a pEndo library plasmid using a plasmid maxiprep kit.
To screen meganuclease genes subjected to random mutagenesis, follow Subheading 3.3.2, Steps 9-30.
4. Notes
We optimize the codon usage of a meganuclease gene for bacterial expression, using DNAworks (http://helixweb.nih.gov/dnaworks/), and synthesize the ORF (containing a start codon 5′-ATG-3′ and a stop codon 5′-TAA-3′) with extra sequences at both termini, which are required for insertion of the endonuclease gene into pET-21d(+) by Gibson Assembly. We purchase two gBlocks, each of which contains approximately a half of the ORF with approximately 20 base pairs of an overlap region (from Integrated DNA Technologies), and assemble these two fragments with the NcoI/NotI-digested pET-21d(+) plasmid using 2 × Gibson Assembly Master Mix.
Amino-acid residues included in a ‘contact module’ at every 3 consecutive base-pair positions are deposited in the LAHEDES webserver (http://homingendonuclease.net), and can be found by clicking the name of a homing endonuclease in the ‘Homing Endonuclease Browser’ under the ‘Browse’ menu.
If PCR fails, increase a temperature during template-primer annealing up to 65°C.
Use new TAE buffer when PCR fragments to be purified are separated on an agarose gel.
Washed a spin column twice (with 750 μl of Buffer PE and with 250 μl of the same buffer), and incubate it with supplied Buffer EB for 2 minutes at room temperature before centrifugation.
Use a positive-displacement pipettor (e.g. MICROMAN from Gilson) to add an accurate amount of surfactant.
If no DNA band is specifically amplified from a library that is subjected to in vitro selection, carry out ligation with different DNA adaptor(s). The efficiencies of PCR amplification of meganuclease genes linked to DNA adaptors are greatly dependent on sequences of adaptors and meganucelase genes.
If screening at 42°C fails, reduce temperature to 37°C.
To amplify 3′-terminal half of engineered meganuclease genes, use the MidRev primer and a forward primer that is annealed to a nucleotide sequence corresponding to a loop connecting two half domains. To generate a fragment containing 5′-terminal half of variant endonuclease genes, use the Up4 primer and a primer that is reverse complement of the forward primer used to PCR-amplify 3′-terminal half genes. The resulting PCR products share an approximately 20 base-pair region that can be hybridized in the next round of PCR.
Sequences of oligonucleotides to be inserted into pCcdB are as follows: 5′-CGTGT-(2 copies of a target site)-A-3′ and 5′-GATCT-(2 copies of a reverse complement target site)-A-3′ (to be inserted between AflII and BglII sites), and 5′-CTAGC-(2 copies of a target site)-CCGC-3′ and 5′-GG-(2 copies of a reverse complement target site)-G-3′ (to be inserted between NheI and SacII sites).
We use chemical competent cells of this E. coli strain that are prepared by a standard protocol.
We routinely purify pCcdB plasmids using spin miniprep kit from cells grown overnight in 3.5 mL of the LB medium containing 33 μg/mL chloramphenicol and 0.5% glucose.
Primers used are as follows: 5′-CTT TAA GAA GGA GAT ATA CCC ATG-(5′ terminal sequence of a meganuclase gene sequence)-3′ and 5′-CCA ATT AAC CAA TTC TGA GCG GCC GCT TA-(reverse complement of a 3′ terminal meganuclease gene sequence)-3′. A start codon (5′-ATG-3′) and a stop codon (5′-TAA-3′) are underlined.
A cuvette is chilled on ice before electroporation.
Adjust the dilution rate and the volume of sterilized water to spread at least 3-fold excess of cells over the complexity of a library on the selective plates.
The survival rate of cells harboring the intact pCcdB plasmid on the selective plates is generally very low (<0.1%). If a subset of variant endonucleases encoded by a library are capable of eliminating this reporter plasmid in cellulo, the survival rate (calculated by dividing the number of cells that form colonies on the selective plates by that on the control plates) is significantly high over the background level in the second round of selection.
References
- 1.Kolb AF, Coates CJ, Kaminski JM, Summers JB, Miller AD, Segal DJ. Site-directed genome modification: nucleic acid and protein modules for targeted integration and gene correction. Trends in biotechnology. 2005;23:399–406. doi: 10.1016/j.tibtech.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188:773–782. doi: 10.1534/genetics.111.131433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nature reviews. Genetics. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 4.Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333:1843–1846. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- 5.Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li T, Huang S, Jiang WZ, Wright D, Spalding MH, Weeks DP, Yang B. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic acids research. 2011;39:359–372. doi: 10.1093/nar/gkq704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnould S, Perez C, Cabaniols JP, Smith J, Gouble A, Grizot S, Epinat JC, Duclert A, Duchateau P, Paques F. Engineered I-CreI derivatives cleaving sequences from the human XPC gene can induce highly efficient gene correction in mammalian cells. Journal of molecular biology. 2007;371:49–65. doi: 10.1016/j.jmb.2007.04.079. [DOI] [PubMed] [Google Scholar]
- 10.Stoddard BL. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure. 2011;19:7–15. doi: 10.1016/j.str.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Segal DJ, Meckler JF. Genome engineering at the dawn of the golden age. Annual review of genomics and human genetics. 2013;14:135–158. doi: 10.1146/annurev-genom-091212-153435. [DOI] [PubMed] [Google Scholar]
- 12.Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, Paques F. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Current gene therapy. 2011;11:11–27. doi: 10.2174/156652311794520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brunet E, Simsek D, Tomishima M, DeKelver R, Choi VM, Gregory P, Urnov F, Weinstock DM, Jasin M. Chromosomal translocations induced at specified loci in human stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:10620–10625. doi: 10.1073/pnas.0902076106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee HJ, Kweon J, Kim E, Kim S, Kim JS. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome research. 2012;22:539–548. doi: 10.1101/gr.129635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sollu C, Pars K, Cornu TI, Thibodeau-Beganny S, Maeder ML, Joung JK, Heilbronn R, Cathomen T. Autonomous zinc-finger nuclease pairs for targeted chromosomal deletion. Nucleic acids research. 2010;38:8269–8276. doi: 10.1093/nar/gkq720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoddard BL. Homing endonuclease structure and function. Quarterly reviews of biophysics. 2005;38:49–95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 17.Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, Scharenberg AM. Tracking genome engineering outcome at individual DNA breakpoints. Nature methods. 2011;8:671–676. doi: 10.1038/nmeth.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daboussi F, Zaslavskiy M, Poirot L, Loperfido M, Gouble A, Guyot V, Leduc S, Galetto R, Grizot S, Oficjalska D, et al. Chromosomal context and epigenetic mechanisms control the efficacy of genome editing by rare-cutting designer endonucleases. Nucleic acids research. 2012;40:6367–6379. doi: 10.1093/nar/gks268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao H, Smith J, Yang M, Jones S, Djukanovic V, Nicholson MG, West A, Bidney D, Falco SC, Jantz D, et al. Heritable targeted mutagenesis in maize using a designed endonuclease. The Plant journal : for cell and molecular biology. 2010;61:176–187. doi: 10.1111/j.1365-313X.2009.04041.x. [DOI] [PubMed] [Google Scholar]
- 20.Grizot S, Smith J, Daboussi F, Prieto J, Redondo P, Merino N, Villate M, Thomas S, Lemaire L, Montoya G, et al. Efficient targeting of a SCID gene by an engineered single-chain homing endonuclease. Nucleic acids research. 2009;37:5405–5419. doi: 10.1093/nar/gkp548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takeuchi R, Lambert AR, Mak AN, Jacoby K, Dickson RJ, Gloor GB, Scharenberg AM, Edgell DR, Stoddard BL. Tapping natural reservoirs of homing endonucleases for targeted gene modification. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13077–13082. doi: 10.1073/pnas.1107719108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boissel SJ, Astrakhan A, Jarjour J, Adey A, Shendure J, Stoddard BL, Certo MT, Baker D, Scharenberg AM. MegaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. 2013 doi: 10.1093/nar/gkt1224. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeuchi R, Choi M, Stoddard BL. Efficient engineering of multiple meganucleases and MegaTALs using bioinformatics and in vitro compartmentalization. 2013 Submitted. [Google Scholar]
- 24.Miller OJ, Bernath K, Agresti JJ, Amitai G, Kelly BT, Mastrobattista E, Taly V, Magdassi S, Tawfik DS, Griffiths AD. Directed evolution by in vitro compartmentalization. Nature methods. 2006;3:561–570. doi: 10.1038/nmeth897. [DOI] [PubMed] [Google Scholar]
- 25.Tawfik DS, Griffiths AD. Man-made cell-like compartments for molecular evolution. Nature biotechnology. 1998;16:652–656. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- 26.Zheng Y, Roberts RJ. Selection of restriction endonucleases using artificial cells. Nucleic acids research. 2007;35:e83. doi: 10.1093/nar/gkm410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doyon JB, Pattanayak V, Meyer CB, Liu DR. Directed evolution and substrate specificity profile of homing endonuclease I-SceI. Journal of the American Chemical Society. 2006;128:2477–2484. doi: 10.1021/ja057519l. [DOI] [PubMed] [Google Scholar]
- 28.Taylor GK, Petrucci LH, Lambert AR, Baxter SK, Jarjour J, Stoddard BL. LAHEDES: the LAGLIDADG homing endonuclease database and engineering server. Nucleic acids research. 2012 doi: 10.1093/nar/gks365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor GK, Petrucci LH, Lambert AR, Baxter SK, Jarjour J, Stoddard BL. LAHEDES: the LAGLIDADG homing endonuclease database and engineering server. Nucleic acids research. 2012;40:W110–116. doi: 10.1093/nar/gks365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molina R, Redondo P, Stella S, Marenchino M, D’Abramo M, Gervasio FL, Epinat JC, Valton J, Grizot S, Duchateau P, et al. Non-specific protein-DNA interactions control I-CreI target binding and cleavage. Nucleic acids research. 2012;40:6936–6945. doi: 10.1093/nar/gks320. [DOI] [PMC free article] [PubMed] [Google Scholar]